

Abstract
Aims
Sacubitril/valsartan is indicated for the treatment of heart failure and reduced ejection fraction (HFrEF). Furosemide, a loop diuretic commonly used for the treatment of HFrEF, may be coadministered with sacubitril/valsartan in clinical practice. The effect of sacubitril/valsartan on the pharmacokinetics and pharmacodynamics of furosemide was evaluated in this open label, two‐period, single‐sequence study in healthy subjects.
Methods
All subjects (n = 28) received 40 mg oral single‐dose furosemide during period 1, followed by a washout of 2 days. In period 2, sacubitril/valsartan 200 mg (97/103 mg) was administered twice daily for 5 days and a single dose of 40 mg furosemide was coadministered on day 6. Serial plasma and urine samples were collected to determine the pharmacokinetics of furosemide and sacubitril/valsartan and the pharmacodynamics of furosemide. The point estimates and the associated 90% confidence intervals for pharmacokinetic parameters were evaluated.
Results
Coadministration of furosemide with sacubitril/valsartan decreased the maximum observed plasma concentration (Cmax) [estimated geometric mean ratio (90% confidence interval): 0.50 (0.44, 0.56)], area under the plasma concentration–time curve (AUC) from time 0 to infinity [0.72 (0.67, 0.77)] and 24‐h urinary excretion of furosemide [0.74 (0.69, 0.79)]. When coadministered with sacubitril/valsartan, 0–4‐h, 4–8‐h and 0–24‐h diuresis in response to furosemide was reduced by ~7%, 21% and 0.2%, respectively, while natriuresis was reduced by ~ 28.5%, 7% and 15%, respectively. Post hoc analysis of the pivotal phase III Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure trial (PARADIGM‐HF) indicated that the median furosemide dose was similar at baseline and at the end of the study in the sacubitril/valsartan group.
Conclusions
Sacubitril/valsartan reduced plasma Cmax and AUC and 24‐h urinary excretion of furosemide, while not significantly affecting its pharmacodynamic effects in healthy subjects.
Keywords: drug–drug interaction, furosemide, pharmacodynamics, pharmacokinetics, sacubitril/valsartan
What is Already Known about this Subject
Sacubitril/valsartan demonstrated a superior reduction in mortality and morbidity compared with enalapril, resulting in the regulatory approval for the treatment of patients with heart failure and reduced ejection fraction (HFrEF).
Based on in vitro results, sacubitril/valsartan has the potential to inhibit organic anion transporter 3 (OAT3) in vivo.
Furosemide, an OAT3 substrate, is a loop diuretic commonly prescribed for symptomatic relief in patients with HFrEF and likely to be coadministered with sacubitril/valsartan.
What this Study Adds
When coadministered with furosemide, sacubitril/valsartan reduced the maximum observed plasma concentration, the area under the plasma concentration–time curve, and 24‐h urinary excretion of furosemide.
While there was no relevant change in 24‐h diuresis, furosemide‐associated 24‐h natriuresis was reduced.
The average daily dose of furosemide was unchanged from baseline until the end of the Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure trial (PARADIGM‐HF) in patients treated with sacubitril/valsartan.
Introduction
http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1613 are the standard of care for the treatment of heart failure with reduced ejection fraction (HFrEF) 1. However, in spite of an incremental reduction in the risk of cardiovascular death with ACE inhibitors, the morbidity and mortality of patients with HFrEF remain high 2. Recent phase III clinical trial data [Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure trial (PARADIGM‐HF)] demonstrated a superior reduction in mortality and morbidity following treatment with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=7857/http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3937 (LCZ696) compared with http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=6322, resulting in the regulatory approval of this drug combination for the treatment of patients with HFrEF in the USA, Europe and many other countries worldwide 3, 4. Sacubitril/valsartan is a novel angiotensin receptor–http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1611&familyId=740&familyType=ENZYME inhibitor (ARNI), inhibiting neprilysin, an endopeptidase (EC 3.4.24.11) which degrades http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4890 (NPs) and other vasoactive substrates, and blocking the http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=34. Following oral administration, sacubitril/valsartan delivers systemic exposure to the pro‐drug sacubitril (AHU377) and the AT1 receptor blocker (ARB) http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=3937 5. Sacubitril is rapidly hydrolysed by carboxyl esterase 1 to the active neprilysin inhibitor sacubitrilat (LBQ657) 6.
Loop diuretics are commonly prescribed for the treatment of patients with congestive heart failure 7, 8. http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=4839 inhibits the luminal Na+‐K+‐2Cl− transporter in the proximal renal tubule, thereby facilitating natriuresis and diuresis 8, 9, 10. As >80% of patients with HFrEF are using loop diuretics (primarily furosemide), sacubitril/valsartan and furosemide are expected to be frequently coadministered 3, 11, 12, 13, 14, 15, 16. Furosemide is a known substrate for http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1027 (OAT3) which is involved in the renal excretion of furosemide. In vitro studies have shown that sacubitrilat and valsartan have the potential to inhibit OAT3 [half‐maximal inhibitory concentration (IC50) 15.2 ± 5.6 μM and 1.11 ± 0.33 μM, respectively]. Hence, coadministration of sacubitril/valsartan may reduce urinary furosemide excretion and thereby affect the pharmacodynamic effects of furosemide. Alternatively, it is possible that coadministration of sacubitril/valsartan may also have an impact on the absorption of furosemide.
The present study was therefore designed to evaluate the potential impact of sacubitril/valsartan on furosemide pharmacokinetics and pharmacodynamics. Furthermore, the dose of furosemide was reviewed in patients treated with sacubitril/valsartan and furosemide in the PARADIGM‐HF study to evaluate the potential clinical relevance.
Methods
Study design
This was an open‐label, two‐period, single sequence, single‐centre study in healthy subjects on a standardized diet (Figure 1). In the first period, subjects received a single oral morning dose of 40 mg furosemide followed by serial plasma and urine sample collections to evaluate the pharmacokinetics and pharmacodynamics of furosemide. Following a washout period of approximately 2 days in the first period, 200 mg sacubitril/valsartan (97 mg/103 mg) was administered twice daily for 5 days (days 3 to 7) in the second period. On day 7, serial plasma samples were collected to assess the steady‐state pharmacokinetics of sacubitril/valsartan analytes. On day 8, 40 mg furosemide was coadministered with the morning dose of 200 mg sacubitril/valsartan, and serial plasma and urine samples were collected to assess the pharmacokinetics and pharmacodynamics of furosemide and steady‐state pharmacokinetics of sacubitril/valsartan analytes.
