

Abstract
Background and Purpose
The liver X receptor (LXR) agonist T317 reduces atherosclerosis but induces fatty liver. Metformin activates energy metabolism by activating AMPKα. In this study, we determined if interactions between metformin and T317 could inhibit atherosclerosis without activation of hepatic lipogenesis.
Experimental Approach
Apolipoprotein E‐deficient mice were treated with T317, metformin or both agents, in a high‐fat diet for 16 weeks. Then, samples of aorta, liver, macrophage and serum were collected to determine atherosclerotic lesions, fatty liver, lipid profiles and expression of related proteins. Techniques used included immunohistochemistry, histology, qRT‐PCR and Western blot.
Key Results
T317 inhibited en face and aortic root sinus lesions, and the inhibition was further enhanced by addition of metformin. Co‐treatment with metformin and T317 increased lesion stability, by increasing collagen content, and reducing necrotic cores and calcification. Formation of macrophages/foam cells and their accumulation in arterial wall were inhibited by the co‐treatment, which was accompanied by increased ABCA1/ABCG1 expression, reduced monocyte adhesion and apparent local proliferation of macrophages. Metformin blocked T317‐induced fatty liver by inhibiting T317‐induced hepatic LXRα nuclear translocation and expression of lipogenic genes and by activating AMPKα. Moreover, co‐treatment with T317 and metformin improved triglyceride metabolism by inducing expression of adipose triglyceride lipase, hormone‐sensitive lipase, PPARα and carnitine acetyltransferase and by inhibiting acyl‐CoA:diacylglycerol acyltransferase 1 expression.
Conclusions and Implications
Co‐treatment with T317 and metformin inhibited the development of atherosclerosis without activation of lipogenesis, suggesting that combined treatment with T317 and metformin may be a novel approach to inhibition of atherosclerosis.
Abbreviations
- ABCA1/G1
ATP‐binding cassette transporter A1/G1
- ACC1
acetyl‐CoA carboxylase 1
- AICAR
5‐aminoimidazole‐4‐carboxamide ribonucleotide
- ALT
alanine aminotransferase
- ALP
alkaline phosphatase
- AMPKα
AMP‐activated protein kinase α
- AST
aspartate aminotransferase
- ATGL
adipose triglyceride lipase
- CAT
carnitine acetyltransferase
- CHD
coronary heart disease
- DGAT1
acyl‐CoA:diacylglycerol acyltransferase 1
- FASN
fatty acid synthase
- HSL
hormone‐sensitive lipase
- ICAM‐1
intercellular adhesion molecule 1
- LXR
liver X receptor
- NgBR
Nogo‐B receptor
- SCD‐1
stearoyl‐CoA desaturase 1
- SMA
smooth muscle α‐actin
- SREBP1c
sterol‐responsive element binding protein 1c
- TG
triglyceride
- VCAM‐1
vascular cell adhesion molecule‐1
Introduction
Atherosclerosis is one of the underlying causes of coronary heart disease (CHD). It develops chronically due to lipid metabolism disorders and chronic inflammation (Patel et al., 2015). Lipid‐laden macrophages or foam cells are the prominent part of atherosclerotic lesions (Moore and Tabas, 2011). Differentiation of macrophage/foam cells depends on both cellular lipid accumulation and cholesterol efflux. Although macrophage scavenger receptors can enhance uptake of modified LDL, the expression of molecules responsible for cholesterol efflux, such as the ATP‐binding cassette transporters A1 (http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=756) or G1 (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=155#791), inhibits foam cell formation (Tall et al., 2008; Dai et al., 2016).
Expression of ABCA1 and ABCG1 can be activated by the liver X receptor (LXR), a ligand‐activated nuclear transcription factor (Zhao and Dahlman‐Wright, 2010). LXR contains two isoforms, http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=602&familyId=89&familyType=NHR and http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=601&familyId=89&familyType=NHR, with distinct functions, in that LXRα mainly activates hepatic lipogenesis while LXRβ induces macrophage cholesterol metabolism (Zhao and Dahlman‐Wright, 2010). Synthetic LXR ligands, such as http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2755) and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2754, can activate both LXRα and LXRβ. Therefore, administration of T317, which has full LXR functions, to pro‐atherogenic mice significantly inhibits atherosclerosis while substantially inducing fatty liver and hypertriglyceridemia (Schultz et al., 2000; Joseph et al., 2002; Chisholm et al., 2003; Terasaka et al., 2003). These adverse lipogenic effects limit the application of LXR ligands for the treatment of atherosclerosis. Based on the differences of tissue expression profiles and functions between LXRα and LXRβ, efforts have been made to identify selective LXRβ modulators which can reduce atherosclerosis without lipogenic effects. Unfortunately, the progress of this search has been seriously hampered because of the high homologies for ligand and DNA binding domains between LXRα and LXRβ (Ratni and Wright, 2010).
http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4779 is a first‐line treatment for Type 2 diabetes (Maruthur et al., 2016). The anti‐diabetic actions of metformin include suppressing hepatic glucose production, increasing insulin sensitivity, enhancing peripheral glucose uptake and releasing insulin‐suppressed fatty acid oxidation (Hundal et al., 2000; Collier et al., 2006; Rena et al., 2013; Madiraju et al., 2014). Metformin also induces the activity of AMP‐activated protein kinase α (http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=474) and this kinase plays a major role in the actions of metformin already outlined(Zhou et al., 2001). In addition to its anti‐diabetes effects, metformin may have other functions including cardioprotection. Although some clinical trials indicate that metformin may reduce CHD events in diabetic patients, such as myocardial infarction, the cardioprotective effects of metformin in non‐diabetic patients are still in doubt (UKPDS Group, 1998aa; Hemmingsen et al., 2012; Johnson et al., 2002). Studies with animal models also suggest that metformin and some other AMPK agonists can inhibit atherosclerosis, mainly in the context of diabetes (Li et al., 2011; Vasamsetti et al., 2015; Wang et al., 2017). The reports summarised above imply that metformin is not a promising strategy for atherosclerosis treatment as a monotherapy.
We have previously reported that addition of a http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=514 inhibitor to a LXR ligand can synergize with a LXR ligand to inhibit atherosclerosis. More importantly, MEK1/2 inhibitors block LXR‐induced lipogenesis, thereby eliminating LXR‐induced undesirable side effects (Chen et al., 2015). In another study, we have found that genetic deletion of hepatic Nogo‐B receptor (NgBR) expression can induce fatty liver and hypertriglyceridemia by a mechanism in which deficiency of hepatic NgBR expression can activate LXRα by inactivating AMPKα (Hu et al., 2016). Based on the findings above, we now have tested the possibility that metformin could selectively antagonize LXRα‐activated lipogenesis and that combined treatment with metformin and T317 could inhibit the development of atherosclerosis, in pro‐atherogenic mice, without activation of lipogenesis.
Methods
Cell culture
All cell lines were purchased from ATCC (Manassas, VA, USA). HepG2 cells, a human hepatic cell line, and primary hepatocytes isolated from apolipoprotein E‐deficient (apoE−/−) mice were cultured in complete DMEM medium containing 10% FBS, 50 μg·mL−1 penicillin/streptomycin and 2 mM glutamine. HepG2 cells (~90% confluence) received treatment in serum‐free medium. HUVECs were cultured in VascuLife basal medium containing VEGF LifeFactors Kit (Lifeline Cell Technology, Frederick, MD, USA). THP‐1 cells, a human monocytic cell line, were cultured in complete RPMI1640 medium containing 10% FBS, 50 μg·mL−1 penicillin/streptomycin and 2 mM glutamine.
HepG2 cells lacking AMPKα, LXRα or LXRβ expression were established using the clustered regulatory interspaced short palindromic repeat (CRISPR)/CRISPR‐associated protein 9 (Cas9) technology (Ran et al., 2013) with the guide oligos which were designed using the online CRISPR Design Tool (http://tools.genome-engineering.org). The sequences of the guide oligos are as follows: LXRα (NR1H3), TCGGCTTCGCAAATGCCGTC; LXRβ (NR1H2), CCCCGGCAGGCATAGCGCC; and AMPKα1, ACCGTTGGCAAACATGAATTGAC. After ligation, HepG2 cells were transfected with the plasmid of pSpCas9 (BB)‐2A‐Puro vector or vector ligated with the guide oligos. The selection of mutated clonal cell lines was completed using the standard protocol, and the knockout of target gene expression was confirmed by Western blots. The cells lacking AMPKα, LXRα or LXRβ expression were defined as Cas9‐AMPKα, Cas9‐LXRα or Cas9‐LXRβ cells, and the corresponding control cells were defined as Cas9‐NS cells.
In vivo studies
All animal care and experimental protocols for in vivo studies conformed to the Guide for the Care and Use of Laboratory Animals published by the NIH (NIH publication no. 85‐23, revised 1996) and were approved by the Ethics Committee of Nankai University. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
ApoE−/− mice (males, ~8‐week‐old, ~22 g bodyweight) with C57BL/6J background and wild‐type C57BL/6J mice (males, ~8‐week‐old, ~22 g bodyweight) were purchased from the Animal Center of Nanjing University (Nanjing, China). The animals were housed in the SPF units of the Animal Center at the College of Life Sciences, Nankai University, at 23 ± 1°C, with a relative humidity of 60–70% and 12 h light/dark cycles. The animals had free access to water and normal chow (NC) or a high‐fat diet (HFD: 21% fat plus 0.5% cholesterol) during the treatment. The mice were kept in standard cages (≤5 mice per cage). Mice were allowed to acclimatize to their housing environment for at least 7 days before experiments. The number of mice was not predetermined by a statistical method, and no mice were excluded for statistical analysis. The treatment during experiment was conducted in a blinded fashion. During the treatment, the animals were checked daily for intake of food and water, and bodyweight.
ApoE−/− mice were randomly divided into four groups (15 per group) and fed HFD, HFD containing T317 (1mg·day−1·kg−1), metformin (100 mg·day−1·kg−1) or T317 plus metformin, for 16 weeks respectively. At the end of experiment, all mice were killed by an overdose of 2,2,2‐tribromoethanol (640 mg·kg−1, i.p. injection), followed by collection of aorta, liver, peritoneal macrophage and blood samples. Serum was prepared to determine levels of total cholesterol (Total‐C), HDL‐C, LDL‐C, very LDL (VLDL)‐C and triglyceride (TG), and activities of aspartate aminotransferase (AST), alkaline phosphatase (ALP) and alanine aminotransferase (ALT) (Chen et al., 2015). Serum glucose levels were determined with the OneTouch glucometer and test strips (LifeScan, Milpitas, CA, USA). Serum TNF‐α, IL‐1β and IL‐6 levels were determined by ELISA using the commercially available kits purchased from ABclonal Inc. (Wuhan, China).
