

Abstract
Comprehensive and accurate evaluation of data quality and false-positive biomarker discovery is critical to direct the method development/optimization for quantitative proteomics, which nonetheless remains challenging largely due to the high complexity and unique features of proteomic data. Here we describe an experimental null (EN) method to address this need. Because the method experimentally measures the null distribution (either technical or biological replicates) using the same proteomic samples, the same procedures and the same batch as the case-vs-contol experiment, it correctly reflects the collective effects of technical variability (e.g., variation/bias in sample preparation, LC–MS analysis, and data processing) and project-specific features (e.g., characteristics of the proteome and biological variation) on the performances of quantitative analysis. To show a proof of concept, we employed the EN method to assess the quantitative accuracy and precision and the ability to quantify subtle ratio changes between groups using different experimental and data-processing approaches and in various cellular and tissue proteomes. It was found that choices of quantitative features, sample size, experimental design, data-processing strategies, and quality of chromatographic separation can profoundly affect quantitative precision and accuracy of label-free quantification. The EN method was also demonstrated as a practical tool to determine the optimal experimental parameters and rational ratio cutoff for reliable protein quantification in specific proteomic experiments, for example, to identify the necessary number of technical/biological replicates per group that affords sufficient power for discovery. Furthermore, we assessed the ability of EN method to estimate levels of false-positives in the discovery of altered proteins, using two concocted sample sets mimicking proteomic profiling using technical and biological replicates, respectively, where the true-positives/negatives are known and span a wide concentration range. It was observed that the EN method correctly reflects the null distribution in a proteomic system and accurately measures false altered proteins discovery rate (FADR). In summary, the EN method provides a straightforward, practical, and accurate alternative to statistics-based approaches for the development and evaluation of proteomic experiments and can be universally adapted to various types of quantitative techniques.
Keywords: false altered proteins discovery rate (FADR), experimental null (EN), quantitative proteomics, ion-current-based quantification
Graphical abstract
INTRODUCTION
Recent advances in mass spectrometry (MS) technology greatly advance quantitative proteomics. Labeling and label-free methods represent two major categories of proteomics quantitative techniques. Although labeling methods help to achieve accurate quantification, the methods fall short in that the number of quantifiable replicates is limited by the number of isotopic reagents,1,2 and that labeling procedure may introduce additional variations and that the labeling reagents are costly. By comparison, label-free methods have the potential to quantify a large number of biological replicates in one set, although some technical challenges remain.3,4
High quantitative accuracy and precision are critically important to discover the significantly altered proteins in profiling experiments and correctly estimate the protein ratios between groups. Current proteomics technologies frequently suffer from suboptimal quantitative accuracy and precision due to the biases and variations in experimental and data-processing procedures. The problem is often more pronounced in label-free proteomics because label-free methods do not employ internal standards to account for the aforementioned bias and variations. For label-free quantification, numerous factors may affect the quality of quantification. First, data-processing methods, such as the selection of quantitative features, quantification algorithms, and database-searching approaches, may significantly impact the quantitative accuracy and precision. Second, experimental design may greatly affect the outcome of quantitative proteomics. For instance, sample size (i.e., number of replicates in each group) could markedly impact analytical accuracy and precision of relative quantification.5 For quantification using technical replicates, increased number of technical replicates improves the quality of quantification by alleviating technical variability. In the event that biological replicates are employed, which is often critical for clinical and pharmaceutical application, small sample size renders the analysis liable to bias and variation due to biological variability. The utilization of sufficient biological samples per group will greatly alleviate the problem of both biological and technical variability. Running more replicates is more time-consuming, costly, and often more technically demanding, which necessitates the identification of an optimal sample size for a specific proteomic project. Another example is that the order in which the samples are analyzed in one experiment set could influence quantitative precision and accuracy.4 Finally, many case-specific features (i.e., sample complexity, protein distribution, and interferences from biological matrix and background noise) can exert profound yet perplexing effects on the label-free quantification.6,7 The effects of these factors should be extensively evaluated in specific proteomic projects to achieve high quantitative accuracy and precision and high sensitivity to determine protein changes between groups. Nonetheless, this need has not been adequately addressed largely due to the lack of a practical and universal tools.
Another important issue for proteomic quantification is the estimation and control of false-positive discovery of altered proteins. In label-free proteomic comparison, a large number of proteins will be compared using statistical tests. Because of the nature of multiple testing problems and other factors associated with experimental biases and variations, false-positives are inevitable as the probability of falsely rejecting the null hypothesis is sizable. It is desirable to control false-positives effectively to avoid following the wrong biological lead. Some statistics efforts have been developed and widely used in high-throughput discovery-based analysis such as genomics and transcriptomics;8,9 however, false-positive control methods are less used in proteomics studies6,10 partially due to the fact that p values of most proteomics test statistics are calculated based on either asymptotic distribution or central limit theorem that requires relatively large sample sizes. In practice, typical proteomics studies have limited sample sizes that render many false-positive control methods problematic.10–12
Here we described an experimental null (EN) method, which quantitatively estimates the effects of experimental and data-processing procedures on the quality of proteomic quantification to guide method development and optimization and experimentally measures the null distribution in a specific proteomic project to accurately estimate and control FADR. By performing EN analysis using the same proteomic samples and in the same batch of the experiment, this method can correctly assess the collective effects of technical variably (e.g., variation and bias in sample preparation, LC–MS analysis, and data processing) and project-specific features (e.g., characteristics of the proteome and biological variation). In this study, the performance of EN method was assessed in a number of proteomics systems as well as concocted samples where true identities of positives/negatives are known.
