

Abstract
Energy production in myocardial cells occurs mainly in the mitochondrion. Although alterations in mitochondrial functions in the senescent heart have been documented, the molecular bases for the aging-associated decline in energy metabolism in the human heart are not fully understood. In this study, we examined transcription profiles of genes coding for mitochondrial proteins in atrial tissue from aged (≥65 years old) and comorbidities-matched adult (<65 years old) patients with preserved left ventricular function. We also correlated changes in functional activity of mitochondrial oxidative phosphorylation (OXPHOS) complexes with gene expression changes. There was significant alteration in the expression of 10% (101/1,008) of genes coding for mitochondrial proteins, with 86% downregulated (87/101). Forty-nine percent of the altered genes were confined to mitochondrial energetic pathways. These changes were associated with a significant decrease in respiratory capacity of mitochondria oxidizing glutamate and malate and functional activity of complex I activity that correlated with the downregulation of NDUFA6, NDUFA9, NDUFB5, NDUFB8, and NDUFS2 genes coding for NADH dehydrogenase subunits. Thus, aging is associated with a decline in activity of OXPHOS within the broader transcriptional downregulation of genes regulating mitochondrial energetics, providing a substrate for reduced energetic efficiency in the senescent human atria.
Keywords: myocardium, gene expression, oxidative phosphorylation, ATP
The mitochondrion is the main site of energy production in myocardial cells. Aging-related cardiac structural and functional alterations, including changes in mitochondrial energetic capacity and oxidative stress, play a major role in increasing the susceptibility of the heart to mechanical and electrical dysfunction, as reflected by the increasing prevalence of both atrial fibrillation and heart failure in the aging population (1,2). Research has shown that aging induces broad transcriptional changes in genes coding for mitochondrial functions in animal models, along with downregulation of functional activities of proteins involved in energy metabolism in animal hearts (3,4). Although changes in cardiac energetics in humans have previously been shown to be associated with heart failure (5) and atrial fibrillation (1), the component resulting from age-related changes in mitochondrial energetics has not yet been fully elucidated in nondiseased human hearts and the molecular bases for aging-associated decline in energy metabolism in the human heart are not fully defined.
The purpose of our study was to identify aging-related transcriptional changes in genes coding for mitochondrial proteins involved in substrate metabolism, oxidative phosphorylation system (OXPHOS), and the tricarboxylic acid (TCA) cycle, and to correlate these with the functional changes in the OXPHOS in human atrial appendage tissue from adult and aged patients free of atrial pathologies.
Methods
Patient Selection
Right (RAA) and left (LAA) atrial appendage tissue was harvested from adult (<65 years, n = 24) and aged (≥65 years, n = 26) patients undergoing open heart surgery. Patients with congenital, structural, or functional atrial disease—including persistent or permanent atrial fibrillation, classes III/IV heart failure, advanced mitral valve disease—and those undergoing emergency bypass surgeries or requiring inotropic support were excluded to avoid confounding effects of disease on myocardial function and gene expression. Clinical history, echocardiogram, and electrocardiogram were used to exclude patients with structural or electrical heart disease or systemic illnesses. Within 5–10 min after surgical resection, atrial tissue was either used for mitochondrial isolation or snap-frozen in liquid nitrogen, and stored at –80°C for gene, protein expression, and functional assessment of the OXPHOS. The research study was conducted in accordance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans. The study was approved by the Mayo Clinic and Aurora Health Care Institutional Review Boards for human subject research, written informed consent was obtained from subjects, and all patients’ privacy rights were observed.
Mitochondrial Isolation
Mitochondria were isolated from the LAA by differential centrifugation as described previously (4). Tissue was placed into ice-cold isolation buffer containing (in mM) 200 mannitol, 50 sucrose, 5 KH2PO4, 1 EGTA, 5 MOPS (pH 7.3), 0.2% BSA, and homogenized using OMNI GLH-115 Polytron homogenizer (OMNI International, Kennesaw, GA). Mitochondria were washed in isolation buffer containing neither EGTA nor BSA. Protein concentration was determined with a BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA).
Mitochondrial Respiration
Mitochondrial respiration was measured at room temperature using the Mitocell 200 micro respirometry system comprising a 782 oxygen meter, 1302 oxygen electrode, and MT200 respirometer (Strathkelvin Instruments, Scotland). Mitochondria were incubated in a mitochondrial assay buffer (in mM) of 250 sucrose, 5 MgCl2, 2 KH2PO4, 0.02 EGTA, and 10 Tris–HCl, with a pH of 7.3, in the presence of substrates for complex I (5 glutamate and 2.5 malate), complex II (5 succinate and 0.002 rotenone), or complex IV (6 ascorbate, 0.3 N'N'N'N'-tetramethyl-p-phenylenediamine (TMPD) and 1 µg antimycin A/ml) (6). The rate of basal respiration was measured in the presence of substrates; the rates of ADP-dependent respiration (state 3) and respiration upon depletion of ADP (state 4) were determined after addition of 0.2 mM ADP; the rate of uncoupled respiration was determined in the presence of 30 nM carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP).
Mitochondrial OXPHOS Enzymatic Activity
Frozen RAA tissue sample (~50 mg) was homogenized in ice-cold buffer (1:20 w/v) containing (in mM) 100 KCl, 5 MgCl2, 2 EGTA, and 50 Tris/HCl (pH 7.5), and homogenized using an OMNI GLH-115 Polytron homogenizer (OMNI International). The homogenate was centrifuged at 1,000×g for 15 min at 4°C; the supernatant was aliquoted and stored at –80°C until processed. The functional activity of mitochondrial OXPHOS complexes I–V was measured in tissue homogenate as previously described (1,7).