Figure 1.
Study design. b.i.d., twice daily; PD, pharmacodynamics; PK, pharmacokinetics
Subjects were kept on a standardized diet for calorie, sodium and potassium intake from 2 days before dosing until pharmacokinetic and pharmacodynamics assessments were complete (day 9). Fluid intake was standardized on urine pharmacodynamics collection days (days 1 and 8), and was monitored for 24 h on the day before and the day of urine pharmacodynamics collections (days −1, 1, 7 and 8).
All subjects provided written informed consent before any study assessments were performed. The protocol and informed consent form were approved by an institutional review board [IntegReview Ethical Review Board, Austin, TX, USA (Registration number: IORG0000689)]. The study design adhered to the regulatory guidelines provided by the Food and Drug Administration and European Medicines Agency (CDER 2012 and CHMP 2012) for conducting a drug–drug interaction study, designed in accordance with the International Conference on Harmonization tripartite guideline and was conducted in compliance with the ethical principles specified in the Declaration of Helsinki 17, 18.
Inclusion and exclusion criteria
Healthy male and female (not of child‐bearing potential) subjects aged 18–55 years, with a body weight of ≥50 kg and a body mass index (BMI) of 18–30 kg m–2 were included in the study. The subjects were required to be in good health as determined by medical history, physical examination, vital signs, laboratory tests and electrocardiogram (ECG). Key exclusion criteria were use of tobacco products in past 3 months, clinically significant abnormalities in the ECG, use of investigational drugs at the time of enrolment or prescription drugs within the 4 weeks before dosing, and donation of 400 ml or more blood within 8 weeks of initial dosing. All subjects provided written informed consent before the initiation of any study‐related evaluations.
Pharmacokinetic assessments
Sacubitril/valsartan and its analytes reach steady state within 3 days following twice‐daily (b.i.d.) dosing 19; hence, pharmacokinetic assessment of sacubitril/valsartan and its analytes were carried out at steady state (after 5 days of sacubitril/valsartan b.i.d. dosing). Blood samples were collected for pharmacokinetic assessments at predose and at several time points through 12 h (0.5, 1, 1.5, 2, 3, 4, 6, 8, 10 and 12 h) postdose by direct venipuncture or via an indwelling cannula inserted into a forearm vein. The volume of blood collected pertaining to administration of furosemide or sacubitril/valsartan alone and in combination were 2 ml and 4 ml, respectively for all the sampling time points. The collected blood samples were centrifuged and the separated plasma was stored at or below −20°C until shipment for further analysis.
Urine samples were collected and pooled for predefined sampling intervals (0–2, 2–4, 4–8, 8–12 and 12–24 h). An aliquot of 1 ml was obtained from each fraction and stored at or below −20°C until shipment for further analysis.
Sacubitril, sacubitrilat and valsartan were measured in plasma samples using a validated liquid chromatography (LC)–mass spectrometry/mass spectrometry (MS/MS) method. Following protein precipitation, reconstituted samples underwent high‐performance liquid chromatography (HPLC) with gradient elution followed by MS/MS detection using an API 4000 mass spectrometer. The lower limits of quantification (LLOQ) were 1 ng ml–1, 20 ng ml–1, and 10 ng ml–1 for sacubitril, sacubitrilat and valsartan, respectively 20. Within‐study quality control samples (QCs) showed an assay precision [% coefficient of variation (CV)] and accuracy (bias %) of 2.6–4.7% and −1.6‐5.0% for sacubitril, 1.2–5.5% and 1.3–6.0% for sacubitrilat, and 3.8–6.8% and 0.7–9.7% for valsartan; all met the acceptance criteria (±15%).
Plasma and urine furosemide concentrations were measured using a validated LC–MS/MS method. Plasma and urine samples were pretreated with 1% H3PO4 prior to being extracted by solid‐phase extraction, undergoing HPLC with gradient elution, and detection by MS/MS using an API 4000 mass spectrometer. The LLOQ for plasma and urine furosemide concentrations were 10 ng ml–1 and 50 ng ml–1, respectively. Within‐study QCs showed %CV in the range 0.5–4.2%, bias in the range −1.8% to 4.0% for plasma furosemide; and precision in the range 1.7–2.8%, accuracy in the range −2.0–6.0% for urine furosemide; all met the acceptance criteria (±15%).
Plasma pharmacokinetic parameters analysed for furosemide included area under the plasma concentration–time curve from time 0 to 24 h postdose (AUC0–24); AUC from time 0 to the time of the last quantifiable concentration (AUClast); AUC from time 0 to infinity (AUCinf); maximum observed plasma concentration after drug administration (Cmax); time to achieve Cmax (Tmax); and apparent plasma terminal elimination half‐life (t½), and for sacubitril/valsartan analytes included AUC from time 0 to the end of the dosing interval, tau (AUCtau); steady‐state Cmax (Cmax,ss); steady‐state Tmax (Tmax,ss). The urine pharmacokinetic parameters assessed were the amount of furosemide excreted in the urine from time 0 to 4 h, and 0 to 24 h after furosemide administration (Ae0–4, Ae0–24), and renal furosemide clearance (CLr).
Pharmacodynamic assessments
Similar to the urine sampling for pharmacokinetic analysis, a 5 ml aliquot of urine sample was collected for each sampling period and stored at 4°C for sodium, potassium and creatinine pharmacodynamics assessment. Urine volume and sodium, potassium and creatinine excretion were evaluated in urine samples collected from time 0 to 4 h, 4 to 8 h, and 0 to 24 h (Volume0–4, Volume4–8 and Volume0–24, respectively). In addition, creatinine concentrations were determined for each sampling period and each 24‐h period. Estimated glomerular filtration rate (eGFR) was calculated using the modification of diet in renal disease formula [eGFR (ml min–1/1.73 m–2) = 175 × (serum creatinine)–1.154 × (age)–0.203 × (0.742 if female) × (1.212 if African American)].
Safety assessments
Safety assessments included monitoring vital signs (supine systolic and diastolic blood pressure, pulse rate), 12‐lead ECGs, clinical laboratory values and reporting of adverse events (AEs) and serious adverse events (SAEs) with their severity and relationship with study drugs.
Statistical analyses
A total of 28 subjects were planned to be enrolled in order to obtain 24 subjects with completed assessments in this study. With 24 completer subjects, it was expected that the 90% confidence interval (CI) of the pharmacokinetic parameter ratios were within 84–119% of the observed mean for valsartan AUC and Cmax, within 92–109% of sacubitrilat AUC and Cmax, and within 81–124% of the observed ratio for furosemide AUC and Cmax, with 90% coverage probability. Sample size calculations were performed using Power Analysis and Sample Size (PASS) 12 software (NCSS LLC, Kaysville, UT, USA) based on log‐transformed pharmacokinetic parameters.