METHODS
Experimental Strategies Evaluation Using Null Proteomics Samples
Two types of null proteomics sets, one representing technical replicates and the other representing biological replicates, were used for the evaluation of different experimental strategies. For technical replicates analysis, repetitive analyses of pooled peripheral blood mononuclear cells (PBMCs) from HIV-1 positive patients, rat liver, or E. coli (details see later) samples were used; for biological replicates analysis, individual rat brain samples from wild-type animals of the same source, identical age, breed, and feeding background were used.
FADR Evaluation Using Spiked-in Sample Sets
To enable an accurate evaluation of the performance to estimate FADR, we prepared two spike-in samples by spiking one proteome (at small portions, representing altered proteins) into another proteome of different species (larger portion, representing unchanged background proteins). Corresponding to the evaluation of technical and biological replicates, we employed two different sets (shown in Figure 4A): (i) Technical replicates: repetitive analysis of two samples prepared by spiking medium abundance human plasma proteins (details in the Supplementary Methods) to E. coli extracts with 1.5-fold difference between group. (ii) Mimicked biological replicates: E. coli proteins (changed proteins) were spiked into rat liver proteins (constant background) at various levels with a mean of 2-fold between the two groups (N = 10 per group) (Figure 4A).
Figure 4.

Estimation of FADR by EN method using concocted proteomic samples. (A) Details of the designs of the concocted samples. Sample set A is for technical replicates and sample set B is for mimic biological replicates. (B) Correlation of the FADR estimated by EN method versus actual FADR (calculated based on known true-positives/negatives). Details are shown in the Methods section.
Protein Extraction and Digestion
Cell or tissue samples used in this study were homogenized in an ice-cold lysis buffer (50 mM Tris-formic acid, 150 mM NaCl, 0.5% sodium deoxycholate, 2% SDS, 2% NP-40, complete protease inhibitor, pH 8.0) using a Polytron homogenizer (Kinematica AG, Switzerland). Homogenization was performed for a 5–10 s burst at 15 000 rpm, followed by a 20 s cooling period until the foam was settled. This procedure was repeated for 10 times. The mixture was then sonicated on ice for ~1 min, followed by centrifugation at 140 000g for 30 min at 4 °C. The supernatant was carefully transferred to a fresh tube and the protein concentrations were measured using BCA protein assay (Pierce, Rockford, IL). The resulted samples were stored at −80 °C until analysis. A precipitation/on-pellet-digestion protocol was employed as previously described for enzyme digestion process.4,13–15
Nano LC–MS/MS Analysis
The nano-RPLC (reverse-phase liquid chromatography) system consisted of a Spark Endurance autosampler (Emmen, Holland) and an ultrahigh pressure Eksigent (Dublin, CA) Nano-2D Ultra capillary/nano-LC system. Mobile phases A and B were 0.1% formic acid in 2% acetonitrile and 0.1% formic acid in 88% acetonitrile, respectively. Four µL of samples was loaded onto a reversed-phase trap (300 µm ID × 1 cm) with 1% mobile phase B at a flow rate of 10 µL/min, and the trap was washed for 3 min. A series of nanoflow gradients (flow rate at 250 nL/min) was used to back-flush the trapped samples onto the nano-LC column (75 µm ID × 75 cm) for separation. The nano-LC column was heated at 52 °C to improve both chromatographic resolution and reproducibility. An LTQ/ Orbitrap XL hybrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA) was used for protein identification. The parameters for MS are shown in our previous publications.4,13–15
Database Search and MS2-Based Protein Quantification
The raw data files were searched against the Swiss-Prot protein database using the Sequest HT algorithm embedded in Proteome Discoverer 1.4 (Thermo-Scientific). A total of 20 229, 25 023, 10 474, 33 682, and 32 116 entries were presented, respectively, in human database, concatenated rat E. coli database, concatenated human-E. coli database, and reviewed and unreviewed rat and reviewed mouse–rat combined protein database. The search parameters used were as follows: 20 ppm tolerance for precursor ion masses and 0.6 Da for fragment ion masses. Two missed cleavages were permitted for fully tryptic peptides. Carbamidomethylation of cysteines was set as a fixed modification, and a variable modification of methionine oxidation was allowed. The false discovery rate (FDR) was determined by using a target-decoy search strategy.16 Scaffold 4.117 (Proteome Software, Portland, OR), which is capable of handling large-scale proteomic data sets, was used to validate MS2-based peptide and protein identification. The peptide and protein FDR was controlled at 0.1 and 1%, respectively, for all sample sets to ensure stringent identification. Validated peptides were grouped into individual protein clusters by Scaffold.
Protein Quantification Using Ion-Current-Based Strategy
Spectral Counts (SpC) and Total MS2 ion current (MS2-TIC) quantification of proteins were performed under the same protein/peptide identification criteria as previously described. The “Quantitative Analysis” module embedded in Scaffold Q+ (Proteome Software, Portland, OR) was employed. The “weighted spectra” method was chosen for SpC-based quantitative analysis, while “total TIC” method were chosen for MS2-TIC-based quantitative analysis. 0.5 and 10 000 were used to substitute the missing values, respectively, for SpC and MS2-TIC. The quantitative analysis by ion current (IC), also known as MS1, was performed by two steps: procurement of area-under-the-curve data for peptides using SIEVE v2.1 (Thermo Scientific, San Jose, CA) and then a sum-intensity method to aggregate the quantitative data from peptide level to protein level as we previously described.18 SIEVE is a label-free proteomics quantification software that performs chromatographic alignment and global intensity-based MS1 feature extraction.19 The software processes chromatographic alignment between LC–MS runs using the ChromAlign algorithm.20 Quantitative “frames” were defined based on m/z and retention time of peptide precursors in the aligned runs. Peptide IC areas were calculated for individual replicates in each frame. MS2 fragmentation scans associated with each frame were matched to identified proteins (cf. the database search and data validation procedure) by the locally developed R script (Supplementary Methods in the SI). Then, the data were subject to filtering, normalization, and quantification previously described.18 IC-based quantification results using linear mixed model (LMM) were obtained by R package MsStats2.21 The significantly altered proteins were defined by the rational ratio cutoff obtained from the EN method, and a p-value filter <0.05 (by student t test) was also applied.