Western Blot Analysis
Frozen tissue (~40 mg) from the RAA was homogenized in RIPA lysis buffer (Abcam, Cambridge, MA) with protease inhibitor cocktail freshly added (Cell Signaling Technology, Danvers, MA). Protein extracts (20 µg) were separated on NuPAGE precast 4–12% Bis–Tris gel (Thermo Fisher Scientific) and transferred to polyvinylidene difluoride membranes (Thermo Fisher Scientific). The membranes were blocked in 5% BSA that was dissolved in Tris-buffered saline containing 0.2% Tween-20. After the blocking, the membranes were immunoblotted using primary human MitoProfile® Total OXPHOS antibody cocktail (Abcam) against complex I subunit nicotinamide adenine dinucleotide hydrogen dehydrogenase (ubiquinone) (NDUFB8), complex II 30kDa (SDHB), complex III core protein 2 (UQCRC2), complex IV subunit II (COX2), complex V alpha subunit (ATP5A), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH - 14C10) HRP-conjugated rabbit monoclonal antibody against GAPDH (Cell Signaling Technology). The secondary goat anti-mouse IgG-HRP antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was used against the primary human OXPHOS antibodies. Proteins were visualized by SuperSignal™ West Pico chemiluminescent substrate and monitored using UltraQuant® v6.0 software in molecular imaging systems (UltraLum, Claremont, CA). Densitometric evaluation of protein bands was performed using ImageJ software (National Institutes of Health [NIH], http://rsb.info.nih.gov/ij/). Bands corresponding to the mitochondrial complexes were normalized to the density of respective GAPDH band.
Total RNA Isolation and Quantification
Frozen RAA tissue (~20 mg) was homogenized in TRIzol® Reagent (Thermo Fisher Scientific). Total RNA was isolated following the TRIzol® Reagent protocol as previously described (4) and quantification performed using spectrophotometric analysis (absorbance-emission A260/A280). The integrity of the samples was assessed qualitatively on an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Each sample was then used for microarray analysis and verification of selected genes by real-time reverse transcription polymerase chain reaction (RT-PCR). The total amount of RNA extracted was not significantly different between the two age groups (5.1 ± 1.1 µg in aged vs 6.8 ± 5.0 µg in adult, per gram of tissue).
Gene Expression Profiling
Total isolated RNA (100 ng) from individual samples was converted to cDNA using the Two-Cycle cDNA synthesis kit and Target Labeling Protocols (Affymetrix, Santa Clara, CA). The obtained cDNA was then purified by phase lock gel (Eppendorf, Westbury, NY) followed by phenol/chloroform extraction and used as a template for the in vitro transcription reaction. cRNA was then synthesized and labeled using RNA transcript labeling reagent (Affymetrix). Labeled cRNA was fragmented and hybridized onto GeneChip® Human Genome U133 Plus 2.0 microarrays (Affymetrix) according to the manufacturer’s protocol. The quality of the fragmented biotin-labeled cRNA in each experiment was evaluated before hybridization as previously described (4).
Microarray Data Analysis
The images of the hybridized microarrays were scanned and quantitatively analyzed by a GeneChip 3000 scanner (Affymetrix). Intensity values for each probe cell in the arrays were calculated by GeneChip® software, and flags were assigned to each probe set declaring Present, Marginal, or Absent call (Detection Call Algorithm). Probe cell intensities were used to calculate an average intensity for each set of probe pairs representing a gene, which directly correlated with the amount of cRNA. Data analysis and normalization were performed by GeneSpring GX bioinformatics software (Agilent Technologies). Quality control (QC) was performed on the normalized intensity values after excluding the probe sets with absent call in 100% of the arrays. After applied QC filtering to diminish background noise created by low-intensity gene probes, genes were clustered into the two age group conditions (a and b) using the following cosine correlation (r) equation:
The data were then plotted in a hierarchical clustering heat map and volcano plots to identify genes that were significantly up- or downregulated in the aging atria. Statistical analysis of the microarray gene expression was performed using Welch’s t-test and a multiple testing correction formula, the Benjamini–Hochberg false discovery rate, which together reported a corrected (Corr) p value for each gene. The hierarchical clustering for conditions and entities was performed using Euclidean distance metric and Ward’s linkage algorithm. Statistical significance was set at p(Corr) < .05.
Real-Time Reverse Transcription Polymerase Chain Reaction
Real-time RT-PCR was performed to quantify gene expression levels using TaqMan® (Applied Biosystems, Foster City, CA) as previously described (4). Gene transcript levels were subsequently measured according to instructions for the Applied Biosystems Assay-on-Demand Gene Expression Product.
Gene expression assays for NDUFA6, NDUFB8, SDHD, UQCRC2, COX7A2L, ATP5G1, PDHA1, CS, and ACO2 coding for NADH dehydrogenase 1 alpha subcomplex subunit 6 and beta subunit 8 (complex I), succinate dehydrogenase subunit D (complex II), ubiquinol-cytochrome c reductase core protein 2 (complex III), cytochrome c oxidase subunit VIIa polypeptide 2 (complex IV), ATP synthase subunit C isoform 1 (complex V), pyruvate dehydrogenase alpha 1, citrate synthase, and aconitase 2, respectively, were selected using TaqMan® Gene Expression assay search tool (Applied Biosystems). Results were normalized to a pre-designed TaqMan® Endogenous Control, Eukaryotic 18S rRNA Endogenous Control (Applied Biosystems), which was used to normalize against differences in RNA isolation, RNA degradation, and the efficiencies of the reverse transcription and PCR reactions. All samples were run in triplicate and quantitated using an absolute standard curve, normalizing each gene signal to the 18S signal.