The effects of multiple doses of sacubitril/valsartan on single‐dose furosemide pharmacokinetics were determined using log‐transformed furosemide pharmacokinetic parameters (AUClast, AUCinf and Cmax) on day 8 (period 2) compared with those on day 1 (period 1). Similarly, to investigate the effects of a single dose of furosemide on the pharmacokinetics of sacubitril/valsartan analytes following multiple doses of sacubitril/valsartan, log‐transformed sacubitrilat and valsartan pharmacokinetics parameters AUCtau and Cmax,ss on day 8 (coadministration of sacubitril/valsartan and furosemide) were compared with those on day 7 (administration of sacubitril/valsartan alone). Geometric mean ratio (GMR) and 90% CI of the ratio were calculated by back‐transforming the estimated mean and 90% CI of the treatment difference. All pharmacokinetic parameters were determined by standard noncompartmental methods using Phoenix® Version 6.2.1.
Sodium excretion (natriuresis), potassium excretion (kaliuresis) and urinary volume (diuresis) were analysed using analysis of variance, with treatment and subject as fixed factors. Adjusted treatment means and treatment difference, along with 95% CI, were estimated from the model for all pharmacodynamic parameters.
Post hoc analysis of furosemide dose in the PARADIGM‐HF study
The PARADIGM‐HF study demonstrated a reduced risk of mortality and heart failure (HF) hospitalizations and prevention of clinical progression for sacubitril/valsartan compared with the ACE inhibitor enalapril in patients with HFrEF 3. Briefly, in this double‐blind trial, 8442 patients with New York Heart Association class II–IV HF, reduced ejection fraction (<35/40%) and a plasma B‐type natriuretic peptide (BNP) level of ≥150 pg ml–1 or N‐terminal proBNP (NT‐proBNP) level of ≥600 pg ml–1 were randomized to receive either sacubitril/valsartan 200 mg b.i.d. or 10 mg enalapril b.i.d., in addition to recommended therapy, including diuretics such as furosemide 3, 21. Eligible patients pre‐exposed to an ACE inhibitor or ARB at a dose equivalent to enalapril 10 mg b.i.d. for at least 4 weeks and on a stable dose of beta‐blockers (if tolerated) and a mineralocorticoid receptor antagonist (MRA) (if indicated) entered a single‐blind enalapril run‐in period (to achieve a target dose of 10 mg b.i.d.) for 2 weeks followed by a sacubitril/valsartan run‐in phase (100 mg up‐titrated to 200 mg b.i.d.) of 4–6 weeks. Patients who tolerated enalapril and sacubitril/valsartan at the target dose were randomized 1:1 to double‐blind treatment with either enalapril or sacubitril/valsartan. Dose adjustment of diuretics such as furosemide was allowed as per study protocol and as per clinical practice, to control HF symptoms 21. The median follow‐up time in the trial was 27 months 3. In this post hoc analysis, the baseline dose of furosemide was compared with the last post‐baseline dose of furosemide reported as a concomitant medication in the PARADIGM‐HF study 3. Patients with postbaseline furosemide doses that were not quantifiable (e.g. reported as ‘as needed’) were not included in this comparison. Median difference was reported for the analysis. For all analysis, SAS programming was used.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY 22, and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 23, 24, 25.
Results
Subjects
A total of 28 healthy subjects were enrolled and all subjects completed the study. The majority of the subjects were male (n = 24; 85.7%), with a mean age of 34.5 years, a mean weight of 75.9 kg and a mean BMI of 25.1 kg m–2. The majority of the subjects were black (n = 15; 53.6%) with the next largest group being Caucasian (n = 11; 39.3%) and then Native Americans (n = 2; 7.1%). The subject demographics and baseline characteristics are presented in Table 1.
Table 1.
Demographics of study subjects
| Parameter | n = 28 |
|---|---|
| Age (years) | 34.5 (9.26) |
| Gender, male, n (%); | 24 (85.7) |
| Race, n (%) | |
| Caucasian | 11 (39.3) |
| Black | 15 (53.6) |
| Native American | 2 (7.1) |
| Ethnicity, n (%) | |
| Hispanic/Latino | 9 (32.1) |
| Other | 19 (67.9) |
| Weight (kg) | 75.9 (8.09) |
| BMI (kg m –2 ) | 25.1 (2.86) |
Data are mean (standard deviation) unless otherwise specified;
BMI, body mass index
Pharmacokinetics
The mean plasma concentration–time profiles following multiple doses of sacubitril/valsartan 200 mg b.i.d. with or without coadministration of single‐dose 40 mg furosemide are presented in Figure 2. Following oral administration of sacubitril/valsartan alone, peak plasma concentrations of sacubitril, sacubitrilat and valsartan were observed at a median Tmax,ss of 0.52, 2.00 and 1.75 h, respectively (Table 2). There were no relevant changes in Tmax when sacubitril/valsartan was coadministered with furosemide (Table 2). The Cmax,ss values were comparable for sacubitril, sacubitrilat and valsartan when sacubitril/valsartan was coadministered with furosemide compared with sacubitril/valsartan alone (1810.04 ± 796.68 ng ml–1 vs. 1743.68 ± 801.26 ng ml–1 for sacubitril; 12 728.21 ± 2420.19 ng ml–1 vs. 11 835.71 ± 2262.61 ng ml–1 for sacubitrilat; and 4253.57 ± 1521.39 ng ml–1 vs. 3862.50 ± 1714.88 ng ml–1 for valsartan) (Table 2). The pharmacokinetics of sacubitril and sacubitrilat remained similar when sacubitril/valsartan was administered either alone or with furosemide, as the 90% CI of AUCtau and Cmax,ss of both analytes remained within 0.80–1.25 range. By contrast, both the AUCtau and Cmax,ss of valsartan increased marginally by 16% when coadministered with furosemide [90% CI for AUCtau (1.01, 1.31) and Cmax,ss (1.01, 1.32)] (Table 3).
Figure 2.