RESULTS AND DISCUSSION
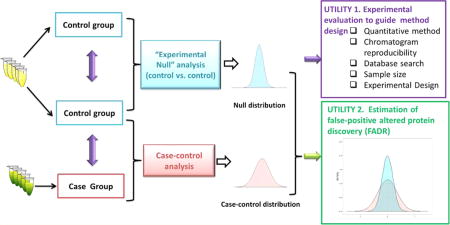
The schematic describing the rationale of the EN method is shown in Figure 1. Two control groups (either technical or biological replicates depending on the requirement of the project) were prepared, analyzed by LC–MS and quantitatively compared using exactly the same protocols as those for the comparison of case-vs-contol groups to obtain an EN data set. In case the variation in the case group is markedly higher than that in the control group, the case-vs-case comparison can be used to measure null distribution and thus to obtain a most conservative evaluation.
Figure 1.

Schematic of experimental null (EN) method and its utilities in guiding experiment design/development and FADR estimation.
The EN data set provides two important utilities: (1) Measurement of the quantitative precision and accuracy (i.e., because the true ratio value of all proteins is 1.0) is achieved by a specific experimental strategy in a specific proteomic system, which greatly facilitates the design and optimization of the experimental and data processing procedures. (2) Because the EN data set represents the null distribution devoid of true-positive proteins, it can be used to estimate the level of false-positive discovery of altered proteins arising from biological or technical variations. Similar strategies have been used by our lab to estimate FADR and to determine the optimal cutoff threshold (e.g., folds of changes) for the discovery of altered proteins in various pharmaceutical and clinical applications.14,18,22,23 Nevertheless, a systematic assessment of this method, for example, whether it provides an accurate estimation of FADR, has not been conducted thus far.
Here we extensively appraise the performance of EN method in the previously described two aspects. To show a proof of concept, we assessed the effects of sample-size, experimental design, reproducibility of chromatographic separation, database searching approaches, and quantitative strategies on label-free proteomic quantification. A wide range of clinical, preclinical, and cellular proteomes in both biological and technical replicates were analyzed by the EN method to demonstrate its universal applicability. To ensure a reliable investigation, we employed highly stringent criteria to define the quantifiable proteins, including strict criteria for peptide identification resulting in low peptide FDR, strict criteria for peak detection and frame generation, and the fact that each quantifiable protein was required to have quantitative data from at least two unique peptides that both met the above two criteria (Methods). For FADR evaluation, two spiked-in proteomics sample sets (respectively, representing the investigations using technical and mimic biological replicates) with known positives and negatives were employed.
To avoid confusion, we define some terms used in this paper as the following: “Precision” is the reproducibility for quantification of the relative ratio of a protein between two groups, while “accuracy” refers to the closeness of a measured protein ratio to the true value. “FDR” denotes the false-positive rate in protein/peptide identification, and “FADR” is the false-positive rate in discovery of altered proteins between groups.
EN Method to Optimize Quantitative Strategies and Experimental Setup
Accuracy and Precision of Different Label-Free Strategies
Two distinct quantitative approaches are widely used for label-free proteomics quantification, employing quantitative features acquired from either area-under-the-curve of MS1 IC7,24–26 or MS2 product ion signals, for example, SpC and MS2-TIC27,28 to measure relative protein abundances. While MS2-based approaches conduct quantification at protein level, IC-based approaches require the aggregation of quantitative data from peptide-level to protein-level to calculate protein abundance.29 In this regard, aggregation methods based on sum-intensity (sum-IC), median intensity(median-IC), and linear mixed model (LMM-IC) are mostly used.10,21
Here we utilized EN method to evaluate the quantitative accuracy and precision by previously mentioned MS1- and MS2-based approaches. Technical replicate analysis of pooled E. coli samples was used as the test system. Eight repetitive analyses of a pooled sample were randomly assigned to two groups (N = 4/group), which were then compared using different quantitative strategies. In total, 688 proteins were quantified with high confidence (Supplementary Table 1 in the SI). The intragroup precisions for quantification of individual proteins by the five approaches are shown in Figure 2A. While the means of coefficient of variation (CV%) within group by all three IC-based methods are <10%, those by SpC and MS2-TIC were much higher (~20 and ~40%, respectively). Moreover, all IC-based approaches resulted in substantially better quantitative accuracy than those based on MS2 (i.e., MS2-TIC and SpC, Figure 2B). In terms of quantitative accuracy, only 27 and 14% of proteins were quantified within ±20% deviation from true value (ratio = 1.0) range for TIC-MS2 and SpC, respectively; by comparison, >90% of proteins fell within the same threshold by the three IC-based methods. Median-IC approach showed significantly higher mean deviation and CV% of deviations than LMM-IC and Sum-IC. These results corroborate well with our recent observations in other evaluation systems.18,30 Only Sum-IC approach was employed in the following investigations because of its superior accuracy and precision than MS2-based approaches and the fact that it is the most prevalently used approach among the IC-based methods.
Figure 2.

Use of EN method to evaluate the effects of different experimental strategies and parameters on precision (i.e., within-group variability, CV %) and quantitative accuracy(i.e., deviation from true ratio, 0 at log 2 scale). (A,B) Effects of different quantitative strategies including ion current (IC)-based methods (median-IC, LMM-IC, and Sum-IC) and MS2-based methods (MS2-TIC and SpC). (C,D) Effects of the quality of chromatographic separation and (E,F) effects of database selection.