Statistical Analysis
Mean ± standard error (SE) was applied to describe quantitative variables, and percentages were used for qualitative variables. The Kolmogorov–Smirnov test was used to evaluate the normality of continuous variable distribution. Bivariate comparison of the aged and adult groups was performed using the t-test for normally distributed variables or the nonparametric Kruskal–Wallis test for the variables that did not follow the normal distribution. To reveal association between quantitative variables, linear regression analysis was applied. The statistical analysis was performed using SAS software (version 9.2, SAS Institute, Cary, NC) and SigmaPlot (version 12.3, Systat Software, Inc, San Jose, CA). Gene expression analysis was performed for global profiling and comparison of expression between adult and aged patients, and statistical significance for each gene was set at p < .05.
Results
Clinical Characteristics
The clinical characteristics of the patients included in the study are shown in Table 1. The mean ages of the adult and aged group were 51 ± 2 years (n = 24) and 74 ± 1 years (n = 26), respectively. The adult and aged patients were well matched for the presence of risk factors for cardiovascular diseases, comorbidities, and cardiac function. No significant differences were observed in sex distribution, body mass index, hypertension, hyperlipidemia, coronary artery disease, history of heart failure, prior myocardial infarction, type of surgery performed, or major medications. The patients from both groups had preserved left ventricular ejection fraction, and there were no patients with a history of diabetes mellitus or atrial fibrillation.
Table 1.
Clinical Characteristics of Patients in the Study
| Adult | Aged | p-value | |
|---|---|---|---|
| Number of patients | 24 | 26 | |
| Age (y) | 51 ± 2 | 74 ± 1 | <.001 |
| (29–62) | (65–83) | ||
| Sex (male/female) | 15/9 | 15/11 | .59 |
| BMI > 30 | 11 (48%) | 7 (27%) | .20 |
| Coronary artery disease | 18 (78%) | 24 (92%) | .18 |
| History of MI | 6 (26%) | 6 (23%) | .81 |
| Hypertension | 14 (61%) | 19 (73%) | .38 |
| Hyperlipidemia | 19 (83%) | 20 (77%) | .63 |
| Diabetes mellitus | 0 (0%) | 0 (0%) | |
| Atrial fibrillation | 0 (0%) | 0 (0%) | |
| History of smoking | 16 (67%) | 12 (46%) | .10 |
| LA size < 4.5 cm | 19 (68%) | 21 (78%) | .42 |
| LVEF (%) | 63 ± 1 | 59 ± 2 | .07 |
| Beta blockers | 16 (70%) | 16 (62%) | .56 |
| ACE/ARB inhibitors | 11 (48%) | 10 (39%) | .52 |
| Calcium blockers | 4 (17%) | 5 (19%) | .87 |
| Statins | 15 (65%) | 13 (50%) | .29 |
| CABG | 17 (74%) | 23 (88%) | .21 |
| AVR | 3 (13%) | 7 (27%) | .23 |
| MVR | 1 (4%) | 3 (12%) | .75 |
| Other | 0 (0%) | 1 (4%) |
Note. Data are shown as the number of patients and percent of the total number in parenthesis n(%).. Age and left ventricular ejection fraction (LVEF) are presented as the mean ± SE. ACE I = angiotensin-converting enzyme inhibitors; AVR = aortic valve replacement; ARB = angiotensin II receptor antagonists; BMI = body mass index; CABG = coronary artery bypass grafting; MI = myocardial infarction; MVR = mitral valve replacement. Other surgery: aortic or septal myomectomy.
Aging Reduces the Activity of the Mitochondrial Phosphorylation System
Aging in the human myocardium was associated with a reduction in functional activity of the mitochondrial electron transport chain (ETC, Figure 1A). The activity of ETC complex I as determined by the rotenone-sensitive reduction of ubiquinone-1 was significantly reduced in the aged group compared with the adult group (44.8 ± 20.8 vs 73.0 ± 44.2 nmol/min/mg protein, respectively, p = .03). The functional activities of the remaining four OXPHOS complexes were not significantly altered by age. The rate of reduction of ubiquinone-2 by succinate (complex II) was 49.4 ± 28.7 in the aged group versus 56.2 ± 18.3 nmol/min/mg protein in the adult group (p = .50). Antimycin A-sensitive reduction of oxidized cytochrome c by decylubiquinol (complex III) was 34.8 ± 30.5 in the aged group versus 36.5 ± 11.0 nmol/min/mg of protein in the adult group (p = .88). Complex IV activity, estimated as a decrease in absorbance of ferrocytochrome c by cytochrome c oxidase, was 241.3 ± 127.0 versus 205.3 ± 89.0 nmol/min/mg protein in the adult and aged groups, respectively (p = .45). Oligomycin-sensitive oxidation of NAD+, reflecting F0F1 ATPase (complex V) activity was 48.0 ± 30.7 in the aged group versus 60.6 ± 24.9 nmol/min/mg protein in the adult group (p = .22). In addition, we correlated the effect of age on the activity of OXPHOS complexes I–V, which has been plotted as a function of age in Figure 1B, confirming the age-associated reduction in the activity of complex I (r = 0.42, p = .01). The activities of the remaining OXPHOS complexes were not significantly correlated with age.