C57BL/6J mice were randomly divided into four groups (6 per group) and fed NC, NC containing GW3965 (40 mg·day−1·kg−1), metformin (100 mg·day−1·kg−1) or GW3965 plus metformin, for 2 weeks respectively. At the end of experiment, all mice were killed as described above. Liver and blood samples were collected to determine hepatic lipid content and serum glucose levels.
Lipid content in liver and hepatocytes was determined by Oil Red O staining with liver frozen sections or fixed cells; and by quantitation of triglyceride (TG) levels using the total lipid extract from liver samples or cells respectively. Expression of proteins for TG metabolism was determined by immunofluorescent staining with liver frozen sections (Chen et al., 2015).
The en face aortas were collected and used to prepare aortic root cross sections followed by determination of en face and sinus lesions with Oil Red O staining (Chen et al., 2015). All the images were obtained with a microscope and quantified lesion areas in en face aorta, aortic arch (AA), descending aorta (DA), thoracic aorta (TA), abdominal aorta (Ad A) and aortic root cross sections, respectively, using a computer‐assisted image analysis protocol (Photoshop CS3), by technicians who were blinded to the treatment. The lesion areas were expressed as μm2.
Necrotic core, thickness of fibrous cap, collagen content, calcification and expression of ABCA1, ABCG1, Ki‐67, MOMA‐2, CD68, smooth muscle α‐actin (SMA), intercellular adhesion molecule 1 (ICAM‐1) and vascular cell adhesion molecule‐1 (VCAM‐1) protein in lesion areas were determined by haematoxylin and eosin, Verhoeff‐Van Gieson (VVG), Alizarin Res S and immunofluorescent staining with aortic root cross sections, respectively (Chen et al., 2015).
Determination of foam cell formation in vitro and in vivo
In vitro, peritoneal macrophages collected from apoE−/− mice were plated on cover slips in 24‐well plates. After attachment, cells in serum‐free RPMI1640 medium were incubated with oxidized LDL (oxLDL) for 3 h to induce foam cell formation, followed by T317 or/and metformin treatment for 16 h. Cells were then incubated with medium containing both apo‐AI (5 μg·mL−1) and HDL (for 20 μg·mL−1) for 5 h. After apo‐AI and HDL were removed, cells were stained with Oil Red O solution (Chen et al., 2015).
To measure foam cell formation in vivo, peritoneal macrophages were collected by lavage with PBS from mouse abdominal cavity and plated on cover slips in 24‐well plates. The attached cells were fixed and stained with Oil Red O solution. Cells containing lipid droplets (>10 per cell) were considered as foam cells, and >10 fields per sample were counted.
Western blot and quantitative real‐time RT‐PCR (qRT‐PCR)
Total cellular and nuclear proteins were extracted from cells or a sample of liver (Chen et al., 2012). Protein expression of ABCA1, ABCG1, LXRα and LXRβ in macrophages; fatty acid synthase (FASN), sterol‐responsive element binding protein 1c (SREBP1c), AMPKα, acetyl‐CoA carboxylase 1 (ACC1), phosphorylated ACC1 (pi‐ACC1), acyl‐CoA:diacylglycerol acyltransferase 1 (DGAT1), adipose triglyceride lipase (ATGL), hormone‐sensitive lipase (HSL), http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=86, LXRα and LXRβ in HepG2 cells or in liver samples were determined by Western blot.
Total cellular RNA was extracted from cells or a sample of liver followed by determination of mRNA expression by qRT‐PCR with a reverse transcription kit (New England Biolabs, Ipswich, MA, USA), an SYBR green PCR master mix (Bio‐Rad, Los Angeles, CA, USA) and the primers with sequences listed in Table 1. Expression of stearoyl‐CoA desaturase 1 (SCD‐1), DGAT1, ATGL, HSL, PPARα, carnitine acetyltransferase (CAT), IL‐1β, TNFα, IL‐6, VCAM‐1 and ICAM‐1 mRNA was normalized to GAPDH mRNA in the corresponding samples.
Table 1.
Sequences of primers for qRT‐PCR
| Gene | Sense | Anti‐sense |
|---|---|---|
| ATGL | 5′‐GAGCCCCGGGGTGGAACAAGAT‐3′ | 5′‐AAAAGGTGGTGGGCAGGAGTAAGG‐3′ |
| CAT | 5′‐TGCTGCCAGAACCGTGGT‐3′ | 5′‐TCCAGGGATTGCTGAAGTGG‐3′ |
| DGAT1 | 5′‐GGTGCCCTGACAGAGCAGAT‐3′ | 5′‐CAGTAAGGCCACAGCTGCTG‐3′ |
| HSL | 5′‐TGTCAGGATACAGTGGGACG‐3′ | 5′‐CATGTTGTGGATGAGCCTTG‐3′ |
| ICAM1 | 5′‐AAGGTGTGATATCCGGTAGA‐3′ | 5′‐CCTTCTAAGTGGTTGGAACA‐3′ |
| IL‐1β | 5′‐GACCTTCCAGGATGAGGACA‐3′ | 5′‐AGCTCATATGGGTCCGACAG‐3′ |
| IL‐6 | 5′‐CTGCAGCCACTGGTTCTGT‐3′ | 5′‐CCAGAGCTGTGCAGATGAGT‐3′ |
| PPARα | 5′‐AGTTCGGGAACAAGACGTTG‐3′ | 5′‐CAGTGGGGAGAGAGGACAGA‐3′ |
| SCD‐1 | 5′‐TGGGTTGGCTGCTTGTG‐3′ | 5′‐GCGTGGGCAGGATGAAG‐3′ |
| TNFα | 5′‐TGGCCCAGGCAGTCAGA‐3′ | 5′‐GGTTTGCTACAACATGGGCTACA‐3′ |
| VCAM1 | 5′‐TGGGAAAAACAGAAAAGAGGTG‐3′ | 5′‐GTCTCCAATCTGAGCAGCAA‐3′ |
| GAPDH | 5′‐ACCCAGAAGACTGTGGATGG‐3′ | 5′‐ACACATTGGGGGTAGGAACA‐3′ |
Monocyte adhesion to endothelial cells
HUVECs were plated in 24‐well plates and pretreated with LPS (100 ng·mL−1) for 2 h. Cells were then treated with T317, metformin or T317 plus metformin in the presence of LPS overnight. The carboxyfluorescein succinimidyl ester (CFSE, 5 μmol·L−1)‐labelled THP‐1 cells were then added to HUVECs and co‐incubated for 1 h. The culture medium was aspirated to remove cells in suspension and washed three times with PBS. The numbers of THP‐1 cells adhering to HUVECs were observed with a microscope and counted with Image J (NIH). The number of total adherent cells in control groups (treated with LPS alone) was defined as 1. The changes of adherent cells in other groups by treatment were obtained by dividing the number of adherent cells in the corresponding group with the number of total adherent cells in the control group.
Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Data are presented as mean ± SEM except where indicated, and were generated from at least five independent experiments. The density of each captured image was quantified by a technician (blinded to the treatments) with segmentation colour‐threshold analysis using morphometry software (IP Lab, Scanalytics, Rockville, MD, USA) (Stein et al., 2010). For Western blot assays, the target band was normalized to GAPDH or to Lamin A/C in the corresponding sample to reduce variance. All values (control and test) were normalized to the mean value of the experimental control group. The data were expressed as folds of the control group's mean value. The data in normal distribution, which was determined by the 1‐sample K‐S of non‐parametric test with SPSS 22 software, were analysed by parametric statistics (one‐way ANOVA for more than two groups). For ANOVA, Bonferroni's post hoc test was performed for data with F at P < 0.05 and no significant variance inhomogeneity. Differences between group means were considered to be significant when P < 0.05.
Materials
Rabbit anti‐CD68, FASN, DGAT1, CAT, ATGL, HSL and GAPDH polyclonal antibodies, rat anti‐MOMA‐2 monoclonal antibody and mouse anti‐SMA monoclonal antibody were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Rabbit anti‐SREBP1c and ABCA1 polyclonal antibodies were purchased from Novus Biologicals (Littleton, CO, USA). Rabbit anti‐AMPKα, ACC1 and pi‐ACC1 polyclonal antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Rabbit anti‐ABCG1, ICAM‐1, PPARα, LXRα, LXRβ, Lamin A/C and VCAM‐1 polyclonal antibodies were purchased from Proteintech Group, Inc. (Rosemont, IL, USA). Rabbit anti‐Ki‐67 polyclonal antibody was purchased from Abcam (Cambridge, MA, USA). Goat anti‐rabbit IgG (whole molecule)‐FITC and goat anti‐rabbit IgG (H + L), F(ab) fragment‐Rhodamine antibodies were purchased from Sigma‐Aldrich (St Louis, MO, USA). TG assay kit was purchased from Wako Chemicals (Neuss, Germany). GW3965, T317, 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) and metformin were purchased from Cayman Chemical (Ann Arbor, MI, USA) and AK Scientific, Inc. (Union City, CA, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org/, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c).
Results
Metformin enhances the inhibition of atherosclerosis induced by T317
To determine if metformin could antagonize the lipogenesis induced by T317 while enhancing inhibition of atherosclerosis, apoE−/− mice were fed HFD or HFD containing T317, metformin or both T317 and metformin for 16 weeks. During the treatment, we routinely checked food and water intake, and body weight. We observed no differences between control group (HFD) and the groups receiving metformin, T317 or metformin plus T317.
At the end of treatment, we measured the aortic lesions. Compared with mice fed HFD alone, the en face aortic lesions in mice receiving T317 or metformin alone were inhibited by 43% and 30% (Figure 1A, B). These lesions were further inhibited by about 65% after co‐treatment with metformin and T317, suggesting that metformin enhanced the inhibition of atherosclerosis induced by the LXR agonist. Meanwhile, in different segments of aortas (Figure 1C), the results showed that T317 alone inhibited lesions in the AA, DA, TA and Ad A (Figure 1D). Metformin alone also inhibited lesions in the TA and Ad A. However, metformin inhibited lesions in the AA with a much weaker effect than T317, while having no effect on lesions in other parts of the aorta.
Figure 1.