Effect of Chromatographic Reproducibility on Accuracy and Precision
For IC-based quantification of a peptide, the retention time of the peptide is an important index to locate the IC peak of the peptide among parallel runs and to eliminate isomeric interferences.31 Therefore, alignment of chromatograms, which corrects mild chromatographic variations and coordinates peptide peaks among multiple LC–MS runs, has become a critical component for IC-based analysis.7,13 Nonetheless, if the chromatographic reproducibility was too low, even alignment algorithm cannot properly adjust the variations in peptide retention times among many runs,2 which may negatively impact the quality of relative quantification. Such effects have not been previously investigated.
In our pilot study, we found the reproducibility of chromatographic separation to profoundly affect the accuracy and precision. To examine this relationship, we employed the EN method to quantitatively evaluate the effect of chromatographic separation on accuracy and precision and to identity the optimal cutoff of alignment score (given by the alignment algorithm as an indicator of chromatographic reproducibility20), which enables high-quality quantification while maximizing the useable sample runs. It was found the alignment score of 0.7 was a good cutoff when using Chrom-align algorithm. An example for the comparison of optimal versus suboptimal separation reproducibility is shown in Figure 2C,D. Two LC– MS data sets, each containing six replicate analyses of a pooled rat liver sample that were separated into two groups (N = 3/ group), were selected for this evaluation. In one data set, alignment scores of all six runs were >0.7 (the set of “optimal reproducibility”), while in the other data set, four runs had scores >0.7 and two had scores <0.7 (“suboptimal reproducibility”). Detailed quantification data are in Supplementary Table 2 in the SI. The set with optimal reproducibility showed markedly better accuracy and precision (p = 0.0008) for quantification of the 1054 proteins over the set with suboptimal reproducibility (Figure 2C,D). This example demonstrated that suboptimal chromatographic reproducibility may substantially deteriorate the accuracy and precision of IC-based MS1 quantification, even when only a relatively small portion of all runs in a group have reproducibility problem, thereby underscoring the importance of consistent LC separation.
Effect of Database Selection
Database selection is not only important for reliable protein identification but also may profoundly affect proteomic quantification because quantification of incorrectly identified peptides may severely undermine quantification.32 Our preliminary study indicated that the selection of database may impact quantification, and such influence appeared to be more pronounced in species for which complete, reviewed protein database is not available. For these species, either concatenated database using reviewed sequences from multiple species or nonreviewed, more complete single-species database including both reviewed and unreviewed rat proteins was used. Two groups of LC–MS runs, each for the analysis of rat livers from five control animals (N = 5/group, biological replicates), were compared obtain the EN data set. Quantitative results of the two previuosly mentioned databases were compared, with the search parameters and FDR cutoff thresholds otherwise the same. Searching with the combined database resulted in 156 more proteins than using the single-species database (1060 vs 904). Detailed quantification data are in Supplementary Table 3 in the SI. Interestingly, while the quantitative precision achieved by the two databases is similar (Figure 2E), the quantitative accuracy by the combined database is significantly lower than that using the single-species database (p < 0.01, Figure 2F). One possible explanation is that some rat proteins identified based on mouse entries in the combined database contain fewer identified peptides than the counterparts identified using the complete (albeit non-reviewed) rat database; fewer quantifiable peptides per protein therefore led to inferior quantitative accuracy. Therefore, the results underscored the importance of using of single-species and complete database.
EN Method to Determine Rational Ratio Cutoff for Reliable Quantification
In proteomics studies, statistics test are used to determine the significantly altered proteins between groups. As observed in our works and others,4,6,22 the p values and fold of change do not always correlate well in proteomic quantification, especially for proteins with small changes (e.g., many proteins exhibit an excellent p value, albeit extremely small fold of changes). In practice, a fold-change cutoff (i.e., cutoff of the minimum fold of change that is considered significant) is often implemented on top of statistic test to eliminate proteins with small changes, which not only removes the very small alternations that are not likely to be biologically relevant but also is shown to reduce false-positive discovery.18
However, a rational determination of the ratio cutoff has largely been overlooked. In most events, the selection of the cutoff was merely based on the perception of the researchers as to what is the lowest threshold of change that is considered “biologically significant”. It is important to determine the rational ratio cutoff that warrants reliable quantification in a proteomic project. For instance, in many biological systems it is critical to characterize mild changes of marker proteins;23 it is essential to have the capacity of determining the minimal protein fold change that can be quantified with confidence to develop and optimize a method capable of quantifying the desired extents of fold changes. A rational fold-change cutoff is closely related to both the proteomic quantification method and the interindividual biological/technical variations, which are, in turn, governed by the combinatory effects of specific experiments, data-processing procedures, and characteristics of a specific proteomic system. Because the EN method can correctly assess this effect via estimating the null distribution using the same quantitative method in the same proteomic system as for the case-vs-contol set, here it was utilized to determine the rational cutoff for reliable relative quantification
Evaluation of the Effect of Sample Size
Sample size has a substantial effect on the reliability of proteomic quantification. Increased sample size substantially alleviates the effect of biological or technical variability and thereby enhances the reliability of quantification and the ability to perceive smaller ratio changes with increased statistical power.33 Running more replicates is often more time-consuming, costly, and technically challenging. Therefore, it is necessary to determine an optimal sample size that provides sufficient accuracy and capacity to quantify the desired extent of protein changes. Although statistical methods can be used to calculate the proper sample size needed to reach the desired statistical power based on prior information on the null distribution, our pilot study showed that these approaches did not work well for label-free proteomics due to the complexity of proteomic experiments and the fact that typically small sample sizes are used (<10/ group, data not shown).