Figure 1.

Functional activities of mitochondrial oxidative phosphorylation complexes in the right atria of adult and aged patients. (A) Data are expressed as nmol/min/mg protein and presented as mean ± SE, *p < .05. (B) Correlation between functional activity of complexes I–V and age; n = 18 for adults and n = 17 for aged.
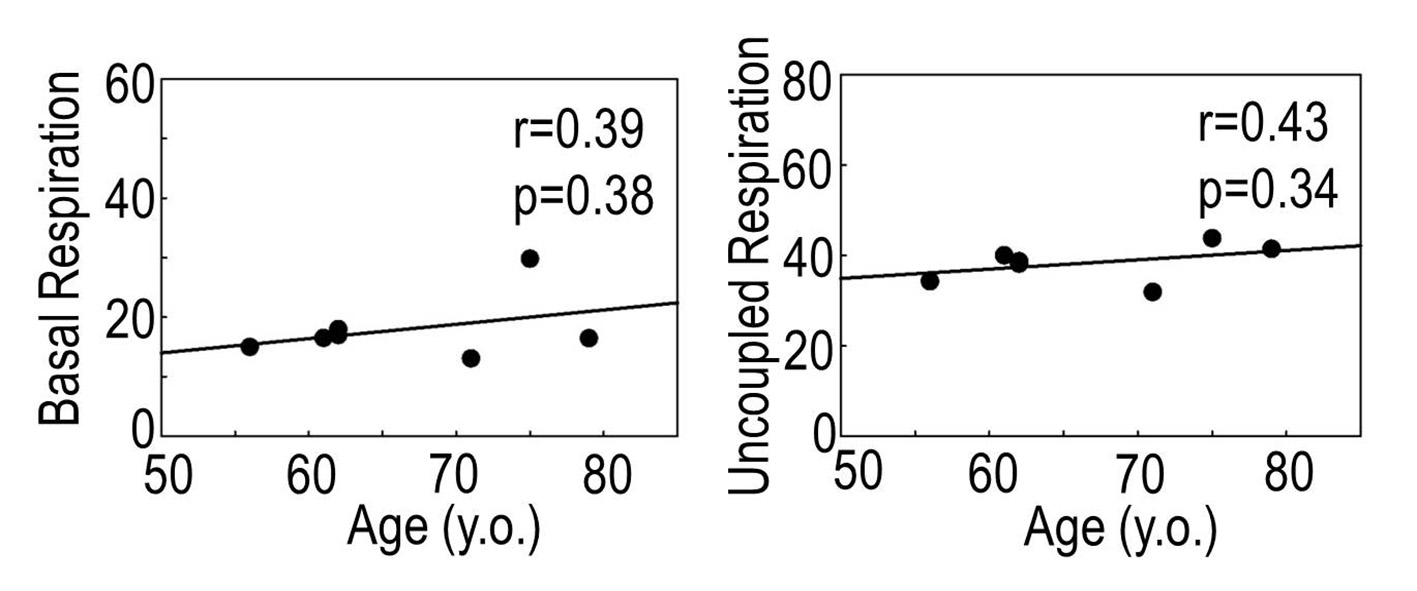
These results were further confirmed on isolated mitochondria from different age groups measuring oxygen consumption rate in the presence of substrates supporting complexes I, II, and IV. As summarized in Supplementary Figures 1–3, the basal respiration rate of mitochondria oxidizing NADH-dependent substrates (glutamate and malate) was not altered with age (Supplementary Figure 1), however, state 3 respiration was diminished in mitochondria from the aged patients, demonstrating a significant correlation with age (r = 0.83, p = .01). The respiratory control ratio (state 3/state 4), which indicates the degree of coupling between the rate of electron transport in the ETC (respiration) and phosphorylation of ADP (ATP production) (r = 0.80, p = .02), and uncoupled respiration (r = 0.89, p = .003) were significantly affected by age (Supplementary Figure 1). No age-related alterations were observed in the rate of oxygen consumption by mitochondria oxidizing succinate (Supplementary Figure 2) or ascorbate and TMPD (Supplementary Figure 3).
Thus, with aging, the efficiency of the mitochondrial OXPHOS pathway declines along with a significant reduction in the functional activity of complex I in human aging atria.
Aging-Related Decrease in Oxidative Phosphorylation Proteins Expression Level
The effect of aging-associated changes in the OXPHOS was also confirmed at the protein level by Western blots using an antibody cocktail against specific subunits of the OXPHOS complexes (Figure 2A and B). There was statistically significant age-related reduction in the protein expression level of complex I NDUFB8 subunit (r = 0.65, p = .01). Although complex III UQCRC2 subunit (r = 0.63, p = .05) and complex IV COX2 subunit (r = 0.62, p = .06) protein levels were lower in older patients, these changes did not reach the statistical cut-off (p < .05) for significance (Figure 2B). Non-significant correlation was found between age and the protein levels of complex II SDHB and complex V ATP5A subunits (Figure 2).
Figure 2.