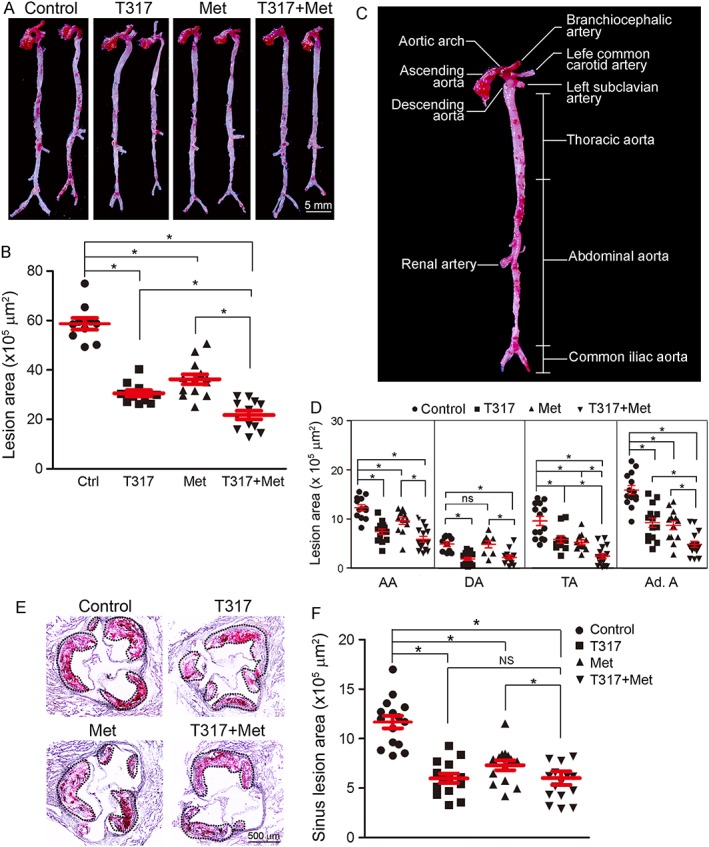
Metformin enhances the inhibition of atherosclerosis by T317. ApoE−/− mice in four groups (15 per group) received the following treatment for 16 weeks: Control, HFD; T317, HFD containing T317 (equivalent to 1 mg · kg−1·day−1); Met, HFD containing metformin (100 mg · kg−1·day−1); and T317 + Met, HFD containing both T317 and metformin. After treatment, aortas were collected for the following assays: lesions in en face aortas were determined by Oil Red O staining (A) and quantified by a computer‐assisted image analysis protocol (B). The lesion areas in different segments of aortas were also quantified (C, D). (E, F) Lesions in aortic root cross sections were determined by Oil Red O staining (E) and quantified (F). Lesion areas were expressed as μm2. *P < 0.05, significantly different as indicated; NS/ns: not significantly different (n = 15).
Similarly, T317 alone inhibited sinus lesions (Figure 1E, F) and the co‐treatment with metformin further enhanced the inhibition induced by T317. Taken together, the data in Figure 1 suggests that the addition of metformin could further enhance the inhibition of the development of atherosclerosis by T317 alone.
Co‐treatment with T317 and metformin enhances lesion plaque stability and decreases mineralization
The size of the necrotic core and the thickness of the fibrous cap affect the vulnerability of plaques. The necrotic core (Figure 2A), defined as the area containing cholesterol crystal and apoptotic foam cells or/and vascular smooth muscle cells (VSMCs), was reduced by T317, metformin or their co‐treatment (Figure 2A, B). In contrast, the thickness of fibrous cap (Figure 2A) which consists mainly of VSMCs and extracellular matrix to cover the necrotic core, was increased by the treatments (greater than threefold; Figure 2A, C).
Figure 2.
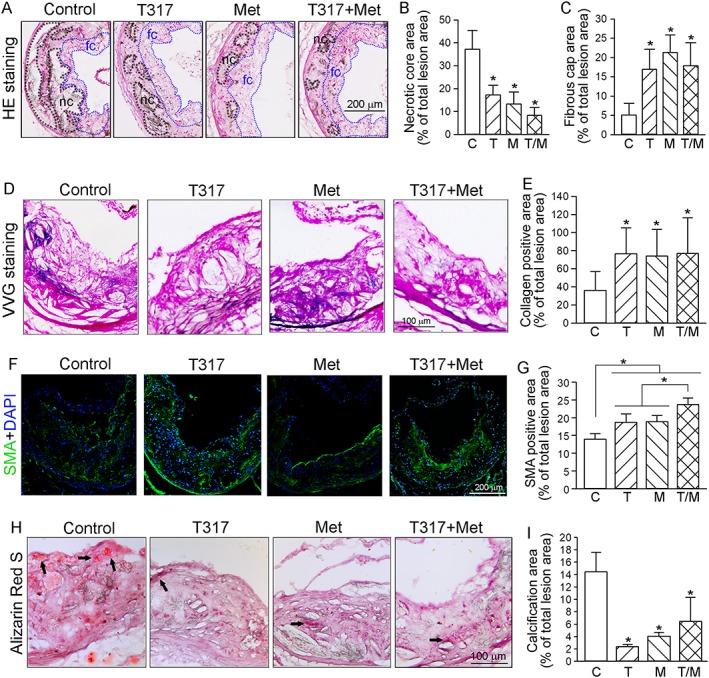
Co‐treatment of metformin and T317 maintains structural integrity of aortic wall and inhibits calcium deposits in aortic lesion areas. The aortic root cross sections were assessed by the following assays: (A–C) Haematoxylin and eosin staining (A) followed by quantitative analysis of necrotic core area (B) and fibrous cap area (C). nc: necrotic cores marked by a black dashed line; fc: fibrous cap marked by a blue dashed line; (D, E) VVG staining (D) followed by quantitative analysis of collagen content (E); (F, G) immunofluorescent staining for expression of SMA (F) with quantification of SMA positive areas (G); (H, I) Alizarin Red S staining for calcification (H, indicated by black arrows) and quantification of calcification positive areas (I). *P < 0.05, significantly different from control or as indicated (n = 15). C, control; T, T317; M, metformin; T/M, T317 plus metformin.
Collagen content also plays an important role in stabilization of lesion plaques and prevention of plaque rupture (Adiguzel et al., 2009). VVG staining of aortic root cross sections (Figure 2D, E) demonstrated that T317, metformin or their co‐treatment increased the area staining positively for collagen similarly (approximately twofold).
VSMCs constitute a major cell type in lesion caps and their presence stabilizes the plaque. Immunofluorescent staining with antibody to SMA (the marker for VSMCs) (Figure 2F, G) showed that SMA expression is at a low level in the cap areas of lesions in the control group. T317 substantially increased SMA expression in most lesion areas while metformin mainly increased it in the caps. The co‐treatment with T317 and metformin further increased SMA expression in lesions with greater effect on the caps.
Vascular calcification is a high‐risk factor for myocardial infarction in CHD patients (Rennenberg et al., 2009). We used Alizarin Red S staining of aortic root cross sections to determine the level of calcification and observed a prominent staining in the lesion areas of control mice (Figure 2H) indicating vascular mineralization. However, T317, metformin or their co‐treatment substantially inhibited such calcification in aortic root sections (Figure 2H, I). Taken together, these data showed that T317 and metformin, together, enhanced plaque stability by increasing collagen and VSMC content, while decreasing necrotic cores and calcification.
The anti‐atherogenic mechanisms underlying the effects of co‐treatment with T317 and metformin
Abnormal lipid profiles are a major risk factor for atherosclerosis. As shown in Table 2, treatment with T317 alone substantially increased Total‐C levels (by ~34%) which was mainly due to increased VLDL‐C levels (by ~61%), whereas metformin alone had little effect on serum lipid profiles. However, in the co‐treated group, the addition of metformin attenuated the serum lipids increased by T317 alone, particularly the VLDL‐C levels, suggesting that metformin can antagonize T317‐activated lipogenesis. In addition, the reduced ratio of HDL‐C to LDL‐C by T317 (39% in control group vs. 27% in T317 group) was partly restored by metformin (33%) which further confirmed the amelioration of lipid profiles by metformin.
Table 2.
Metformin inhibits T317‐induced hypertriglyceridemia in apoE−/− micea
| Treatment | Control | T317 | Metformin | T317 + Met |
|---|---|---|---|---|
| Bodyweight (g) | ||||
| 0 week | 22.73 ± 1.76 | 21.1 ± 1.98 | 22.07 ± 2.25 | 21.13 ± 2.68 |
| 16 weeks | 28.62 ± 2.14 | 28.78 ± 2.56 | 28.94 ± 3.93 | 27.93 ± 3.52 |
| Total‐C (mM) | 14.81 ± 5.14 | 19.78 ± 3.57* | 15.60 ± 3.32 | 15.28 ± 4.18** |
| HDL‐C (mM) | 2.08 ± 0.32 | 1.84 ± 0.17* | 2.23 ± 0.28 | 1.73 ± 0.14* |
| LDL‐C (mM) | 5.32 ± 1.33 | 6.90 ± 1.19* | 5.81 ± 1.14 | 5.30 ± 1.43** |
| VLDL‐C (mM) | 7.26 ± 0.83 | 11.67 ± 1.32* | 7.48 ± 1.03 | 7.93 ± 0.96** |
| Triglycerides(mM) | 0.41 ± 0.08 | 0.58 ± 0.13* | 0.32 ± 0.07* | 0.49 ± 0.04** |
Male apoE−/− mice received the treatment, indicated in Figure 1. Serum levels of Total‐C, VLDL‐C, LDL‐C and HDL‐C, and TG were measured. The results are expressed as means ± SD (n = 15; for VLDL‐C only, n=5).
P < 0.05, significantly different from control;
P < 0.05, significantly different from T317 alone.
To assess the effects of treatment on macrophage/foam cell accumulation in lesion areas, we used immunofluorescent staining on aortic root cross sections with anti‐CD68 antibody. CD68 expression was substantially reduced by T317, metformin or their co‐treatment, with the greatest reduction following the co‐treatment (Figure 3A). We then used Oil Red O staining to measure lipid accumulation in the peritoneal macrophages collected from mice at the end of 16 week treatment. As shown in Figure 3B, HFD feeding resulted in ~50% of peritoneal macrophages being differentiated into foam cells. Treatment with T317 or metformin alone decreased the rate of differentiated foam cells to 23 or 29%, and the co‐treatment further decreased it to 18%, suggesting a marked inhibition of macrophage lipid accumulation and foam cell formation by the co‐treatment.