To identify the optimal sample size, we employed the EN method to evaluate the effect of the numbers of technical and biological replicates on proteomic quantification. Assessment of the effect on technical replicates was performed by repetitive analysis of pooled human PBMC samples at different sample sizes (N = 1–9/group). The histograms of the log 2 ratio of the quantified proteins under different sample sizes are shown in Figure 3A. The quantitative accuracy increased as the variance decreaseed along with the sample size, as reflected by the much tighter ratio distribution around the true value (log 2 ratio = 0) at higher replicate numbers. This increased accuracy enhanced the ability to quantify more subtle changes, and some typical profiles are shown in Figure 3B. Here we define the rational cutoff for reliable quantification as the absolute log 2 ratio value below which 95% of total quantified proteins fall, which is measured by the control-vs-control comparison; this threshold estimates the extent of protein ratio changes that can be reliably quantified in a specific proteomic system by a specific experimental procedure. As shown in Figure 3B, the rational cutoff decreases with the increased sample size. Using this relationship, a proper sample size can be determined; for example, when it is required to determine protein changes as low as 0.25 (log 2 scale), five technical replicates per group are sufficient; three replicates is the minimum for protein changes down to 0.3 (log 2 scale). Detailed data are shown in Supplementary Table 4 in the SI.
Figure 3.

EN method to determine the effect of sample size and experimental design on the capacity to determine protein abundance changes. Histograms of the log 2 ratio of the quantified proteins under different sample sizes for analyzing (A) technical replicates, (C) biological replicates, and (E) different experimental design (true value = 0 at log 2 scale). (B,D,F) Plot for protein % above log 2 ratios. Dashed vertical line denotes the rational ratio cut off for reliable quantification, which is defined as the ratio, where 95% of the proteins quantified in the EN data set fall below.
For the experiments using biological replicates, it is important to identify the proper number of biological subjects per group that are needed to achieve confident quantification down to a desired fold change. Because this value is dependent on both the experimental method and the characteristics of the specific proteomic sample set (e.g., protein abundance distribution in each sample and interindividual variability), the EN method is a valuable tool for this purpose. Rat brain samples from individual control animals (with identical age, breed, and feeding schedules) were used for the EN evaluation of sample size. As shown in Figure 3C, the protein log 2 ratio distribution was tighter along with the increased biological replicates. Corresponding to this observation, the rational cutoff decreased substantially with the increased number of subjects (Figure 3D). In this particular proteomic system, a minimum of seven and five biological replicates per group are necessary to achieve a rational cutoff down to 0.25 (log 2 scale) and 0.35 (log 2 scale), respectively. Detailed data are in Supplementary Table 5 in the SI.
Evaluation of the Effect of Experimental Design
Experimental design often markedly affects the quality of a quantitative proteomic investigation. For example, samples in an experimental set are often analyzed in a random order to reduce analytical biases.4 Nonetheless, the effect of the order in which the samples are analyzed has not been quantitatively characterized. Here we investigated two sequences for sample analysis using the EN method.”Sequential” and “alternating” sequences with N = 5/group were selected from the same set of technical replicate runs. “Sequential” refers to the sequential assignment of groups A and B so that all samples in the same groups were prepared and analyzed sequentially (e.g., runs 1–5 are group A, while runs 6–10 are group B), while “alternating” refers to alternating assignment (i.e., odd-number runs are group A and even-number runs are group B).
As shown in Figure 3E,F, the alternating set resulted in remarkably better accuracy for protein quantification and lower ratio cutoff for confident quantification. Detailed data are provided in Supplementary Table 6 in the SI. This is likely because the drift in ionization efficiency and chromatographic behaviors over time could result in significant analytical bias in the “sequential” design but much less so in “alternating” design. Moreover, it was found that strictly random order resulted in inferior performance compared with “alternating”, probably because of the small number of replicates typically used in proteomics studies (data not shown). As a result, alternating analysis order may be superior to sequential or random order when using relatively small replicate numbers (e.g., N = <5).
EN Method for FADR Estimation
Using the EN method, the empirical null distribution for the specific proteome experiments of interest can be estimated accurately; it can be employed to estimate the FADR and to determine the optimal threshold for the discovery of altered proteins with a target FADR. Although statistics-based FADR control methods have been successfully employed in transcriptomics and genomics studies, most of these methods have been demonstrated to be suboptimal and are therefore seldom employed in the proteomics field.6,36 Previously, the EN or similar methods have been used by our lab and others to estimate the FADR in specific proteomic investigations as an alternative to statistical methods.4,14,23,33,37,38 This strategy is based on the assumption that the level of false-positives in the control-vs-control data set (i.e., the null data set) reflects that in the case-vs-contol data set with similar null distribution.
Here we examined whether the EN method can accurately estimate FADR. For accurate evaluation, we concocted two sample sets with known identities of true-positives and -negatives, by spiking one proteome into another at various ratios. (Details are in the Supplementary Methods in the SI.) One set is for the evaluation of technical replicates (sample set A), which consists of replicate analysis of two concocted samples (N = 4 analysis for each) prepared by spiking a small amount of human plasma protein mixtures (variable proteins and true-positives; details in Methods section) to E. coli protein mixture (i.e., serve as a constant proteome background, true negatives) with a 1.5-fold difference in human plasma proteins levels (Figure 4A). Sample set B was designed to mimic relative quantification using biological replicates (Figure 4B). E. coli protein mixtures (true-positives) were spiked into rat liver proteins (true negatives) at varied ratios in each of the 20 samples (N = 10 samples/group); the mean ratio of the true-positives is 2.0 between the two groups (p < 0.01).