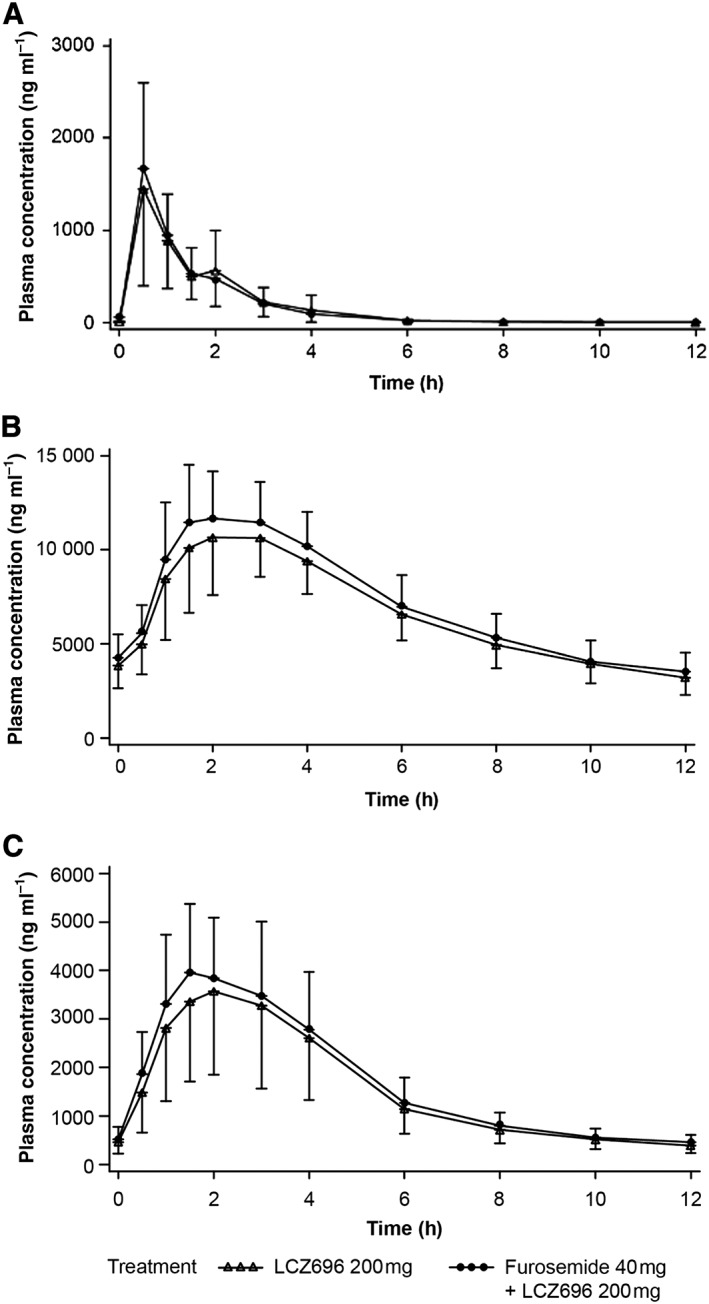
Plasma concentration–time profile of (A) sacubitril, (B) sacubitrilat and (C) valsartan following administration of 200 mg twice‐daily sacubitril/valsartan with or without furosemide (pharmacokinetic analysis set)
Table 2.
Plasma pharmacokinetic parameters (pharmacokinetic analysis set) analyses of sacubitril/valsartan
| Treatment | |||
|---|---|---|---|
| Analyte | Parameter | Sacubitril/valsartan + Furosemide | Sacubitril/valsartan alone |
| Sacubitril | AUCtau (h*ng ml–1) | 2328.82 (518.06) | 2333.44 (580.17) |
| Cmax,ss (ng ml–1) | 1810.04 (796.68) | 1743.68 (801.26) | |
| Tmax,ss (h) a | 0.50 [0.50–2.02] | 0.52 [0.50–2.03] | |
| Sacubitrilat | AUCtau (h*ng ml–1) | 86093.76 (17083.16) | 79595.08 (16363.48) |
| Cmax,ss (ng ml–1) | 12728.21 (2420.20) | 11835.71 (2262.61) | |
| Tmax,ss (h) a | 2.00 [1.50–4.00] | 2.00 [1.08–4.00] | |
| Valsartan | AUCtau, (h*ng ml–1) | 20953.01 (7392.18) | 18908.85 (8242.45) |
| Cmax,ss (ng ml–1) | 4253.57 (1521.39) | 3862.50 (1714.88) | |
| Tmax,ss (h) a | 1.50 [1.00–4.00] | 1.75 [1.00–4.00] | |
Data represent mean (standard deviation) unless otherwise specified. Sacubitril/valsartan alone: sacubitril/valsartan 200 mg twice daily; sacubitril/valsartan + furosemide: sacubitril/valsartan 200 mg twice daily + furosemide 40 mg single dose. AUCtau, area under the plasma concentration–time curve from time 0 to the end of the dosing interval tau (h*ng ml–1); Cmax,ss, maximum observed plasma concentration after drug administration at steady state (ng ml–1); Tmax,ss, time to achieve maximum observed plasma concentration after drug administration at steady state (h)
Data represent median (minimum – maximum)
Table 3.
Statistical analysis of plasma pharmacokinetic parameters of sacubitril/valsartan
| Geometric LS means of treatments | |||||
|---|---|---|---|---|---|
| Analyte | Parameter | Sacubitril/valsartan + furosemide | Sacubitril/valsartan alone | Estimated geometric mean ratio | 90% CI of the ratio |
| Sacubitril | AUCtau (h*ng ml–1) | 2278.91 | 2254.95 | 1.01 | (0.95, 1.06) |
| Cmax,ss (ng ml–1) | 1657.33 | 1588.29 | 1.04 | (0.91, 1.19) | |
| Sacubitrilat | AUCtau (h*ng ml–1) | 84644.93 | 78124.70 | 1.08 | (1.06, 1.10) |
| Cmax,ss (ng ml–1) | 12525.47 | 11635.58 | 1.08 | (1.03, 1.12) | |
| Valsartan | AUCtau (h*ng ml–1) | 19715.90 | 17050.14 | 1.16 | (1.01, 1.31) |
| Cmax,ss (ng ml–1) | 4006.10 | 3457.00 | 1.16 | (1.01, 1.32) | |
Sacubitril/valsartan alone: sacubitril/valsartan 200 mg twice daily; sacubitril/valsartan + furosemide: sacubitril/valsartan 200 mg twice daily + furosemide 40 mg single dose. AUCtau, area under the plasma concentration–time curve from time 0 to the end of the dosing interval tau (h*ng ml–1); CI, confidence interval; Cmax,ss, maximum observed plasma concentration after drug administration at steady state (ng ml–1); LS, least squares
The mean plasma concentration–time profiles following single‐dose administration of 40 mg furosemide with or without coadministration of 200 mg sacubitril/valsartan bid are presented in Figure 3. Furosemide was absorbed rapidly with a median Tmax of 2 h and a Cmax of 1063.54 ng ml–1. Coadministration of sacubitril/valsartan did not significantly alter the Tmax, but decreased the Cmax (geometric least square means 975.36 vs. 488.84 ng ml–1), AUClast (2460.65 vs. 1707.22 ng*h ml–1) and AUCinf (2552.95 vs. 1840.86 ng*h ml–1) of furosemide significantly by 50%, 31% and 28%, respectively, when coadministered with sacubitril/valsartan (Table 4). The plasma clearance of furosemide (CL/F) increased when coadministered with sacubitril/valsartan (Table 4). The urinary excretion of furosemide decreased by 26% (Ae0–24), while the renal clearance (CLr) of furosemide was not altered when furosemide was coadministered with sacubitril/valsartan (Table 4).