Figure 3.
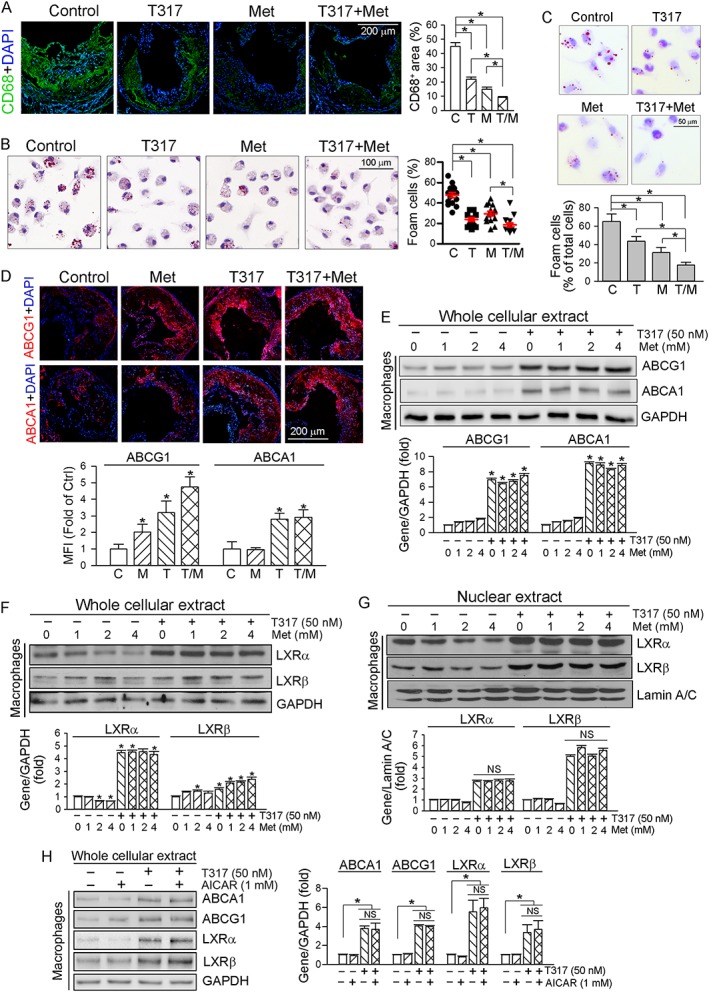
The anti‐atherogenic mechanisms of co‐treatment with T317 and metformin. (A) Aortic root cross sections were examined by immunofluorescent staining with anti‐CD68 antibody, and the density of fluorescence in images was quantified. (B) Peritoneal macrophages collected from mice used in Figure 1 were stained with Oil Red O to assess formation of foam cells (>10 lipid droplets per cell, >10 fields per sample). (C) Peritoneal macrophages isolated from untreated apoE−/− mice were incubated in serum‐free medium containing oxLDL (50 μg·mL−1) for 3 h and then treated with T317 (50 nM), metformin (Met, 0.5 mM) or both for 16 h respectively. Cells were then switched to medium containing apo‐AI (5 μg·mL−1) and HDL (20 μg·mL−1) and incubated for 5 h followed by Oil Red O staining. *P < 0.05, significantly different as indicated (n = 10); (D) aortic root cross sections were stained with anti‐ABCA1 and ABCG1 antibodies respectively. *P < 0.05, significantly different from control (n = 6); (E–H) peritoneal macrophages isolated from apoE−/− mice received indicated treatment overnight. Expression of ABCA1 and ABCG1 (E, H) and LXRα/β in total cellular extract (F, H) and nuclear extract (G) was determined by Western blot. *P < 0.05, significantly different from control or as indicated; NS: not significantly different (n = 5). C, control; T, T317; M, metformin; T/M, T317 plus metformin.
In vitro, we isolated peritoneal macrophages from apoE−/− mice fed a normal chow and induced macrophage/foam cell differentiation by incubating cells with oxLDL. The lipid‐loaded cells were then treated with T317 or/and metformin followed by incubation with apo‐AI and HDL. As shown in Figure 3C, T317 or metformin alone reduced lipid content in cells, which was further decreased by the co‐treatment, confirming the in vivo inhibitory effect of co‐treatment on foam cell formation.
To determine if the inhibition of macrophage/foam cell formation in lesion area could be attributed to increased expression of the transporters ABCA1 or ABCG1, aortic root cross sections were stained with anti‐ABCA1 or anti‐ABCG1 antibody. Figure 3D shows that metformin alone moderately induced ABCG1, but not ABCA1 expression. T317 potently induced both ABCA1 and ABCG1 expression, and this level of induction was not affected by metformin. The effects of T317 and metformin on ABCA1 and ABCG1 expression in macrophages were further analysed by in vitro experiments which showed that, while T317 substantially induced macrophage ABCA1 and ABCG1 expression, metformin had no significant effects (Figure 3E).
Metformin alone slightly reduced LXRα but slightly activated LXRβ expression in macrophages (Figure 3F). In contrast, T317 strongly induced macrophage LXR expression and nuclear translocation, particularly the nuclear translocation, and the induction was not affected by metformin (Figure 3F, G). In addition, we determined that regulation of macrophage ABCA1/G1 and LXRα/β expression by T317 was not affected by AICAR, another AMPKα agonist (Figure 3H).
The accumulation of macrophage/foam cells in the artery wall can be influenced by many different factors, such as adhesion of monocytes to endothelial cell layer, infiltration and local proliferation of macrophages. We initially measured the expression of VCAM‐1 and ICAM‐1, two important molecules for monocyte adhesion, in lesion areas of aortic root cross sections. We observed that expression of both was inhibited by metformin, T317 and their co‐treatment in vivo (Figure 4A). Production of inflammatory cytokines can cause endothelial injury to activate expression of adhesion molecules. The long‐term (16 week) treatment of apoE−/− mice with metformin, T317 or their combination substantially decreased serum levels of TNF‐α, IL‐1β and IL‐6 (Figure 4B), indicating that the inhibition of monocyte adhesion might be related to the anti‐inflammatory effects of the treatment.
Figure 4.
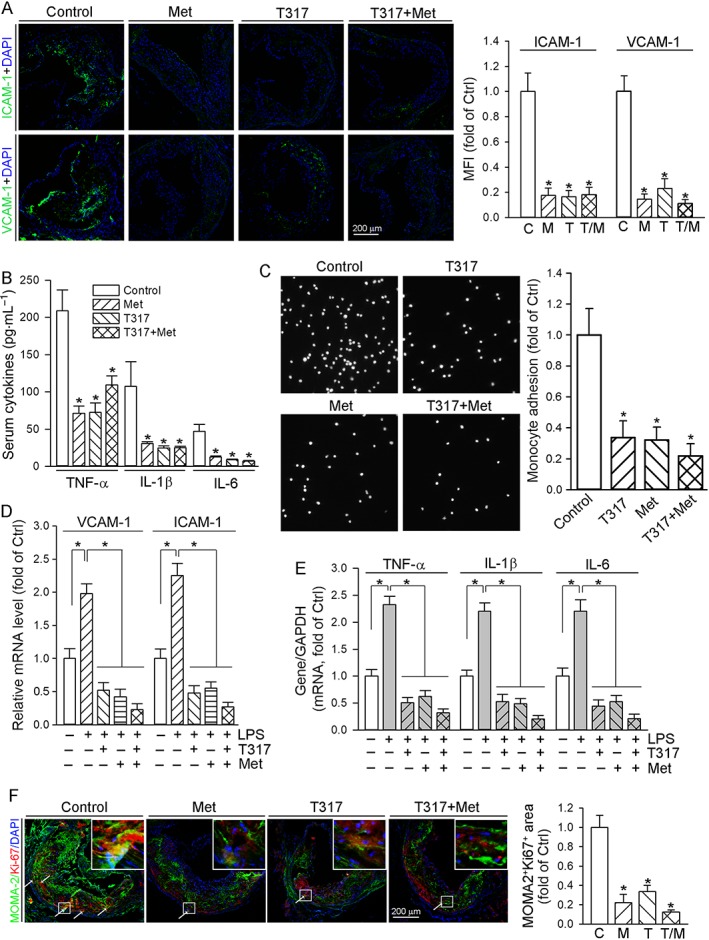
T317 and / or metformin inhibit monocyte adhesion to ECs, production of inflammatory cytokines and possible local macrophage proliferation. (A) Aortic root cross sections from mice used in Figure 1 were stained with anti‐VCAM‐1 or anti‐ICAM‐1 antibody, with quantitative analysis of fluorescent density. (B) Mouse serum Levels of TNF‐α, IL‐1β and IL‐6 were determined in samples of mouse serum (collected as shown in Figure 1). *P < 0.05, significantly different from control (n = 15); (C) HUVECs in 24‐well plates were pretreated with LPS (100 ng·mL−1) for 2 h followed by addition of T317 (50 nM), metformin (2 mM) or T317 plus metformin overnight. CFSE‐labelled THP‐1 cells were then added to HUVECs and co‐incubated for 1 h. The number of adherent THP‐1 cells were determined, using a microscope. The number of adherent THP‐1 cells in control group (treated with LPS alone) was defined as 1. The fold changes were obtained by calculating the ratio of adherent cells in the corresponding group to that in the control group, *P < 0.05, significantly different from control group (n = 6). (D, E) HUVECs were treated with metformin (2 mM), T317 (50 nM) or metformin plus T317 in the absence or presence of LPS (100 ng·mL−1) overnight. Expression of VCAM‐1 and ICAM‐1 mRNA (D), TNFα, IL‐1β and IL‐6 mRNA (E) was determined by qRT‐PCR with total cellular RNA, respectively, *P < 0.05, significantly different as indicated (n = 6); (F) the aortic root cross sections from mice used in Figure 1 were conducted co‐immunofluorescent staining with anti‐Ki‐67 and MOMA‐2 antibodies with quantitative analysis of MOMA‐2+Ki‐67+ areas. *P < 0.05, significantly different from control group (n = 6). C, control; T, T317; M, metformin; T/M, T317 plus metformin.