The FADR under a given set of cutoff thresholds (Supplementary Methods in the SI) was estimated by uniformly applying the thresholds to both the case-vs-control and control-vs-control (i.e., EN) sets using a homemade R script (Supplementary Method in the SI). The FADR was defined as the ratio of the number of false-positives in the EN set over the positives in the case-vs-contol set using the same cutoff value. The FADR estimated by EN method is compared against true FADR, which is calculated as observed false-positive/ (true-positive + false-positive) in the case-vs-contol experiment, and the results are shown in Figure 4B,C. Five estimated FADR levels, 2, 5, 10, 15, and 20%, were investigated. A good correlation between the estimated versus true FADR was achieved for both technical and biological replicates (R2 > 0.9 for both). The EN method was found to achieve a markedly more accurate estimation of FADR than using the Student’s t test, Limma-modified t test, and Fisher-exact test followed by Benjamini–Hochberg correction (Supplementary Figure in the SI). Detailed proteomics data for sample sets A and B are shown in Supplementary Tables 7 and 8 in the SI, respectively. Results of estimated FADR and actual FADR calculation using different strategies are provided in Supplementary Tables 9 and 10 in the SI.
Using the EN method for FADR estimation is analogous to the use of a decoy database to determine FDR for protein/ peptide identification in two ways. First, both the decoy database and the EN data set (control-vs-control) are devoid of true-positive; therefore, like any protein/peptide identified from the decoy database is false-positive by the searching practice, the “differentially altered” proteins discovered from the EN data set represent false-positives caused by technical/ biological variations. Second, in the target-decoy searching strategy, the underlying assumption is that the extent of false-positives in target and decoy is roughly equal, which is also the case for the EN method: the null distribution in EN data set should resemble the distribution of negatives in the case/ control data set because the negative distribution resembles the true null distribution in the long run. We examined this assumption by comparing the true null distributions (as the identities of true negatives are known) in the sample sets A and B against experimentally measured null distribution in the EN set. As evident from Figure 5A,B, the EN distribution of protein log 2 ratios overlaps well with the true negative distribution in the case/control set in both sample sets. These results demonstrated that the EN method can accurately estimate null distribution in complex proteomes.
Figure 5.

Assessment of the underlying assumption of EN method by comparing the null distribution measured by EN method versus the true null distribution (negative distribution) in the case-vs-contol sample set for (A) sample set A and (B) sample set B. Excellent agreement was observed for both sample sets.
CONCLUSIONS
For proteomic profiling, comprehensive evaluation of the quality of proteomic quantification as well as false-positive discovery of altered proteins is critical to direct method development and optimization, but it is also challenging largely due to the high complexity and unique features of proteomic data. Here we developed an EN method to address this need, which experimentally measures the actual null distribution in specific proteomic projects regardless of the statistical method used. The null sample set, for example, control-vs-control, is prepared and analyzed along with the case-vs-contol set using the same experimental and data processing strategy.
On the basis of the fact that the true ratio of all proteins is 1.0 between the two null groups, one important utility of the EN data set is to evaluate the quality of quantification and thus to identify the optimal experimental and data-processing strategies. To show a proof of concept, we employed the EN method to evaluate the quantitative accuracy and precision using different experimental parameters and in various proteomic systems. It was observed that chromatographic reproducibility and the choices of quantitative features and databases profoundly impact the accuracy and precision of proteomic quantification. Moreover, the EN method was used to assess the effects of sample sizes and experimental designs on the ability to reliably quantify subtle protein changes. It was observed that increased sample size resulted in higher quantitative accuracy and improved ability to quantify small changes for both technical and biological replicates; these data can be used to choose the proper sample size according the requirement of the project. Furthermore, on the basis of the evaluation by EN method, an alternating sample analysis sequence is preferred for experiments using relatively small sample size (e.g., N ≤ 5).
Because the EN method measures the null distribution of a specific proteomic project, it can also be utilized to estimate and control the false-positive discovery of altered proteins in the case/control set. In this study, the performance of EN method was assessed using two concocted sample sets mimicking proteomic quantification using respectively technical and biological replicates. The true-positives/negatives in these sets span a wide concentration range and thus enable the evaluation of quantification at different protein levels. It was found the EN method was able to correctly reflect the null distribution in the case/control sets, therefore enabling a relatively accurate estimation of FADR levels in the case/ control set.
In summary, the EN method provides an empirical, straightforward, and promising alternative to statistical approaches that mostly address the “multiple hypothesis testing” problem, which sometimes may not be sufficiently robust for proteomic profiling, where the typical sample sizes are relatively small (e.g., N < 10/group). This method can also be adapted to proteomic investigations based on isotope-labeling strategies. Finally, as the EN method experimentally measures the null distribution, it can also serve as a tool for the development of statistical efforts in the proteomic field.
Supplementary Material
Acknowledgments
We acknowledge NIH grants U54HD071594 (J.Q.) and HL103411 (J.Q.), Center of Protein Therapeutics Industrial Award (J.Q.), and American Heart Association (AHA) award 12SDG9450036 (J.Q.).
ABBREVIATIONS
- CV
coefficient-of-variation
- EN
experimental null
- FDR
false discovery rate
- FADR
false altered proteins discovery rate
- IC
ion current
- MS
mass spectrometry
- SpC
spectrum counts
- MS2-TIC
total MS2 ion current
- LMM
linear mixed model
Footnotes
ASSOCIATED CONTENT
Supplementary Figure S1. Comparison of performance of FADR estimation by EN method and Benjamini–Hochberg. Supplementary Tables 1–10. Quantitative strategies comparison, comparison of effect of chromotographic reproducibility, comparison of effect of database selection, EN method to determine the effect of sample size in technical and biological sample sets, EN method to determine the effect of experimental design, estimation of FADR by EN method using concocted sample set a (technical replicates), estimation of FADR by EN method using concocted sample set b (biological replicates), detailed statistics of estimated FADR and true FADR calculation in sample sets a and b. details of supplementary methods. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.5b00200.