Age-associated changes in protein expression level of mitochondrial OXPHOS complexes I–V. (A) Immunoblots of representative subunits of complexes (C I–C V) using MitoProfile Total OXPHOS human antibody cocktail (Abcam, Cambridge, MA). (B) Age-related correlation in densitometry of bands corresponding to mitochondrial complexes normalized to the density of glyceraldehyde 3-phosphate dehydrogenase (GAPDH); n = 5 for adults and n = 5 for aged.
Aging Alters Expression Profile of Genes Coding for Proteins that Regulate Cardiac Mitochondrial Energetics
The differences in gene expression pattern in the RAA tissue from the adult (n = 10) and aged (n = 10) patient groups were analyzed using GeneSpring GX (Agilent Technologies). Hierarchical clustering heat map revealed a condition clustering pattern where some of the adult (A1–A6) and aged (O7–O10) patient samples clustered within the same age group (Figure 3A). The majority of genes coding for mitochondrial proteins from the aged patients (O2–O10) clustered together except for one patient (O1). Four patients (A7–A10) from the adult group clustered with the aged group.
Figure 3.

Aging-associated alterations in mitochondrial gene transcripts. (A) Hierarchical clustering and heat map of genes coding for proteins regulating mitochondrial energetics from adult (A1–A10) and aged patients (O1–O10). Green indicates downregulation; red, upregulation; and black, no change in the level of expression of genes. (B) Volcano plot illustrating the relative changes in expression of genes coding for mitochondrial proteins from aged and adult patients (p(Corr) < .05). There were 101 genes coding for mitochondrial proteins that were significantly different in the aged group compared with the adult group (blue dots). (C) Number of significantly downregulated (87 genes, green bar) and upregulated (14 genes, red bar) genes in the aged atrias compared with adults and their distribution according to their molecular function with 49% of the altered genes coded for proteins regulating mitochondrial energetic pathways (30% for substrate metabolism, 13% for oxidative phosphorylation (OXPHOS), and 6% for tricarboxylic acid cycle (TCA), and the remaining 51% for other mitochondrial functions including apoptosis (Apopt) and detoxification (Detox). (D) Expression values for selected genes coding for subunits of mitochondrial proteins regulating energetics from right atrial appendages of adult (n = 10, white dots) and aged (n = 10, black dots) patients are shown as dot-plots highlighting the clustering of gene expression within each age group with the statistical significance of age-related changes between the two groups shown with their p-values, n = 10 for adults and n = 10 for aged.
Differences in selected genes coding for mitochondrial proteins in aged atria compared with adults is plotted as a volcano plot according to their p-value and fold change (Figure 3B). Out of 1,008 genes coding for mitochondrial proteins in humans, 101 (10%) genes were differentially expressed in the atria of patients in the aged group compared with patients in the adult group (p(Corr) < .05). None of the mitochondrial DNA-coded genes were altered between the groups. The differences were all due to nuclear DNA-coded genes. The majority (86%) of these genes (87/101) was downregulated (Figure 3C, green bar), while 14 genes (14%) were upregulated (Figure 3C, red bar) (Supplementary Table 1). Forty-nine percent of the altered genes were confined to pathways regulating mitochondrial energetics, including genes coding for substrate metabolism (30%), OXPHOS (13%), and TCA cycle (6%, Figure 3C). The clustering of gene expression values within each age group for selected altered genes of mitochondrial proteins, including NDUFA6 (NADH: dehydrogenase ubiquinone 1 alpha subcomplex 6), ATP5G1 (ATP synthase, H+-transporting, mitochondrial F0 complex subunit C1), ACO2 (aconitase 2), IDH2 (isocitrate dehydrogenase 2 (NADP+), ME2 (malic enzyme 2), and PDHA1 (pyruvate dehydrogenase alpha 1), are shown as dot-plots in Figure 3D, which demonstrates tight grouping of expression within the same age group and significant differences between the two age groups (p(Corr) < .05).
Aging Decreases the Expression of Genes Regulating the Oxidative Phosphorylation and Substrate Metabolism Pathways in Human Atria
Microarray data showed that the expression of genes coding for several subunits of the mitochondrial OXPHOS complexes was downregulated with aging. Out of 78 genes that code for subunits of complexes I–IV of the ETC, the expression of ten genes (13%) was significantly reduced in aged atria (Figure 4, colored green). These included five genes coding for subunits of complex I, one gene each for complexes II and IV, two genes for complex III, and one gene coding for a chaperone complex I assembly protein (TIMMDC1) as summarized in Figure 4 and Supplementary Table 1. Also, out of 21 genes known to code for the mitochondrial F0F1 ATP synthase complex (complex V of OXPHOS), one gene, ATP5G1, that codes for subunit c isoform 1 of the F0 subcomplex was significantly downregulated (Figure 4 ; colored green). The effect of aging on the expression profile of genes regulating the TCA cycle and substrate metabolism with fold change and p-values is summarized in the Supplementary Table and illustrated in Figure 4. Out of the 15 genes that code for components of the TCA cycle, six genes were downregulated—CS, ACO2, FH, IDH2, ME2, and PDHA1, coding for citrate synthase, aconitase 2, fumarate hydratase, isocitrate dehydrogenase 2, malic enzyme 2, and pyruvate dehydrogenase alpha 1.
Figure 4.