To obtain direct evidence for the inhibition of monocyte adhesion to endothelial cells (ECs) by metformin, T317 or their co‐treatment, we pretreated HUVECs with LPS to induce inflammation and measured expression of VCAM‐1/ICAM‐1, in the absence or presence of metformin, T317 or T317 plus metformin. We then added the CFSE‐labelled THP‐1 monocytes to the HUVECs and measured the numbers of adherent monocytes. As shown in Figure 4C, treatment of HUVECs with metformin, T317 or their combination clearly reduced the number of THP‐1 cells adhering to HUVECs. Consistent with the effects in vivo, we observed that metformin, T317 or their co‐treatment also inhibited LPS‐induced expression of VCAM‐1 and ICAM‐1 (Figure 4D), and inflammatory cytokines (TNF‐α, IL‐1β and IL‐6) (Figure 4E) in HUVECs.
To determine the effect of treatment on local macrophage proliferation indirectly, we carried out immunofluorescent staining on cross sections of aortic root with anti‐Ki‐67 antibody. Compared with samples from the control group, Ki‐67 expression was reduced by metformin and T317 with a greater effect by metformin, implying the possible inhibition of cell proliferation (Figure 4F). In addition, we determined that decreased MOMA‐2 (the marker for macrophages) expression can partially overlay with decreased Ki‐67 expression (Figure 4F) suggesting that it is possible that the treatments in vivo inhibited local macrophage proliferation within plaques. Taken together, reduction of monocyte adhesion/infiltration and possible local macrophage proliferation might provide other important anti‐atherogenic mechanisms which are related to anti‐inflammatory effects of the treatments.
Co‐treatment with T317 and metformin blocks induction of fatty livers by T317
T317 is known to induce fatty liver and hypertriglyceridemia by activating LXRα, the transcription factor activating expression of lipogenic genes including SREBP1c and FASN. Therefore, administration of T317 alone to apoE−/− mice reduced liver colour (Figure 5A) and increased the ratio of liver to body weight (Figure 5B), indicating marked lipid accumulation in the liver. Although metformin alone had little effect on liver size and colour, it substantially blocked the changes of both induced by T317 (Figure 5A, B). Oil Red O staining demonstrated that T317 alone increased hepatic lipid accumulation and that this increase was substantially attenuated by metformin (Figure 5C). Quantitation of TG levels in the liver showed that T317 increased hepatic TG levels and that this increase was totally blocked by metformin (Figure 5D). Levels of serum ALT and ALP, but not of AST, were elevated by T317. However, metformin blocked and attenuated T317‐increased serum ALT and ALP levels, respectively (Figure 5E). Also, the serum VLDL‐C and TG levels increased by T317 were restored to normal levels by metformin (Table 2).
Figure 5.
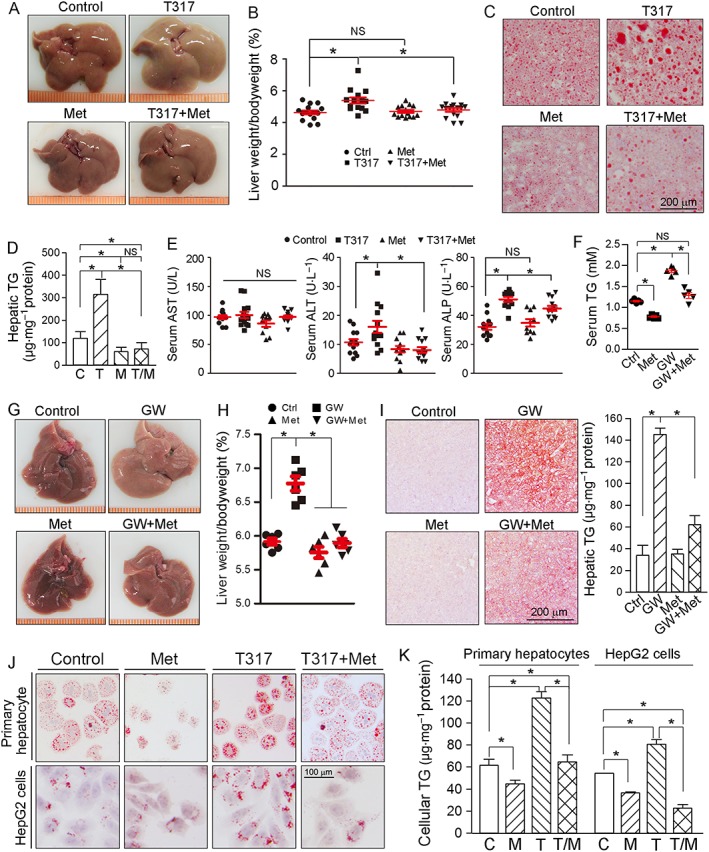
Metformin blocks LXR‐induced fatty liver and hepatic lipogenesis. At the end of 16 week treatment, serum and liver samples were collected from the mice used in Figure 1 and assayed as follows: (A) liver photos; (B) ratio of liver to body weight; (C, D) Oil Red O staining of liver frozen sections (C) and TG quantitative analysis with total liver lipid extract (D); (E) serum AST, ALT and ALP levels by enzymic methods. *P < 0.05, significantly different as indicated; NS: not significantly different (n = 15). (F–I) C57BL/6J mice (male, ~8‐week‐old) in four groups (n = 6 per group) received the following treatment for 2 weeks: Control, normal chow (NC); GW3965, NC containing GW3965 (GW, 40 mg · kg−1·day−1); Met, NC containing metformin (100 mg · kg−1·day−1); and GW + Met, NC containing both. After treatment, serum and liver samples were collected for the following assays: (F) serum TG level; (G) liver photos; (H) ratio of liver weight to bodyweight; (I) Oil Red O staining of liver frozen sections and TG quantitative analysis with total liver lipid extract. *P < 0.05, significantly different as indicated (n = 5); (J, K) HepG2 cells and primary hepatocytes isolated from apoE−/− mouse liver were treated with metformin (Met, 2 mM); T317 (50 nM) or T317 plus metformin overnight respectively. After treatment, cellular lipid content was determined by Oil Red O staining with intact cells (J) and TG quantitation with total cellular lipid extract (K), *P < 0.05, significantly different as indicated (n = 5). C, control; T, T317; M, metformin; T/M, T317 plus metformin.
To determine if the anti‐lipogenic effect of metformin could be exhibited with other LXR agonists, we treated C57BL/6J mice with GW3965, another synthetic LXR agonist, or GW3965 plus metformin, for 2 weeks. As observed with the longer‐term treatment of apoE−/− mice with T317, GW3965 also increased serum TG levels, the liver to body weight ratio and hepatic lipid content. However, GW3965‐induced fatty liver and hypertriglyceridemia were inhibited by metformin (Figure 5F–I). These data suggest that metformin would protect animals against fatty liver and hypertriglyceridemia, induced by other LXR agonists.
We found that T317 induced lipid accumulation in primary hepatocytes isolated from apoE−/− mouse livers. However, the accumulation was substantially blocked by metformin (upper panel, Figure 5J; left panel, Figure 5K). Furthermore, T317 increased lipid content in HepG2 cells, a human hepatic cell line, and this increase was also blocked by metformin (lower panel, Figure 5J; right panel, Figure 5K). Thus, inhibition of T317‐activated lipogenesis by metformin could be a species‐independent effect.
The mechanisms underlying the anti‐fatty liver effects of the combination of T317 and metformin
In macrophages, metformin alone had little effect on LXRα/β expression and also did not affect the activation of LXRα/β by T317 (Figure 3F, G). In contrast, metformin selectively inhibited T317‐activated LXRα, but not LXRβ, in mouse liver or hepatocytes. As shown in Figure 6A, B, T317 substantially activated both LXRα and LXRβ expression. In contrast, metformin alone moderately inhibited while blocking T317‐induced LXRα expression in mouse liver. Meanwhile, metformin had no effect on T317‐induced LXRβ expression in mouse liver. Therefore, the combination of metformin and T317 can selectively attenuate T317‐induced hepatic lipogenesis. Indeed, T317‐activated expression of SREBP1c and FASN was substantially reduced by metformin (Figure 6C). Other relevant enzymes, such as ACC1, particularly its phosphorylation status (pi‐ACC/ACC), and SCD‐1 also influence fatty acid synthesis. T317 increased both ACC1 and pi‐ACC1 to the same extent and thus the ratio of pi‐ACC1 to ACC1 was not changed. Metformin blocked T317‐induced ACC1 expression while increasing pi‐ACC levels. Therefore, the ratio of pi‐ACC1 to ACC1 was greatly increased by the co‐treatment (Figure 6C, D). In addition, T317‐activated expression of mRNA for SCD‐1 was blocked by metformin (Figure 6E). Taken together, the results above suggest that metformin inhibits T317‐induced lipogenesis by selectively inactivating LXRα and expression of lipogenic genes.
Figure 6.
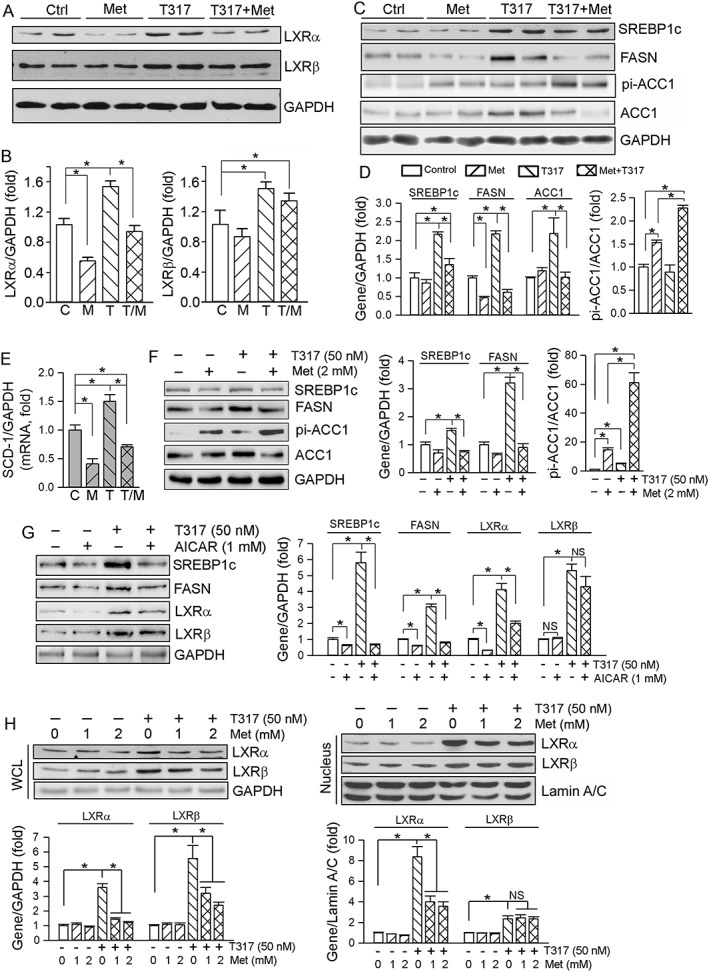
Metformin antagonizes T317‐induced lipogenesis both in vivo and in vitro. (A–E) Total protein and RNA extracted from livers of mice used in Figure 1 were assayed as follows: expression of LXRα and LXRβ protein (A), SREBP1c, FASN, pi‐ACC1 and ACC1 protein (C) was determined by Western blot with quantification of band density (B, D); n = 5. *P < 0.05, significantly different as indicated (B); significantly different from control (D). Expression of SCD‐1 mRNA was determined by qRT‐PCR (E), *P < 0.05 (n = 6); (F–H) HepG2 cells were treated with metformin or AICAR at the indicated concentrations or plus T317 (50 nM) overnight. Expression of SREBP1c, FASN, pi‐ACC1, ACC1 protein in total cellular extract (F, G), LXRα and LXRβ protein in total cellular and nuclear extracts (G, H) was determined by Western blot respectively. *P < 0.05, significantly different as indicated; NS: not significantly different (n = 5). C, control; M, metformin; T, T317; T/M, T317 plus metformin.