The authors declare no competing financial interest.
References
- 1.Mueller LN, Brusniak MY, Mani DR, Aebersold R. An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J. Proteome Res. 2008;7(1):51–61. doi: 10.1021/pr700758r. [DOI] [PubMed] [Google Scholar]
- 2.Zhu W, Smith JW, Huang CM. Mass spectrometry-based label-free quantitative proteomics. J. Biomed Biotechnol. 2010;2010:840518. doi: 10.1155/2010/840518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffin NM, Yu J, Long F, Oh P, Shore S, Li Y, Koziol JA, Schnitzer JE. Label-free, normalized quantification of complex mass spectrometry data for proteomic analysis. Nat. Biotechnol. 2010;28(1):83–89. doi: 10.1038/nbt.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tu C, Li J, Jiang X, Sheflin L, Pfeffer B, Behringer M, Fliesler S, Qu J. Ion current-based proteomic profiling of the retina in a rat model of Smith-Lemli-Opitz Syndrome. Mol. Cell. Proteomics. 2013;12:3583–3598. doi: 10.1074/mcp.M113.027847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwammle V, Leon IR, Jensen ON. Assessment and improvement of statistical tools for comparative proteomics analysis of sparse data sets with few experimental replicates. J. Proteome Res. 2013;12(9):3874–3883. doi: 10.1021/pr400045u. [DOI] [PubMed] [Google Scholar]
- 6.Margolin AA, Ong SE, Schenone M, Gould R, Schreiber SL, Carr SA, Golub TR. Empirical Bayes analysis of quantitative proteomics experiments. PLoS One. 2009;4(10):e7454. doi: 10.1371/journal.pone.0007454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen X, Young R, Canty JM, Qu J. Quantitative proteomics in cardiovascular research: Global and targeted strategies. Proteomics: Clin. Appl. 2014;8(7–8):488–505. doi: 10.1002/prca.201400014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moskvina V, Schmidt KM. On multiple-testing correction in genome-wide association studies. Genet. Epidemiol. 2008;32(6):567–573. doi: 10.1002/gepi.20331. [DOI] [PubMed] [Google Scholar]
- 9.Bertolino F, Cabras S, Castellanos ME, Racugno W. Unscaled Bayes factors for multiple hypothesis testing in microarray experiments. Stat. Methods Med. Res. 2012 doi: 10.1177/0962280212437827. [DOI] [PubMed] [Google Scholar]
- 10.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26(12):1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 11.Chang J, Van Remmen H, Ward WF, Regnier FE, Richardson A, Cornell J. Processing of data generated by 2-dimensional gel electrophoresis for statistical analysis: missing data, normalization, and statistics. J. Proteome Res. 2004;3(6):1210–1218. doi: 10.1021/pr049886m. [DOI] [PubMed] [Google Scholar]
- 12.Hendrickson EL, Xia Q, Wang T, Leigh JA, Hackett M. Comparison of spectral counting and metabolic stable isotope labeling for use with quantitative microbial proteomics. Analyst. 2006;131(12):1335–1341. doi: 10.1039/b610957h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duan X, Young R, Straubinger RM, Page B, Cao J, Wang H, Yu H, Canty JM, Qu J. A straightforward and highly efficient precipitation/on-pellet digestion procedure coupled with a long gradient nano-LC separation and Orbitrap mass spectrometry for label-free expression profiling of the swine heart mitochondrial proteome. J. Proteome Res. 2009;8(6):2838–2850. doi: 10.1021/pr900001t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tu C, Li J, Bu Y, Hangauer D, Qu J. An ion-current-based, comprehensive and reproducible proteomic strategy for comparative characterization of the cellular responses to novel anti-cancer agents in a prostate cell model. J. Proteomics. 2012;77:187–201. doi: 10.1016/j.jprot.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tu C, Li J, Young R, Page BJ, Engler F, Halfon MS, Canty JM, Jr, Qu J. Combinatorial peptide ligand library treatment followed by a dual-enzyme, dual-activation approach on a nanoflow liquid chromatography/orbitrap/electron transfer dissociation system for comprehensive analysis of swine plasma proteome. Anal. Chem. 2011;83(12):4802–4813. doi: 10.1021/ac200376m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elias JE, Haas W, Faherty BK, Gygi SP. Comparative evaluation of mass spectrometry platforms used in large-scale proteomics investigations. Nat. Methods. 2005;2(9):667–675. doi: 10.1038/nmeth785. [DOI] [PubMed] [Google Scholar]
- 17.Searle BC. Scaffold: a bioinformatic tool for validating MS/ MS-based proteomic studies. Proteomics. 2010;10(6):1265–1269. doi: 10.1002/pmic.200900437. [DOI] [PubMed] [Google Scholar]
- 18.Tu C, Sheng Q, Li J, Shen X, Zhang M, Shyr Y, Qu J. ICan: An Optimized Ion Current-Based Quantification Procedure with Enhanced Quantitative Accuracy and Sensitivity in Biomarker Discovery. J. Proteome Res. 2014;13(12):5888–5897. doi: 10.1021/pr5008224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lopez MF, Kuppusamy R, Sarracino DA, Prakash A, Athanas M, Krastins B, Rezai T, Sutton JN, Peterman S, Nicolaides K. Mass spectrometric discovery and selective reaction monitoring (SRM) of putative protein biomarker candidates in first trimester Trisomy 21 maternal serum. J. Proteome Res. 2011;10(1):133–142. doi: 10.1021/pr100153j. [DOI] [PubMed] [Google Scholar]