Schematic representation of genes of complexes coding for the mitochondrial oxidative phosphorylation and tricarboxylic acid cycle (TCA). Genes coding for subunits of the electron transport chain (C I–IV), F0F1 ATP synthase (C V) and TCA that are significantly downregulated in the aging atria are colored green, while subunits not altered with aging are shown in gray. None of the genes coding for subunits of the oxidative phosphorylation pathway were upregulated in the aged hearts. NDUFA6, NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 6.; NDUFA9, NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 9; NDUFB5, NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 5; NDUFB8, NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 8; NDUFS2, NADH dehydrogenase (ubiquinone) 1 beta subcomplex, 2; TIMMDC1, complex I assembly factor TIMMDC1; SDHD, succinate dehydrogenase complex, subunit D; COQ9, coenzyme Q9 homolog (S. cerevisiae); NNT, nicotinamide nucleotide transhydrogenase; Core2, ubiquinol-cytochrome c reductase core protein 2; Cyt b, cytochrome b5 reductase 4; VIIa2, cytochrome c oxidase subunit VIIa polypeptide 2 like; c, ATP synthase, mitochondrial F0 complex, subunit c, isoform 1; FH, fumarate hydratase; MDH2, malate dehydrogenase 2; CS, citrate synthase; ME2, malic enzyme 2, NAD(+)-dependent, mitochondrial; PDHA1, pyruvate dehydrogenase (lipoamide) alpha 1; IDH2, isocitrate dehydrogenase 2 (NADP+), mitochondrial; ACO2, aconitase 2, mitochondrial; AKGDH, alpha-ketoglutarate dehydrogenase; SUCLG, succinyl-CoA ligase, subunit alpha; IMS, inner mitochondrial space; IMM, inner mitochondrial membrane; MM, mitochondrial matrix.
To verify age-related changes in the transcriptional profile of genes determined by microarray analysis, expression of selected genes in adult and aged atrial appendages was assessed by real-time RT-PCR. The expression of NDUFA6, NDUFB8, SDHD, UQCRC2, COX7A2L, ATP5G1, PDHA1, CS, and ACO2 coding for NADH dehydrogenase 1 alpha subcomplex subunit 6 and beta subunit 8 (complex I), succinate dehydrogenase subunit D (complex II), ubiquinol-cytochrome c reductase core protein 2 (complex III), cytochrome c oxidase subunit VIIa polypeptide 2 (complex IV), ATP synthase subunit C isoform 1 (complex V), pyruvate dehydrogenase alpha 1, citrate synthase, and aconitase 2, respectively, was significantly reduced in the aging atrial appendages (Supplementary Figure 4). The mRNA levels of these enzymes or regulatory factors determined by real-time RT-PCR were reduced in the aged atria compared with the adult atria by a similar extent as determined by microarray analysis.
Discussion
Our study identified broad changes in the expression of genes coding for mitochondrial proteins with selective transcriptional downregulation of subunits of complexes I–V of OXPHOS, TCA cycle, and substrate metabolism with age in the human atria from patients free of structural or electrical atrial dysfunction. These changes were associated with age-elicited decline in mitochondrial oxidative phosphorylation associated with a decrease in the levels of OXPHOS proteins and activity of complex I in the respiratory chain.
Aging is characterized by reduced functional reserve and increased susceptibility of the atria to electrical and mechanical dysfunction, including atrial fibrillation, reduced contractility, and increased fibrosis (8), but the molecular bases for such changes are not entirely known. Aging is associated with a diminished capacity of mitochondria to generate ATP, as previously shown in animal models (4,9). This is in agreement with human studies showing that high energy phosphates decline in the human heart with age (10). Correspondingly, mitochondrial oxidative capacity drops by 50% in human hearts with aging (11). Aging reduces the state 3 respiration rate during oxidation of NADH-dependent substrates in heart mitochondria (4). Our findings in atrial tissue from aged patients compared with a comorbidity-matched adult group demonstrated a significant reduction in mitochondrial OXPHOS capacity by using substrates that enter the ETC at complex I (glutamate and malate) but not at complexes II (succinate) or IV (ascorbate and TMPD) (Supplementary Figures 1–3). The difference in decline of mitochondrial respiratory capacity between different metabolic substrates could be due to age-related changes in complex I, substrate transporters or dehydrogenases activities (12) or the OXPHOS supercomplex destabilization in mitochondrial senescence (13). Loss of supercomplexes has been shown to compromise different stoichiometries of the OXPHOS complexes I, III, and IV in interfibrillar mitochondria from aged rats (14), which may result in inefficient electron flux through the ETC affecting mitochondrial energy reserve capacity (15). It appears that supercomplex formation is not required for keeping basal respiratory activity but for respiratory coupling since destabilization of supercomplexes diminishes state 3 respiration thus reducing mitochondrial energetic reserve capacity (13). This needs to be further assessed in human atrial tissue.