As observed in the in vivo study above, metformin or AICAR inhibited the expression of T317‐induced lipogenic genes, such as SREBP1c, FASN and ACC1, in HepG2 cells (Figure 6F, G). Apart from decreasing T317‐induced LXRα, metformin also selectively attenuated T317‐induced nuclear translocation of LXRα in HepG2 cells (Figure 6H, right panel).
We carried out immunofluorescent staining, Western blot and qRT‐PCR assays with liver samples (Figure 7) to assess the contributions of mechanisms other than inhibition of fatty acid synthesis to the amelioration of TG homeostasis by metformin. We first found that expression of DGAT1, the rate‐limiting enzyme for TG synthesis, was inhibited by T317, metformin or their co‐treatment. We then observed that expression of ATGL and HSL, two enzymes catalysing TG hydrolysis (Gaidhu et al., 2009), was activated by metformin or metformin plus T317. Similarly, expression of PPARα, a transcription factor activating free fatty acid oxidation systems, and CAT, one of the PPARα target genes which is involved in fatty acid oxidation, was induced by metformin or metformin plus T317. Taken together, the results in Figures 6 and 7 demonstrate that co‐treatment with metformin and T317 protects against fatty liver by several different actions including inhibition of hepatic fatty acid and TG synthesis and activation of TG hydrolysis and fatty acid oxidation.
Figure 7.
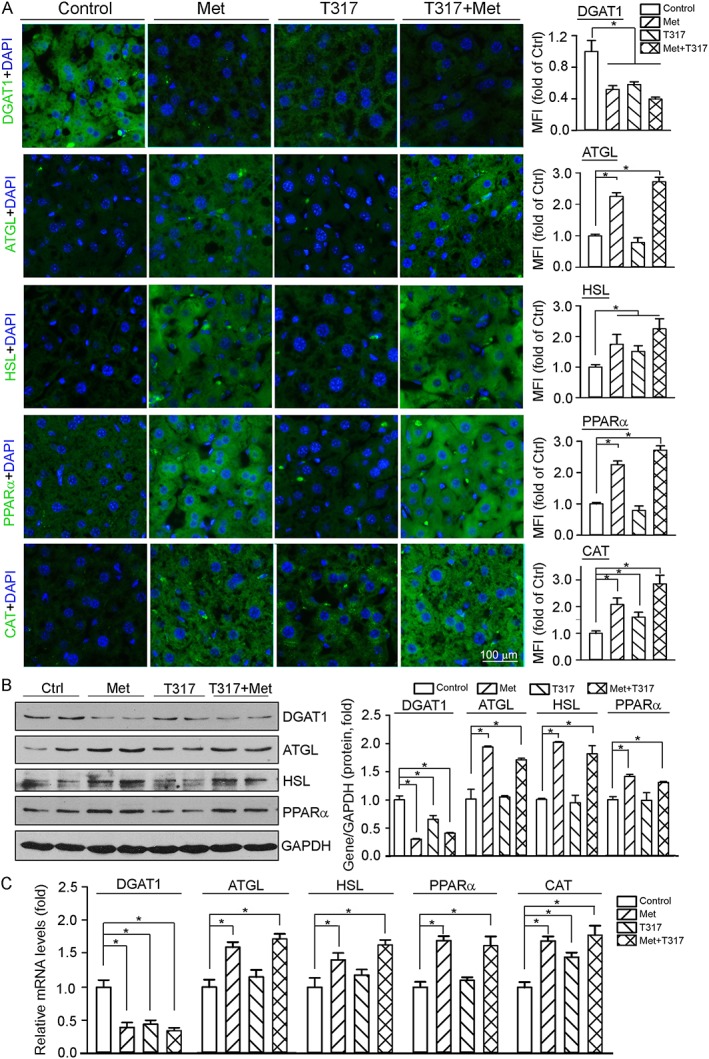
Metformin ameliorates TG metabolism in vivo by regulating expression of genes for TG synthesis and hydrolysis, and fatty acid oxidation. Total protein, RNA and frozen sections were prepared from mouse liver samples used in Figure 1 and completed the following assays respectively: (A) expression of DGAT1, ATGL, HSL, PPARα and CAT protein was determined by immunofluorescent staining with quantification of the mean immunofluorescent intensity (MFI) of images; (B) expression of DGAT1, ATGL, HSL and PPARα protein was determined by Western blot, *P < 0.05, significantly different as indicated (n = 6); (C) expression of DGAT1, ATGL, HSL, PPARα and CAT mRNA was determined by qRT‐PCR, *P < 0.05, significantly different as indicated (n = 6).
The anti‐lipogenic effects of metformin depends on activation of AMPKα and inactivation of LXRα
Our previous study demonstrated that inactivation of AMPKα can selectively activate LXRα (Hu et al., 2016). To search for links between the protective effects of metformin against T317‐induced lipogenesis and AMPKα‐inactivated LXRα, we generated HepG2 cell lines lacking AMPKα (Cas9‐AMPKα), LXRα (Cas9‐LXRα) or LXRβ (Cas9‐LXRβ) expression. Compared with control cells (Cas9‐NS), lack of AMPKα expression moderately increased cellular lipid content at the basal levels (Figure 8A, B). Metformin alone reduced lipid content and blocked T317‐induced lipid accumulation in Cas9‐NS cells, but not in Cas9‐AMPKα cells (Figure 8A, B). Correspondingly, inhibition of T317‐induced SREBP1c/FASN expression and activation of PPARα/ATGL expression by metformin were impaired in the cells lacking AMPKα expression (Figure 8C, D).
Figure 8.
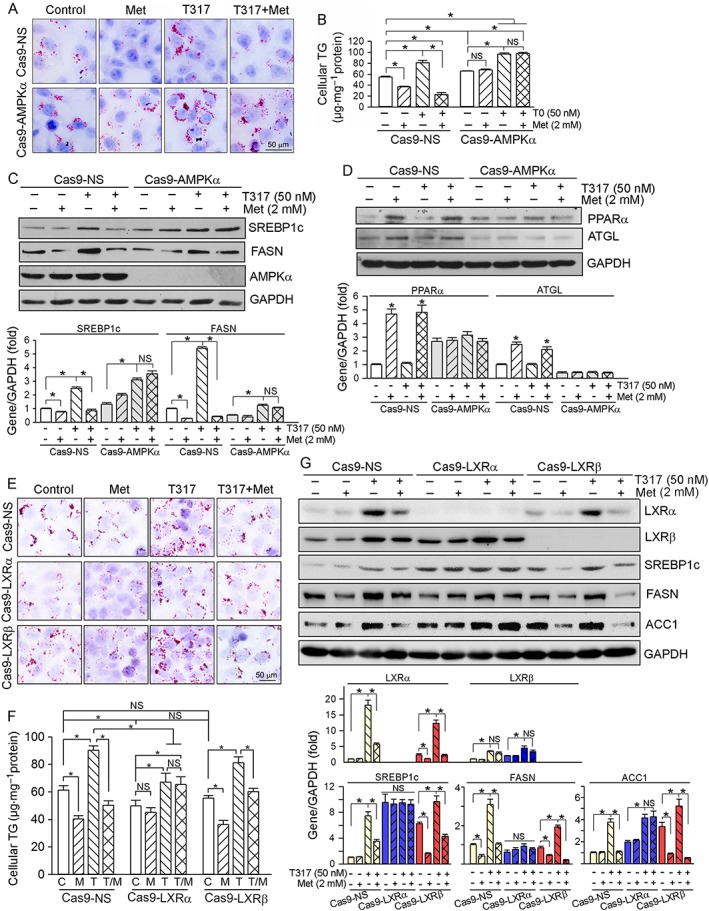
The anti‐lipogenic effects of metformin depend on inactivation of LXRα in an AMPKα‐dependent manner. (A–D) Cas9‐NS and Cas9‐AMPKα cells were treated with metformin (Met, 2 mM), T317 (50 nM) or both overnight. Cellular lipid content was determined by Oil Red O staining with intact cells (A) and TG quantitation with total lipid extract (B). Expression of SREBP1c, FASN and AMPKα (C), PPARα and ATGL (D) was determined by Western blot, respectively, *P < 0.05, significantly different from control; NS: not significant, (n = 5); (E–G): Cas9‐NS, Cas9‐LXRα and Cas9‐LXRβ cells received treatment with metformin (Met, 2 mM), T317 (50 nM) or both overnight followed by determination of cellular lipid content by Oil Red O staining (E) and TG quantitation (F), and expression of LXRα, LXRβ, SREBP1c, FASN and ACC1 protein (G) by Western blot respectively. C, control; M, metformin; T, T317; T/M, T317 plus metformin. *P < 0.05, significantly different as indicated; NS: not significant (n = 5).
As shown in Figure 8E, F, lack of LXRα but not of LXRβ expression moderately reduced cellular lipid at basal levels. Similar to Cas9‐NS cells, the increased TG accumulation by T317 alone in Cas9‐LXRβ cells was inhibited by adding metformin (bottom row, Figure 8E; right panel, Figure 8F). However, lack of LXRα expression attenuated T317‐induced cellular TG levels, which was not affected by metformin either (middle row, Figures 8E; middle panel, Figure 8F). Correspondingly, activation of SREBP1c, FASN and ACC1 expression by T317 was reduced by metformin in Cas9‐NS and Cas9‐LXRβ cells, but not in Cas9‐LXRα cells (Figure 8G). Taken together, the results in Figure 8 suggest that inhibition of T317‐induced lipogenesis by metformin involves the inactivation of LXRα, through an AMPKα‐dependent mechanism.