- 20.Sadygov RG, Maroto FM, Huhmer AF. ChromAlign: A two-step algorithmic procedure for time alignment of three-dimensional LC-MS chromatographic surfaces. Anal. Chem. 2006;78(24):8207–8217. doi: 10.1021/ac060923y. [DOI] [PubMed] [Google Scholar]
- 21.Clough T, Thaminy S, Ragg S, Aebersold R, Vitek O. Statistical protein quantification and significance analysis in label-free LC-MS experiments with complex designs. BMC Bioinf. 2012;13(Suppl 16):S6. doi: 10.1186/1471-2105-13-S16-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qu J, Young R, Page BJ, Shen X, Tata N, Li J, Duan X, Fallavollita JA, Canty JM. Reproducible Ion-Current-Based Approach for 24-Plex Comparison of the Tissue Proteomes of Hibernating versus Normal Myocardium in Swine Models. J. Proteome Res. 2014;13(5):2571–2584. doi: 10.1021/pr5000472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nouri-Nigjeh E, Sukumaran S, Tu C, Li J, Shen X, Duan X, DuBois DC, Almon RR, Jusko WJ, Qu J. Highly multiplexed and reproducible ion-current-based strategy for large-scale quantitative proteomics and the application to protein expression dynamics induced by methylprednisolone in 60 rats. Anal. Chem. 2014;86(16):8149–8157. doi: 10.1021/ac501380s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiener MC, Sachs JR, Deyanova EG, Yates NA. Differential mass spectrometry: a label-free LC-MS method for finding significant differences in complex peptide and protein mixtures. Anal. Chem. 2004;76(20):6085–6096. doi: 10.1021/ac0493875. [DOI] [PubMed] [Google Scholar]
- 25.Gautier V, Mouton-Barbosa E, Bouyssie D, Delcourt N, Beau M, Girard JP, Cayrol C, Burlet-Schiltz O, Monsarrat B, Gonzalez de Peredo A. Label-free quantification and shotgun analysis of complex proteomes by one-dimensional SDS-PAGE/NanoLC-MS: evaluation for the large scale analysis of inflammatory human endothelial cells. Mol. Cell Proteomics. 2012;11(8):527–539. doi: 10.1074/mcp.M111.015230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bondarenko PV, Chelius D, Shaler TA. Identification and relative quantitation of protein mixtures by enzymatic digestion followed by capillary reversed-phase liquid chromatography-tandem mass spectrometry. Anal. Chem. 2002;74(18):4741–4749. doi: 10.1021/ac0256991. [DOI] [PubMed] [Google Scholar]
- 27.Gao J, Friedrichs MS, Dongre AR, Opiteck GJ. Guidelines for the routine application of the peptide hits technique. J. Am. Soc. Mass Spectrom. 2005;16(8):1231–1238. doi: 10.1016/j.jasms.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 28.Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 2004;76(14):4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 29.Carrillo B, Yanofsky C, Laboissiere S, Nadon R, Kearney RE. Methods for combining peptide intensities to estimate relative protein abundance. Bioinformatics. 2010;26(1):98–103. doi: 10.1093/bioinformatics/btp610. [DOI] [PubMed] [Google Scholar]
- 30.Tu C, Li J, Sheng Q, Zhang M, Qu J. Systematic assessment of survey scan and MS2-based abundance strategies for label-free quantitative proteomics using high-resolution MS data. J. Proteome Res. 2014;13(4):2069–2079. doi: 10.1021/pr401206m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neilson KA, Ali NA, Muralidharan S, Mirzaei M, Mariani M, Assadourian G, Lee A, van Sluyter SC, Haynes PA. Less label, more free: approaches in label-free quantitative mass spectrometry. Proteomics. 2011;11(4):535–553. doi: 10.1002/pmic.201000553. [DOI] [PubMed] [Google Scholar]
- 32.Mallick P, Kuster B. Proteomics: a pragmatic perspective. Nat. Biotechnol. 2010;28(7):695–709. doi: 10.1038/nbt.1658. [DOI] [PubMed] [Google Scholar]
- 33.Li Q, Roxas BA. An assessment of false discovery rates and statistical significance in label-free quantitative proteomics with combined filters. BMC Bioinf. 2009;10:43. doi: 10.1186/1471-2105-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carr SA, Anderson L. Protein quantitation through targeted mass spectrometry: the way out of biomarker purgatory? Clin. Chem. 2008;54(11):1749–1752. doi: 10.1373/clinchem.2008.114686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makawita S, Diamandis EP. The bottleneck in the cancer biomarker pipeline and protein quantification through mass spectrometry-based approaches: current strategies for candidate verification. Clin. Chem. 2010;56(2):212–222. doi: 10.1373/clinchem.2009.127019. [DOI] [PubMed] [Google Scholar]
- 36.Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat. Biotechnol. 2006;24(8):971–983. doi: 10.1038/nbt1235. [DOI] [PubMed] [Google Scholar]
- 37.Serang O, Cansizoglu AE, Kall L, Steen H, Steen JA. Nonparametric Bayesian evaluation of differential protein quantification. J. Proteome Res. 2013;12(10):4556–4565. doi: 10.1021/pr400678m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karp NA, McCormick PS, Russell MR, Lilley KS. Experimental and statistical considerations to avoid false conclusions in proteomics studies using differential in-gel electrophoresis. Mol. Cell. Proteomics. 2007;6(8):1354–1364. doi: 10.1074/mcp.M600274-MCP200. [DOI] [PubMed] [Google Scholar]
- 39.Choi H, Nesvizhskii AI. False discovery rates and related statistical concepts in mass spectrometry-based proteomics. J. Proteome Res. 2008;7(1):47–50. doi: 10.1021/pr700747q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.