Figure 3.
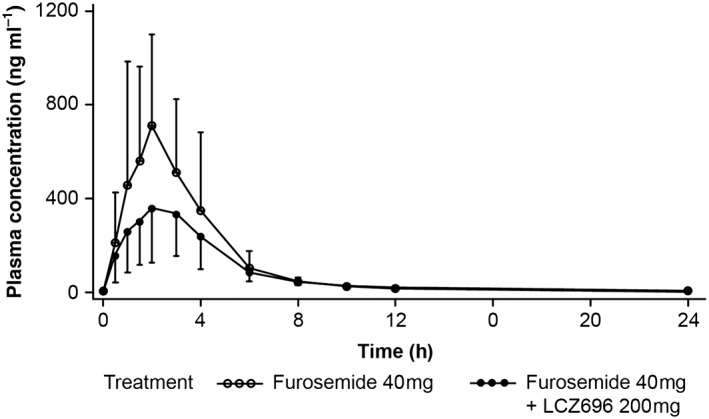
Plasma concentration–time profile of furosemide following single‐dose administration with or without sacubitril/valsartan (pharmacokinetic analysis set)
Table 4.
Plasma and urine pharmacokinetic parameters analyses of furosemide
| Plasma pharmacokinetics | |||||
|---|---|---|---|---|---|
| Analyte | Treatment | ||||
| Parameter | Sacubitril/valsartan + furosemide | Furosemide alone | Estimated geometric mean ratio | 90% CI of the ratio | |
| Furosemide | AUClast (h*ng ml–1) | 1707.22 | 2460.65 | 0.69 | (0.64, 0.74) |
| AUCinf (h*ng ml–1) | 1840.86 (n = 25) | 2552.95 (n = 25) | 0.72 | (0.67, 0.77) | |
| Cmax (ng ml–1) | 488.84 | 975.36 | 0.50 | (0.44, 0.56) | |
| Tmax (h) a | 2.00 [0.50–4.20] | 2.00 [0.50–6.00] | ‐ | ‐ | |
| CL/F | 22840.94 | 15759.15 | ‐ | ‐ | |
| Urine pharmacokinetics | |||||
|---|---|---|---|---|---|
| Furosemide | Ae0–4 (mg) | 5.56 | 8.03 | 0.69 | (0.59, 0.80) |
| Ae0–24 (mg) | 9.30 | 12.54 | 0.74 | (0.69, 0.79) | |
| CLr (ml min–1) | 90.75 | 84.95 | 1.06 | (1.02, 1.11) | |
Data represent geometric least squares mean unless otherwise specified. Furosemide alone: furosemide 40 mg single dose; sacubitril/valsartan + furosemide: sacubitril/valsartan 200 mg twice daily + furosemide 40 mg single dose; Ae0–4, amount excreted in urine from time 0 to 4 h after furosemide administration (mg for pharmacokinetics); Ae4–8, amount excreted in urine from 4 h to 8 h after furosemide administration (mg for pharmacokinetics); Ae0–24, amount excreted in urine from time 0 to 24 h after furosemide administration (mg for pharmacokinetics); AUClast, area under the plasma concentration–time curve from time 0 to the time of the last quantifiable concentration (h*ng ml–1); AUCinf, area under the plasma concentration–time curve from time 0 to infinity (h*ng ml–1); CI, confidence interval; CL/F, plasma clearance of furosemide; CLr, renal furosemide clearance (ml min–1); Cmax, maximum observed plasma concentration after drug administration (ng ml–1); Tmax, time to achieve maximum observed plasma concentration after drug administration (h)
Data represent median (minimum – maximum)
Pharmacodynamics: urinary volume and electrolytes
Sacubitril/valsartan coadministration with furosemide decreased the mean urinary volume within 4 h (adjusted mean difference −119 ml) and 4–8 h following dosing (adjusted mean difference −124 ml). However, there was no change in the urinary volume over 24 h (adjusted mean difference −8 ml) between the groups (Table 5). Following administration of furosemide alone, urinary sodium excretion was 128.87 mmol and 198.68 mmol within 4 h and 24 h of dosing, respectively. Following coadministration of furosemide and sacubitril/valsartan, urinary sodium excretion decreased to 92.15 mmol and 168.23 mmol within 4 h and 24 h of dosing, respectively. The mean urinary excretion of potassium and creatinine was comparable at all time points when furosemide was administered alone or in combination with sacubitril/valsartan (Table 5). No relevant changes in mean (± standard deviation) eGFR from baseline (107.8 ± 10.9 ml min–1 1.73 m–2) were observed when furosemide (103.1 ± 12.5 ml min–1 1.73 m–2) or sacubitril/valsartan (107.7 ± 12.5 ml min–1 1.73 m–2) were administered alone, or when coadministered (105.5 ± 11.5 ml min–1 1.73 m–2).
Table 5.