Discussion
The synthetic LXR ligand T317 reduced atherosclerosis but activated lipogenesis. Although metformin inhibits hepatic steatosis, it has limited cardioprotective actions. In this study, we treated pro‐atherogenic mice with a fixed combination of T317 and metformin and found that this combination substantially inhibited atherosclerosis to a greater extent than T317 alone. More importantly, metformin substantially blocked the induction of fatty liver and hypertriglyceridemia by T317.
Cardiovascular disease is one of the complications of diabetes and the primary basis for the high mortality and morbidity of diabetic patients. Although the United Kingdom Prospective Diabetes Study showed that treatment with metformin reduced some vascular complications including myocardial infarction, conflicting clinical data have also been reported (UKPDS Group 1998b; Ferrannini and DeFronzo, 2015; Riksen and Tack, 2014). Several animal studies also indicate that metformin can inhibit atherosclerosis but only in the context of diabetes (Li et al., 2011; Forouzandeh et al., 2014; Hayashi et al., 2014; Wang et al., 2017).
Metformin can ameliorate glucose metabolism in diabetes. In wild‐type mice, we also determined that metformin reduced blood glucose levels after a short‐term treatment. However, the long‐term (16 week) metformin treatment in our present study, had no effect on blood glucose levels in HFD‐fed apoE−/− mice (Supporting Information Figure S1) indicating that other functions, such as attenuation of hepatic steatosis, anti‐oxidative stress and improvement of EC functions, may be involved in the anti‐atherogenic effects of metformin. Endothelial injury, the initial event in atherogenesis, can be induced by high glucose‐mediated chronic endoplasmic reticulum stress (Dong et al., 2017). Activation of ECs leads to the enhanced expression of adhesion molecules, such as VCAM‐1 and ICAM‐1, which in turn facilitate the adhesion of monocytes to endothelium and subsequent infiltration. Metformin suppresses IL‐1β‐induced release of the pro‐inflammatory cytokines, IL‐6 and IL‐8, to reduce adhesion and infiltration of monocytes by inactivating NF‐κB in ECs (Isoda et al., 2006; Dong et al., 2010). In vitro, metformin inhibits PMA‐induced monocyte/macrophage differentiation. In vivo, the inhibition of angiotensin II‐induced atheromatous plaque formation and aortic aneurysm by metformin in apoE−/− mice was attributed to reduction of monocyte infiltration (Vasamsetti et al., 2015). Consistent with these data, we observed that metformin or T317 inhibited expression of VCAM‐1 and ICAM‐1 in the aortic wall in vivo and LPS‐induced VCAM‐1/ICAM‐1 expression in HUVECs in vitro (Figure 4A, D), suggesting inhibition of monocyte adhesion and infiltration (Figure 4C). Such protection might be partly attributed to inhibition of inflammatory cytokine production by the treatment (Figure 4B, E). In addition, we determined that metformin or T317 may inhibit local macrophage proliferation because the treatment reduced Ki‐67 expression which co‐localized with reduced MOMA‐2 expression (Figure 4F). Therefore, the inhibition of monocyte adhesion and infiltration and possible local macrophage proliferation in the arterial wall may also make contributions to the anti‐atherogenic effects of the combination of metformin and T317.
Treatment of lipid‐loaded macrophages by AICAR increases ABCG1 expression and cholesterol efflux in an LXR‐independent manner, but AICAR has little effect on expression of ABCA1 and scavenger receptors (Li et al., 2010). We also found that metformin moderately activated expression of the transporter ABCG1, but not that of ABCA1 (Figure 3D). Compared with T317, metformin slightly increased LXRβ expression while slightly reducing LXRα expression and had no effect on LXRα/β nuclear translocation in macrophages (Figure 3F, G) suggesting that the effect of metformin on macrophage ABCG1 expression was independent of the LXR pathway.
Although several anti‐atherogenic actions of metformin have been well defined based on cellular and animal studies, the effects of metformin on cardiovascular disease, particularly in non‐diabetic patients, still remain in doubt. For instance, in the Carotid Atherosclerosis: Metformin for insulin ResistAnce study (Preiss et al., 2014), after 18 months of metformin treatment, no effect on distal carotid intima‐media thickness and on several surrogate markers of cardiovascular disease were observed in non‐diabetic patients with high cardiovascular risk. In the GIPS‐III Randomized Clinical Trial study (Lexis et al., 2014), the 4 month metformin treatment did not improve left ventricular function after acute myocardial infarction in non‐diabetic patients. Therefore, as a monotherapy, metformin can't be a promising treatment strategy for cardiovascular disease, particularly in non‐diabetic patients. In addition, metformin exerted no additional protection to that of a statin, on CHD (Preiss et al., 2014), which suggests that metformin should be combined with some other medicine, rather than a statin. In this study, we showed that metformin had weaker inhibitory effect than T317 on en face and sinus lesions (Figure 1), particularly, in the branch points of aortas, such as the ascending aorta and the descending aorta, and metformin had a slight or no effect on lesions in these parts (Figure 1D), which might be related to the nature of the vascular structure in these tissues. The laminar flow in the straight parts of artery, such as the thoracic aorta, can activate AMPKα, while the oscillatory flow in branch points and aortic root can silence AMPKα activity (Guo et al., 2007).
LXR ligands potently inhibit atherosclerosis mainly by activating macrophage cholesterol metabolism through increased expression of ABCA1 and ABCG1 transporters. In this study, metformin had no effect on ABCA1 while slightly activating ABCG1 expression (Figure 3D, E), suggesting that the mild protection of metformin against atherosclerosis was not due to regulation of cholesterol metabolism in macrophages. In contrast, metformin substantially attenuated T317‐induced hepatic lipid accumulation (Figure 5), and restored T317‐increased serum LDL‐C and VLDL‐C levels to normal (Table 2). In addition, metformin inhibited fatty liver and hypertriglyceridemia induced by another LXR agonist(Figure 5F–I), although the anti‐lipogenic effect of metformin did not appear to be clearly dose‐dependent, over a small dose range (Supporting Information Figure S2). Abnormalities of TG metabolism are also considered as risk factors for atherosclerosis as TG can be associated with atherogenic apolipoproteins, such as apoB and apoC‐III. Therefore, activation of lipogenesis partly suppresses the anti‐atherogenic properties of LXR. Treatment with a combination of metformin and T317 removes this suppression, thereby enhancing T317‐inhibited atherosclerosis.
T317 increased hepatic LXR expression and nuclear translocation (Figure 6A, H), which can, in turn, trigger expression of lipogenic genes and development of fatty liver (Figures 5 and 6). Compared with its minimal effects on T317‐activated macrophage LXRα/β expression and nuclear expression (Figure 3F, G), metformin selectively reduced T317‐induced hepatic LXRα expression and nuclear translocation, particularly the nuclear translocation, both in vivo and in vitro (Figure 6A, H). The selective regulation of LXR activity in a cell type‐ and LXR isoform‐dependent manner (Figures 3 and 6) leads to inhibition of LXRα‐induced lipogenesis and hepatic lipid accumulation/fatty liver by the combination of metformin and T317 (Figures 5, 6 and 8), while metformin slightly enhanced LXR‐activated macrophage cholesterol metabolism and LXR‐inhibited foam cell formation (Figure 3). In addition, metformin inhibited TG synthesis and activated TG hydrolysis and fatty acid oxidation (Figure 7) suggesting that metformin regulates TG homeostasis by several actions.
In conclusion, our study demonstrates that addition of metformin to T317 not only enhances the inhibition of atherosclerosis by T317 but also blocks T317‐induced hepatic lipogenesis by selective inactivation of LXRα, suggesting that this combination can function as a novel strategy to inhibit the development of atherosclerosis, in those situations where monotherapy with either LXR or AMPKα agonists is not feasible.
Author contributions
C.M., W.Z., X.Y., Y.L., L.L., K.F., X.Z., S.Y., L.S., M.Y., J.Y., X.L. and W.H. conducted the experiments; R.M., Y.Z. and L.L. provided reagents or/and edited the paper; J.H., Y.C. and Y.D. designed the experiments and wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This http://onlinelibrary.wiley.com/doi/10.1111/bph.13405/abstract acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Effects of metformin and LXR ligands on mouse serum glucose levels. Serum samples collected from the mice used in Figure 1 and Figure 5G were used to determine glucose levels. C: control; M: metformin; T: T317; GW: GW3965; T/M: T317 plus metformin; GW/M: GW3965 plus metformin. *: P < 0.05; NS: not significantly different (n = 5).
Figure S2 Attenuation of T317‐induced fatty liver by metformin is not in a dose‐dependent manner. C57BL/6J mice (male, ~8‐week old) in 8 groups (5/group) received the following treatment for 2 weeks: Control, normal chow (NC); T317, NC containing T317 (1 mpk); Met: NC containing metformin (50, 100 or 200 mpk); and T317 + Met, NC containing T317 (1 mpk) and metformin at the indicated dose. After treatment, liver samples were collected for the following assays: A, liver photos; B, ratios of liver weight to bodyweight; C, D: hepatic lipid content by Oil Red O staining of liver frozen sections (C) and TG quantitative analysis with total liver lipid extract (D). *: P < 0.05 (n = 5).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) Grants 81473204 and 81773727 to J.H., 31770863 to Y.C., 81573427 and 81722046 to Y.D.; the 111 Project of the Ministry of Education of China B08011 to J.H.; the Tianjin Municipal Science and Technology Commission Grants 17JCYBJC25000 to Y.C. and 16JCZDJC34700 to J.H.; and the National Institutes of Health (R01DK112971) to Q.M.
Ma, C. , Zhang, W. , Yang, X. , Liu, Y. , Liu, L. , Feng, K. , Zhang, X. , Yang, S. , Sun, L. , Yu, M. , Yang, J. , Li, X. , Hu, W. , Miao, R. Q. , Zhu, Y. , Li, L. , Han, J. , Chen, Y. , and Duan, Y. (2018) Functional interplay between liver X receptor and AMP‐activated protein kinase α inhibits atherosclerosis in apolipoprotein E‐deficient mice − a new anti‐atherogenic strategy. British Journal of Pharmacology, 175: 1486–1503. doi: 10.1111/bph.14156.