Overall, myocardial ATP production requires proper functioning of the mitochondrial OXPHOS system that is composed of four ETC complexes (complexes I–IV), and the F0F1-ATPase (complex V) within the mitochondrial inner membrane as well as enzymes regulating the TCA cycle and substrate metabolism. Among the OXPHOS complexes, complex I deficiency is the most frequently encountered defect in mitochondrial energetics, as previously described in animal models and human diseases (1,16), and its impairment is also associated with aging (4,17). In our study, alteration in the transcript level of nDNA-encoded genes (NDUFA6, NDUFA9, NDUFB8, NDUFB5, NDUFS2, and TIMMDC1) in the aged human hearts (Figure 4, Supplementary Table 1) was accompanied by a significant reduction in protein level of complex I NDUFB8 subunit (Figure 2) and enzymatic activity (Figure 1) compared with the adult hearts. These results are consistent with previous studies in Fisher 344 rats, in which aging-associated downregulation of genes coding for subunits of mitochondrial complex I were associated with reduced NADH:ubiquinone oxidoreductase activity (4). It is not known how the aging-associated reduction in expression of the various complex I subunits in the human atria demonstrated in our study alters atrial function. The subunit NDUFA6 is a crucial subunit involved in complex I assembly and a target for oxidative-stress-induced damage (18). It is involved in stabilizing the junction between the membrane and matrix arms of complex I, which is critical for complex I activity and stability of the connection between the membrane and matrix arms (19). There also is evidence that NDUFA9 is involved in conformational rearrangement of catalytically active (A) and deactive-dormant (D) states of complex I (20). The A/D transition plays an important role in tissue response to the lack of oxygen and hypoxic deactivation of complex I that occur in mitochondria during ischemia (21). NDUFB8 is primarily thought to be an accessory subunit not involved in catalysis (22). However, peroxynitrite-induced oxidation of NDUFB8 led to alteration in complex I enzymatic activity and mitochondrial bioenergetics in endothelial cells (23), and it has been suggested that polymorphisms in NDUFB8 gene are involved in neurodegeneration (24). NDUFB5 has NADH dehydrogenase and oxidoreductase enzymatic activity and reduction in its expression after systematic application of a tumor necrosis factor-like weak inducer of apoptosis in mice after myocardial infarction was reported to aggravate cardiac dysfunction (25). The protein encoded by the NDUFS2 gene is a core subunit of the iron–sulfur protein fraction of complex I, and mutations in this gene are associated with complex I deficiency and reduced enzymatic activity in Leigh syndrome (26). The TIMMDC1 gene codes for the chaperone protein involved in the assembly of mitochondrial complex I (27). Studies have shown that TIMMDC1 protein is physically associated with multiple factors of mitochondrial complex I assembly as well as soluble and matrix arms. Its depletion affects complex I assembly and leads to a decrease in complex I activity and cellular respiration (28). Although the role of these complex I subunits in the aging human heart has not been studied, the association of the overall decline in the expression of these subunits with reduced complex I activity in the aged study group suggests their potential role in aging-associated mitochondrial impairment. This needs to be further defined. This is important because aging-associated changes in mitochondrial transcriptome and proteins could underlie myocardial functional deficits that not only reduce mitochondrial energetic reserves (4,29) but also increase susceptibility to calcium overload and mitochondrial permeability transition pore opening (29), increasing vulnerability to oxidative damage and stress-mediated myocardial injury (30).
The functional impact of aging in the human atria in our study was mainly observed in the complex I activity. Despite reduction in expression of SDHD (Figure 4, Supplementary Figure 4 and Table 1), a gene coding for subunit D of the OXPHOS complex II, we found no significant changes in enzymatic activities of complex II (Figures 1) in the aged atrial tissue compared with the adult atrial tissue. These findings are similar to previous reports demonstrating no age-associated changes in the activity of complex II in mitochondria from rat hearts (4). We observed an aging-associated decrease in expression of CYB5R4 and UQCRC2 genes coding for the OXPHOS cytochrome b5 reductase 4 and complex III core protein 2, correspondingly (Figure 4, Supplementary Figure 4 and Table 1). The downregulation of UQCRC2 gene resulted in decline of the protein level of the core protein 2 (Figure 2). However, the functional activity of complex III did not change with age (Figure 1). The core protein 2 that lacks the redox center is not directly involved in electron transfer in complex III, but may still play some role in energy transduction, regulation of the enzymatic reaction, or assembly of other subunits into the functional complex and super-complex formed from complexes I, III, and IV (31). Similarly, despite downregulation of the expression of COX7A2L (Figure 4, Supplementary Figure 4 and Table 1) coding for complex IV subunit COX2, and decline in COX2 protein level (32), in aged human myocardium, there was no significant difference in functional activity of cytochrome c oxidase between patients in the adult and aged groups (Figure 1). This is consistent with what was previously observed in aged rat hearts (4), but different from a report by Fannin et al., which described aging-related alterations in cytochrome c oxidase activity limited to interfibrillar mitochondrial subpopulation without impact on subsarcolemmal mitochondria (33). The biochemical activity of complex V has been described to be less affected by age (30) and the functional effect more a reflection of impairment of electron transfer through the ETC rather than a defect in complex V itself (33). A downregulation of nDNA-encoded ATP5G1 gene in the aged human atrial myocardium was observed (Figure 4, Supplementary Figure 4 and Table 1); however, this was not associated with any significant reduction in functional activity or protein expression of complex V (Figures 1 and 2). This is different from the observation in the aged rat ventricular myocardium that displayed downregulation of the transcripts and complex V ATPase activity (4). The effect of aging on complex V is of interest because of its possible role in the regulation of mPTP formation and cell injury, and it will be interesting to assess whether species, or tissue-dependent differences exist in its expression or activity in the aging heart.