Urine pharmacodynamic parameters of furosemide (pharmacodynamic analysis set)
| Analyte | Parameter | ||||
|---|---|---|---|---|---|
| Treatment | Ae0–4 (mmol) | Ae4–8 (mmol) | Ae0–24 (mmol) | CLcr0–24 (ml min –1 ) | |
| Potassium | Sacubitril/valsartan + furosemide | 23.20 (5.82) | 11.83 (4.05) | 62.56 (10.76) | ‐ |
| Furosemide alone | 22.87 (5.60) | 10.04 (3.74) | 60.35 (8.08) | ‐ | |
| Estimated treatment difference (95% CI) | 0.33 (−1.70, 2.37) | 1.79 (0.15, 3.43) | 2.21 (−1.97, 6.39) | ‐ | |
| Sodium | Sacubitril/valsartan + furosemide | 92.15 (25.15) | 20.09 (11.70) | 168.23 (24.58) | ‐ |
| Furosemide alone | 128.87 (42.36) | 21.57 (12.89) | 198.68 (34.95) | ‐ | |
| Estimated treatment difference (95% CI) | −36.71 (−50.34, −23.09) | −1.49 (−8.25, 5.28) | −30.44 (−43.90, −16.99) | ‐ | |
| Creatinine | Sacubitril/valsartan + furosemide | 2.55 (0.48) | 2.75 (0.42) | 14.93 (2.58) | 131.91 (19.52) |
| Furosemide alone | 2.39 (0.50) | 2.94 (0.51) | 15.81 (2.64) | 137.17 (20.20) | |
| Estimated treatment difference (95% CI) | 0.16 (0.06, 0.25) | −0.18 (−0.35, −0.02) | −0.88 (−1.91, 0.15) | ‐ | |
| Urine volume | Parameter | ||||
|---|---|---|---|---|---|
| Volume0–4 (ml) | Volume4–8 (ml) | Volume0–24 (ml) | |||
| Sacubitril/valsartan + furosemide | 1521.43 (537.80) | 461.07 (206.39) | 3807.61 (640.95) | ||
| Furosemide alone | 1640.00 (413.49) | 585.36 (204.30) | 3816.07 (533.04) | ||
| Estimated treatment difference (95% CI) | −118.57 (−308.26, 71.12) | −124.29 (−220.53, −28.04) | −8.46 (−257.61, 240.68) | ||
Data represent mean (standard deviation) unless otherwise specified. Furosemide alone: furosemide 40 mg single dose; sacubitril/valsartan + furosemide: sacubitril/valsartan 200 mg twice daily + furosemide 40 mg single dose; Ae0–4, amount excreted in urine from time 0 to 4 h after furosemide administration (mmol for pharmacodynamics); Ae4–8, amount excreted in urine from 4 h to 8 h after furosemide administration (mmol for pharmacodynamics); Ae0–24, amount excreted in urine from time 0 to 24 h after furosemide administration (mmol for pharmacodynamics); CI, confidence interval; CLcr0–24, creatinine clearance (ml min–1); Volume0–4, volume of urine excreted from time 0 to 4 h after furosemide administration (ml); Volume4–8, volume of urine excreted from 4 h to 8 h after furosemide administration (ml); Volume0–24, volume of urine excreted from time 0 to 24 h after furosemide administration (ml)
Safety and tolerability
The safety analysis set included all 28 enrolled healthy subjects. Of these, six subjects experienced AEs during the study. AEs such as back pain (furosemide), diarrhoea, headache and presyncope (sacubitril/valsartan alone) were each reported by one subject (3.6%). All AEs were mild in severity and resolved without sequelae. A total of four AEs were suspected by the investigator to be related to the study drug, and included blurred vision, back pain, diarrhoea and headache. Two subjects reported AEs when treated with furosemide. There were no deaths or SAEs and none of the subjects had to discontinue the study owing to AEs. Supine systolic and diastolic blood pressure were slightly lower following coadministration of sacubitril/valsartan and furosemide (107.8 ± 9.9/67.8 ± 7.0 mmHg) compared with furosemide (115.5 ± 12.2/71.6 ± 6.1 mmHg) or sacubitril/valsartan (111.1 ± 10.7/67.5 ± 7.9 mmHg) administration alone. Pulse rate remained similar in all the treatment groups (74.9 ± 9.4, 71.0 ± 9.7 and 71.6 ± 9.9 bpm, respectively).
Analysis of furosemide dose in PARDIGM‐HF study
From the safety population of PARADIGM‐HF (n = 8432, 4203 and 4229 patients in the sacubitril/valsartan group and enalapril groups, respectively), there were 2224 and 2209 patients in the sacubitril/valsartan and enalapril groups, respectively, taking furosemide as a standing dose (and not on a per needed basis) (Table 6). The mean/median furosemide doses at baseline were 56.2 mg/40 mg and 57.1 mg/40 mg in the sacubitril/valsartan and enalapril groups, respectively. At the end of the study, 685 and 745 patients in the sacubitril/valsartan and enalapril groups, respectively, were taking standing doses of furosemide and had furosemide doses reported at baseline. The mean/median furosemide doses at the end of the study were 67.6 mg/40 mg and 78.8 mg/60 mg in the sacubitril/valsartan and enalapril groups, respectively. The increase from baseline of the mean furosemide dose was larger in the enalapril group compared with the sacubitril/valsartan treatment group (+21.7 mg with enalapril and +11.4 mg with sacubitril/valsartan). Similarly, an increase from baseline of the median furosemide dose was only observed in the enalapril treatment group (+20 mg) and no median increase was observed in the sacubitril/valsartan group.
Table 6.
Daily dose per patient of furosemide at baseline and end of study for double‐blind period, for those patients taking study drug and furosemide, by treatment group for Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure trial (PARADIGM‐HF) safety population (n = 8432)
| Visit | Statistics | Sacubitril/valsartan (mg) (n = 4203) | Enalapril (mg) (n = 4229) |
|---|---|---|---|
| Baseline | N | 685 | 745 |
| Mean (95% CI) | 56.2 (51.9, 60.5) | 57.1 (53.7, 60.5) | |
| SD | 57.03 | 47.72 | |
| Median | 40.0 | 40.0 | |
| Minimum | 0.0 | 0.0 | |
| Maximum | 1000.0 | 750.0 | |
| End of study | N | 685 | 745 |
| Mean (95% CI) | 67.6 (63.5, 71.7) | 78.8 (73.4, 84.2) | |
| SD | 55.14 | 75.41 | |
| Median | 40.0 | 60.0 | |
| Minimum | 0.0 | 5.7 | |
| Maximum | 500.0 | 1000.0 |
Only oral doses of furosemide are considered in this table. CI, confidence interval; SD, standard deviation
Discussion
Sacubitril/valsartan (LCZ696) has been recently approved for the treatment of HFrEF. Furosemide is one of the most commonly prescribed diuretics in patients with HFrEF; hence, coadministration of sacubitril/valsartan and furosemide is likely to occur in clinical practice. However, there is a potential for a drug interaction because sacubitrilat and valsartan inhibit OAT3 in vitro and furosemide is an OAT3 substrate. Therefore, we evaluated the impact of sacubitril/valsartan on the pharmacokinetics and pharmacodynamics of furosemide in healthy subjects in the present study, demonstrating a reduction in systemic and urinary exposure of furosemide when coadministered with sacubitril/valsartan, resulting in reduced natriuresis. While furosemide exerts diuretic effects both in healthy subjects and in patients with HFrEF, there could be quantitative differences in its action resulting from neurohormonal dysregulation, water and sodium retention, potential renal dysfunction and/or concomitant medication/s in patients with HFrEF. However, a post hoc analysis of the PARADIGM‐HF trial revealed no changes in the median furosemide dose in patients with HFrEF who were randomized to sacubitril/valsartan compared with enalapril, suggesting that no dose adjustment of furosemide is necessary when coadministered with sacubitril/valsartan.