Contributor Information
Jihong Han, Email: jihonghan2008@nankai.edu.cn.
Yuanli Chen, Email: chenyuanli@hfut.edu.cn.
Yajun Duan, Email: yduan@hfut.edu.cn.
References
- Adiguzel E, Ahmad PJ, Franco C, Bendeck MP (2009). Collagens in the progression and complications of atherosclerosis. Vas Med 14: 73–89. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Duan Y, Kang Y, Yang X, Jiang M, Zhang L et al (2012). Activation of liver X receptor induces macrophage interleukin‐5 expression. J Biol Chem 287: 43340–43350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Duan Y, Yang X, Sun L, Liu M, Wang Q et al (2015). Inhibition of ERK1/2 and activation of LXR synergistically reduce atherosclerotic lesions in ApoE‐deficient mice. Arterioscler Thromb Vasc Biol 35: 948–959. [DOI] [PubMed] [Google Scholar]
- Chisholm JW, Hong J, Mills SA, Lawn RM (2003). The LXR ligand T0901317 induces severe lipogenesis in the db/db diabetic mouse. J Lipid Res 44: 2039–2048. [DOI] [PubMed] [Google Scholar]
- Collier CA, Bruce CR, Smith AC, Lopaschuk G, Dyck DJ (2006). Metformin counters the insulin‐induced suppression of fatty acid oxidation and stimulation of triacylglycerol storage in rodent skeletal muscle. Am J Physiol Endocrinol Metab 291: E182–E189. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Brit J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Condorelli G, Mehta JL (2016). Scavenger receptors and non‐coding RNAs: relevance in atherogenesis. Cardiovasc Res 109: 24–33. [DOI] [PubMed] [Google Scholar]
- Dong Y, Fernandes C, Liu Y, Wu Y, Wu H, Brophy ML et al (2017). Role of endoplasmic reticulum stress signalling in diabetic endothelial dysfunction and atherosclerosis. Diab Vascu Dis Res 14: 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Zhang M, Wang S, Liang B, Zhao Z, Liu C et al (2010). Activation of AMP‐activated protein kinase inhibits oxidized LDL‐triggered endoplasmic reticulum stress in vivo . Diabetes 59: 1386–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrannini E, DeFronzo RA (2015). Impact of glucose‐lowering drugs on cardiovascular disease in type 2 diabetes. Eur Heart J 36: 2288–2296. [DOI] [PubMed] [Google Scholar]
- Forouzandeh F, Salazar G, Patrushev N, Xiong S, Hilenski L, Fei B et al (2014). Metformin beyond diabetes: pleiotropic benefits of metformin in attenuation of atherosclerosis. J Am Heart Assoc 3: e001202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidhu MP, Fediuc S, Anthony NM, So M, Mirpourian M, Perry RL et al (2009). Prolonged AICAR‐induced AMP‐kinase activation promotes energy dissipation in white adipocytes: novel mechanisms integrating HSL and ATGL. J Lipid Res 50: 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Chien S, Shyy JY (2007). Regulation of endothelial cell cycle by laminar versus oscillatory flow: distinct modes of interactions of AMP‐activated protein kinase and Akt pathways. Circ Res 100: 564–571. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Kotani H, Yamaguchi T, Taguchi K, Iida M, Ina K et al (2014). Endothelial cellular senescence is inhibited by liver X receptor activation with an additional mechanism for its atheroprotection in diabetes. Proc Natl Acad Sci U S A 111: 1168–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmingsen B, Christensen LL, Wetterslev J, Vaag A, Gluud C, Lund SS et al (2012). Comparison of metformin and insulin versus insulin alone for type 2 diabetes: systematic review of randomised clinical trials with meta‐analyses and trial sequential analyses. BMJ 344: e1771. [DOI] [PubMed] [Google Scholar]
- Hu W, Zhang W, Chen Y, Rana U, Teng RJ, Duan Y et al (2016). Nogo‐B receptor deficiency increases liver X receptor alpha nuclear translocation and hepatic lipogenesis through an adenosine monophosphate‐activated protein kinase alpha‐dependent pathway. Hepatology 64: 1559–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundal RS, Krssak M, Dufour S, Laurent D, Lebon V, Chandramouli V et al (2000). Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes 49: 2063–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isoda K, Young JL, Zirlik A, MacFarlane LA, Tsuboi N, Gerdes N et al (2006). Metformin inhibits proinflammatory responses and nuclear factor‐kappaB in human vascular wall cells. Arterioscler Thromb Vasc Biol 26: 611–617. [DOI] [PubMed] [Google Scholar]
- Johnson JA, Majumdar SR, Simpson SH, Toth EL (2002). Decreased mortality associated with the use of metformin compared with sulfonylurea monotherapy in type 2 diabetes. Diabetes Care 25: 2244–2248. [DOI] [PubMed] [Google Scholar]
- Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA et al (2002). Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci U S A 99: 7604–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Brit J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lexis CP, van der Horst IC, Lipsic E, Wieringa WG, de Boer RA, van den Heuvel AF et al (2014). Effect of metformin on left ventricular function after acute myocardial infarction in patients without diabetes: the GIPS‐III randomized clinical trial. JAMA 311: 1526–1535. [DOI] [PubMed] [Google Scholar]
- Li D, Wang D, Wang Y, Ling W, Feng X, Xia M (2010). Adenosine monophosphate‐activated protein kinase induces cholesterol efflux from macrophage‐derived foam cells and alleviates atherosclerosis in apolipoprotein E‐deficient mice. J Biol Chem 285: 33499–33509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B et al (2011). AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet‐induced insulin‐resistant mice. Cell Metab 13: 376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madiraju AK, Erion DM, Rahimi Y, Zhang XM, Braddock DT, Albright RA et al (2014). Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 510: 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruthur NM, Tseng E, Hutfless S, Wilson LM, Suarez‐Cuervo C, Berger Z et al (2016). Diabetes medications as monotherapy or metformin‐based combination therapy for type 2 diabetes: a systematic review and meta‐analysis. Ann Inter Med 164: 740–751. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Brit J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore KJ, Tabas I (2011). Macrophages in the pathogenesis of atherosclerosis. Cell 145: 341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SA, Winkel M, Ali MK, Narayan KM, Mehta NK (2015). Cardiovascular mortality associated with 5 leading risk factors: national and state preventable fractions estimated from survey data. Ann Intern Med 163: 245–253. [DOI] [PubMed] [Google Scholar]
- Preiss D, Lloyd SM, Ford I, McMurray JJ, Holman RR, Welsh P et al (2014). Metformin for non‐diabetic patients with coronary heart disease (the CAMERA study): a randomised controlled trial. Lancet Diabetes Endocrinol 2: 116–124. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013). Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratni H, Wright MB (2010). Recent progress in liver X receptor‐selective modulators. Curr Opin Drug Discov Devel 13: 403–413. [PubMed] [Google Scholar]
- Rena G, Pearson ER, Sakamoto K (2013). Molecular mechanism of action of metformin: old or new insights? Diabetologia 56: 1898–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennenberg RJ, Kessels AG, Schurgers LJ, van Engelshoven JM, de Leeuw PW, Kroon AA (2009). Vascular calcifications as a marker of increased cardiovascular risk: a meta‐analysis. Vasc Health Risk Manag 5: 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riksen NP, Tack CJ (2014). The cardiovascular effects of metformin: lost in translation? Curr Opin Lipidol 25: 446–451. [DOI] [PubMed] [Google Scholar]
- Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L et al (2000). Role of LXRs in control of lipogenesis. Genes Dev 14: 2831–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein S, Lohmann C, Schafer N, Hofmann J, Rohrer L, Besler C et al (2010). SIRT1 decreases Lox‐1‐mediated foam cell formation in atherogenesis. Eur Heart J 31: 2301–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tall AR, Yvan‐Charvet L, Terasaka N, Pagler T, Wang N (2008). HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab 7: 365–375. [DOI] [PubMed] [Google Scholar]
- Terasaka N, Hiroshima A, Koieyama T, Ubukata N, Morikawa Y, Nakai D et al (2003). T‐0901317, a synthetic liver X receptor ligand, inhibits development of atherosclerosis in LDL receptor‐deficient mice. FEBS Lett 536: 6–11. [DOI] [PubMed] [Google Scholar]
- UKPDS (UK Prospective Diabetes Study) Group (1998a). Effect of intensive blood‐glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 352: 854–865. [PubMed] [Google Scholar]
- UKPDS (UK Prospective Diabetes Study) Group (1998b). Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. BMJ 317: 703–713. [PMC free article] [PubMed] [Google Scholar]
- Vasamsetti SB, Karnewar S, Kanugula AK, Thatipalli AR, Kumar JM, Kotamraju S (2015). Metformin inhibits monocyte‐to‐macrophage differentiation via AMPK‐mediated inhibition of STAT3 activation: potential role in atherosclerosis. Diabetes 64: 2028–2041. [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhang M, Torres G, Wu S, Ouyang C, Xie Z et al (2017). Metformin suppresses diabetes‐accelerated atherosclerosis via the inhibition of Drp1‐mediated mitochondrial fission. Diabetes 66: 193–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Dahlman‐Wright K (2010). Liver X receptor in cholesterol metabolism. J Endocrinol 204: 233–240. [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk‐Melody J et al (2001). Role of AMP‐activated protein kinase in mechanism of metformin action. J Clin Invest 108: 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effects of metformin and LXR ligands on mouse serum glucose levels. Serum samples collected from the mice used in Figure 1 and Figure 5G were used to determine glucose levels. C: control; M: metformin; T: T317; GW: GW3965; T/M: T317 plus metformin; GW/M: GW3965 plus metformin. *: P < 0.05; NS: not significantly different (n = 5).
Figure S2 Attenuation of T317‐induced fatty liver by metformin is not in a dose‐dependent manner. C57BL/6J mice (male, ~8‐week old) in 8 groups (5/group) received the following treatment for 2 weeks: Control, normal chow (NC); T317, NC containing T317 (1 mpk); Met: NC containing metformin (50, 100 or 200 mpk); and T317 + Met, NC containing T317 (1 mpk) and metformin at the indicated dose. After treatment, liver samples were collected for the following assays: A, liver photos; B, ratios of liver weight to bodyweight; C, D: hepatic lipid content by Oil Red O staining of liver frozen sections (C) and TG quantitative analysis with total liver lipid extract (D). *: P < 0.05 (n = 5).