The pharmacokinetic parameters of sacubitril, sacubitrilat and valsartan were unaltered when sacubitril/valsartan was administered alone or in combination with furosemide. A 16% increase in the Cmax of valsartan was observed; however, this observation was not considered to be clinically relevant as the small change in Cmax was less than the coefficient of variation of valsartan pharmacokinetic parameters (35.8%).
Coadministration of sacubitril/valsartan decreased the Cmax of furosemide by 50% and plasma exposure by 28%. Consistent with this observation, the renal excretion of furosemide was decreased by 26%. It should be noted that furosemide is an OAT3 substrate and that both sacubitrilat and valsartan have been shown to inhibit OAT3 under in vitro conditions (IC50 15 μM and 1.1 μM, respectively) 26, 27. If OAT3 is inhibited, then it would be expected that decreased renal excretion is associated with increased systemic exposure to furosemide. Therefore, the observed decrease in the renal excretion of furosemide is unlikely to result from transporter inhibition but could be due to decreased plasma exposure as a result of reduced absorption of furosemide following its coadministration with sacubitril/valsartan. The possible mechanism of the interaction during absorption could be that the solubility (in vivo dissolution) of furosemide may have been affected in the presence of sacubitril/valsartan when coadministered. This hypothesis is consistent with the biopharmaceutical properties of furosemide. Furosemide exhibits highly variable bioavailability due to its poor solubility, and is sensitive to changes in gastric emptying and gastric pH conditions 9. An earlier clinical study, in which the bioavailability of furosemide was also shown to decrease by 26% when coadministered with valsartan, suggests that this observation might be related to the presence of valsartan 28. However, valsartan and furosemide are coadministered in clinical practice without a recommendation for dose adjustment of furosemide 29. Based on these results, it is considered unlikely that sacubitril and sacubitrilat have a significant impact on the physiological action of furosemide.
Coadministration of sacubitril/valsartan and furosemide had no impact on the mean urinary excretion of potassium or creatinine, or on the overall urine volume over 24 h, which is the clinically relevant endpoint for outpatient management using furosemide. However, there was a 37 mmol and 30 mmol decrease in sodium excretion with the combination treatment relative to furosemide administration alone at 4 h and 24 h, respectively, which could potentially have been the result of reduced furosemide exposure.
In order to understand the overall clinical relevance of the observed drug–drug interaction in healthy subjects, a post hoc analysis of data from the PARADIGM‐HF study (in which furosemide is an important concomitant medication) was conducted to understand whether there was an increase in the dose of furosemide within the sacubitril/valsartan group that could be the result of decreased furosemide exposure. This analysis revealed that the increase in the mean dose of furosemide was smaller in the sacubitril/valsartan group compared with the enalapril group, and that the median dose of furosemide increased only in the enalapril group but not in the sacubitril/valsartan group. This suggests that despite significant decreases in the systemic exposure of furosemide and urine sodium excretion in healthy subjects, the interaction between sacubitril/valsartan and furosemide is of minimal clinical relevance in patients with HF. This may partially be attributed to additional renal and/or haemodynamic effects of sacubitril/valsartan. Recent evidence shows that by inhibiting neprilysin, sacubitril/valsartan treatment transiently increases natriuresis and diuresis compared with valsartan in Asian patients with salt‐sensitive hypertension 30. By potentiating the natriuretic peptide signalling in HF patients, it is likely that sacubitril/valsartan may also exert similar effects in HF patients.
However, this analysis has some limitations. The analysis of furosemide dose in the PARADIGM‐HF patients was not prespecified. In addition, the PARADIGM‐HF was not designed to analyse the mutual impact of sacubitril/valsartan and furosemide on their pharmacodynamics and pharmacokinetics. Despite these limitations, the data presented from the present analysis provide important ‘real‐world’ evidence relevant for clinical use.
In summary, coadministration with sacubitril/valsartan significantly decreased systemic exposure to furosemide and 24‐h natriuresis following furosemide administration. However, 24‐h diuresis remained unaltered. In the PARADIGM‐HF study, the median furosemide dose was similar at baseline and at the end of the study in the sacubitril/valsartan group. In addition, the increase over time in the mean furosemide dose was larger in the enalapril group compared with the sacubitril/valsartan group, suggesting that the observed drug interaction may not be clinically relevant in patients with HFrEF, and that no initial dose adjustment of furosemide is necessary when coadministered with sacubitril/valsartan.
Competing Interests
All the authors were employees of Novartis Pharmaceutical Corporation at the time of study conduct and may own company stocks.
This study was supported by Novartis Pharmaceutical Corporation . The authors acknowledge Sabiha A. Mondal (principal investigator), PPD, Austin, TX, USA, for the study conduct. The authors also acknowledge Rohan Mitra, Nagabhushana Ananthamurthy and Sreedevi Boggarapu (Novartis Healthcare Pvt. Ltd., Hyderabad, India) for providing medical writing and editorial support for the development of the manuscript.
Contributors
T.L. was the principal investigator. S.A., U.S., T.L. and G.S. designed the study, performed the research and wrote the manuscript. W.Z. and P.P. analysed the data. All authors reviewed and approved the manuscript.
Ayalasomayajula, S. , Schuehly, U. , Pal, P. , Chen, F. , Zhou, W. , Sunkara, G. , and Langenickel, T. H. (2018) Effect of the angiotensin receptor–neprilysin inhibitor sacubitril/valsartan on the pharmacokinetics and pharmacodynamics of a single dose of furosemide. Br J Clin Pharmacol, 84: 926–936. doi: 10.1111/bcp.13505.
References
- 1. The SOLVD Investigators . Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med 1991; 325: 293–302. [DOI] [PubMed] [Google Scholar]
- 2. Vilela‐Martin JF. Spotlight on valsartan‐sacubitril fixed‐dose combination for heart failure: the evidence to date. Drug Des Devel Ther 2016; 10: 1627–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, et al Angiotensin‐neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371: 993–1004. [DOI] [PubMed] [Google Scholar]
- 4. Packer M, McMurray JJ, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, et al Angiotensin receptor neprilysin inhibition compared with enalapril on the risk of clinical progression in surviving patients with heart failure. Circulation 2015; 131: 54–61. [DOI] [PubMed] [Google Scholar]
- 5. Ksander GM, Ghai RD, deJesus R, Diefenbacher CG, Yuan A, Berry C, et al Dicarboxylic acid dipeptide neutral endopeptidase inhibitors. J Med Chem 1995; 38: 1689–1700. [DOI] [PubMed] [Google Scholar]
- 6. Shi J, Wang X, Nguyen J, Wu AH, Bleske BE, Zhu HJ. Sacubitril is selectively activated by carboxylesterase 1 (CES1) in the liver and the activation is affected by CES1 genetic variation. Drug Metab Dispos 2016; 44: 554–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wargo KA, Banta WM. A comprehensive review of the loop diuretics: should furosemide be first line? Ann Pharmacother 2009; 43: 1836–1847. [DOI] [PubMed] [Google Scholar]
- 8. Roush GC, Kaur R, Ernst ME. Diuretics: a review and update. J Cardiovasc Pharmacol Ther 2014; 19: 5–13. [DOI] [PubMed] [Google Scholar]
- 9. Ponto LL, Schoenwald RD. Furosemide (frusemide). A pharmacokinetic/pharmacodynamic review (Part I). Clin Pharmacokinet 1990; 18: 381–408. [DOI] [PubMed] [Google Scholar]
- 10. Weber KT. Furosemide in the long‐term management of heart failure: the good, the bad, and the uncertain. J Am Coll Cardiol 2004; 44: 1308–1310. [DOI] [PubMed] [Google Scholar]
- 11. Effectiveness of spironolactone added to an angiotensin‐converting enzyme inhibitor and a loop diuretic for severe chronic congestive heart failure (the Randomized Aldactone Evaluation Study [RALES]). Am J Cardiol 1996; 78: 902–907. [DOI] [PubMed] [Google Scholar]
- 12. Philbin EF, Cotto M, Rocco TA Jr., Jenkins PL. Association between diuretic use, clinical response, and death in acute heart failure. Am J Cardiol 1997; 80: 519–522. [DOI] [PubMed] [Google Scholar]
- 13. Cohn JN, Tognoni G, Glazer R, Spormann D. Baseline demographics of the Valsartan Heart Failure Trial. Val‐HeFT Investigators. Eur J Heart Fail 2000; 2: 439–446. [DOI] [PubMed] [Google Scholar]
- 14. Neuberg GW, Miller AB, O'Connor CM, Belkin RN, Carson PE, Cropp AB, et al Diuretic resistance predicts mortality in patients with advanced heart failure. Am Heart J 2002; 144: 31–38. [DOI] [PubMed] [Google Scholar]
- 15. Mielniczuk LM, Tsang SW, Desai AS, Nohria A, Lewis EF, Fang JC, et al The association between high‐dose diuretics and clinical stability in ambulatory chronic heart failure patients. J Card Fail 2008; 14: 388–393. [DOI] [PubMed] [Google Scholar]
- 16. Jentzer JC, DeWald TA, Hernandez AF. Combination of loop diuretics with thiazide‐type diuretics in heart failure. J Am Coll Cardiol 2010; 56: 1527–1534. [DOI] [PubMed] [Google Scholar]
- 17. Center for Drug Evaluation and Research (CDER) UDoHaHS, Food and Drug Administration . Draft guidance for industry, drug interaction studies − study design, data analysis, implications for dosing, and labeling recommendations. Docket No FDA‐2006‐D‐0036 2012.
- 18. Committee for Human Medicinal Products (CHMP) EMA . Guideline on the investigation of drug interactions. London, UK; 2012.
- 19. Ayalasomayajula S, Jordaan P, Pal P, Chandra P, Albrecht D, Langenickel T, et al Assessment of drug interaction potential between LCZ696, an angiotensin receptor neprilysin inhibitor, and digoxin or warfarin. Clin Pharmacol Biopharm 2015; 4: 147. [Google Scholar]
- 20. Gu J, Noe A, Chandra P, Al‐Fayoumi S, Ligueros‐Saylan M, Sarangapani R, et al Pharmacokinetics and pharmacodynamics of LCZ696, a novel dual‐acting angiotensin receptor‐neprilysin inhibitor (ARNi). J Clin Pharmacol 2010; 50: 401–414. [DOI] [PubMed] [Google Scholar]
- 21. McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, et al Dual angiotensin receptor and neprilysin inhibition as an alternative to angiotensin‐converting enzyme inhibition in patients with chronic systolic heart failure: rationale for and design of the Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure trial (PARADIGM‐HF). Eur J Heart Fail 2013; 15: 1062–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S, et al The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res 2017; 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E, et al The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 2017; 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD, et al The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 2017; 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA, et al The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. Br J Pharmacol 2017; 174: S17–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Entresto® EMA SmPC . Available at http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/004062/WC500197536.pdf (last accessed 21 August 2017).
- 27. Yamashiro W, Maeda K, Hirouchi M, Adachi Y, Hu Z, Sugiyama Y. Involvement of transporters in the hepatic uptake and biliary excretion of valsartan, a selective antagonist of the angiotensin II AT1‐receptor, in humans. Drug Metab Dispos 2006; 34: 1247–1254. [DOI] [PubMed] [Google Scholar]
- 28. Bindschedler M, Degen P, Flesch G, de Gasparo M, Preiswerk G. Pharmacokinetic and pharmacodynamic interaction of single oral doses of valsartan and furosemide. Eur J Clin Pharmacol 1997; 52: 371–378. [DOI] [PubMed] [Google Scholar]
- 29. de la Sierra A, Segura J, Gorostidi M, Banegas JR, de la Cruz JJ, Ruilope LM. Diurnal blood pressure variation, risk categories and antihypertensive treatment. Hypertens Res 2010; 33: 767–771. [DOI] [PubMed] [Google Scholar]
- 30. Wang TD, Tan RS, Lee HY, Ihm SH, Rhee MY, Tomlinson B, et al Effects of sacubitril/valsartan (lcz696) on natriuresis, diuresis, blood pressures, and NT‐proBNP in salt‐sensitive hypertension. Hypertension 2017; 69: 32–41. [DOI] [PubMed] [Google Scholar]