Enhanced leakage of electrons from the impaired ETC during respiration leads to increased reactive oxygen species (ROS) production and a decline in the antioxidant system, which has been described to involve nicotinamide nucleotide transhydrogenase (NNT) (18,34). Mitochondrial NNT, a proton pumping transhydrogenase, is considered a key antioxidative enzyme based on its ability to regenerate NADPH from NADH in mitochondria (35). This has an important implication in redox detoxification in the mitochondria as NADPH is used for maintenance of glutathione reductase and thioredoxin reductase generating reduced glutathione and reduced thioredoxin, which are important for normal myocardial functioning during stress. The NNT switches to pro-oxidative mode under pathological heart workload, reversing the direction of the reaction, oxidizing NADH from NADPH to support ATP production by the ETC (18). The combined redox action of reduced glutathione and thioredoxin maintained by NNT protects against ROS overflow from mitochondria in both state 4 and state 3 respirations (36). The reduction in mitochondrial phosphorylation capacity (decline in state 3 with increase in state 4 respiration) observed in our study could lead to increased ROS production in elderly patients. Previously, NNT expression was demonstrated to be reduced more than seven times in aging mice between 5 and 30 months, and this correlated with decline of other genes involved in mitochondrial biogenesis (37). In agreement with these data, we observed substantial decline of NNT gene expression in elderly patients in our study (Figure 4, Supplementary Table 1), but the functional significance needs to be further investigated in view of reduced NNT activity.
Another important player in cardiac mitochondrial energetics is pyruvate dehydrogenase coded by PDHA1 gene, which was downregulated in the aging heart, indicating that the overall conversion of pyruvate to acetyl-CoA catalyzed by this enzyme could be reduced. PDH1A downregulation (Figures 3 and 4, Supplementary Table 1) suggests that the link between glycolysis and TCA cycle is impaired and, therefore, aerobic glucose oxidation through the OXPHOS pathway might be altered. In addition, there was upregulation in Acyl-CoA synthase long-chain family member 3 (Supplementary Table 1), which converts free long-chain fatty acids into fatty acyl-CoA esters and, thereby, plays a role in lipid biosynthesis and fatty acid degradation. The functional consequences of these changes in human atria with aging need to be further investigated and were beyond the scope of this study. This is important in view of recent reports suggesting a switch in mitochondrial metabolism from fatty acid oxidation to glycolysis in humans (38) and animal models (39,40) with aging. Animal models are widely used to study human aging-related diseases (41,42), but it is not clear whether, at the molecular level, cardiac aging in animal models is comparable with human aging. Cardiac senescence is accompanied by decline in mitochondrial function, increased oxidative stress, impairment of protein QC, dysregulated mitochondrial dynamics (fission/fusion), and mitophagy (43), but molecular-level changes in mitochondrial function in human nondiseased heart tissue have not been fully characterized. In this regard, our study provides important information using tissue from human hearts that will help fill some of this gap in knowledge. Age-associated changes in genes coding for OXPHOS complexes were previously described in animal models with downregulation of a higher number of genes coding for mitochondrial proteins and each of the individual complexes of the OXPHOS in aging rat ventricular myocardium (4), which is different from our observation in human atria. The reason behind these differences is not clear but could be related to differences in species, tissue assessed, different metabolic demands of atrial versus ventricular tissue, and other confounding effects of human diseases that have not been seen in inbred animal models.
Another limitation of the published literature is the reporting of changes in human atrial genes mainly from patients who underwent surgery for diseases that can impact atrial structure and function, such as mitral valve disease (44), atrial fibrillation (45), hypertrophic cardiomyopathy (46), or systemic diseases (47). The described changes could, therefore, be due to the effect of disease superimposed on aging-related changes as a wide spectrum of patients belonging to different age groups were included in both the diseased and nondiseased groups. It is very difficult to obtain human cardiac tissue from nondiseased hearts, making it difficult to discern the impact of aging from disease. Studies using cadaveric tissue suffer from variability that could be introduced due to different conditions of tissue acquisition and preservation, duration of ischemia, and storage condition (such as cardioplegia solution constituents, temperature, and duration of storage) (48). The strength of our study is the use of a standardized approach to tissue collection performed on tissue removed from patients. This approach included freezing the tissue within 5–10 min of removal from the body to prevent introduction of variability that could potentially affect studies on transcriptomic changes due to poor reliability of mRNA data. Extra care was taken to exclude all conditions that can potentially affect atrial function and energetics. Only tissue from the RAA was used from aged and adult hearts to avoid regional differences in gene expression, protein level or enzymatic activity. However, the amount of RAA tissue is limited for mitochondrial isolation, and, therefore, only limited information could be obtained. When possible, larger samples from the LAA were used to isolate mitochondria in order to determine differences in age-associated changes in respiratory capacity of isolated mitochondria. We were careful to exclude tissue from all pathological conditions that can alter atrial energetics or mitochondrial function—such as the presence of heart failure, left ventricular dysfunction, left atrial enlargement, mitral valve disease, presence of complicated diabetes, or atrial fibrillation—by careful review of the preoperative electrocardiogram, echocardiogram, laboratory results, and pathological description of the heart by the surgical team. The two groups were matched for gender, comorbidities, medications used, and type of surgery.
A potential limitation of our findings on aging-related differences in the expression of the genes could be the smaller magnitude of differences (less than twofold) for the majority of genes between aged and adult atria. However, these differences showed a tight clustering of values within each age group that were highly significant statistically. As reported by several investigators, the biological consequences of gene expression changes may not depend entirely on the level of expression, as highly expressed genes may depict less than a twofold difference in expression by microarrays, while less-expressed genes may display a greater than twofold change (49). Recently, transcriptomic profiling (41,298 genes) across the whole body in humans revealed 828 (2%) age-associated genes in the heart ventricle (50). The aging-related genes were grouped into 374 (45%) upregulated and 454 (55%) downregulated genes. The downregulated genes were related to mitochondrial biology, including TCA cycle and mitochondrial ETC. Our study results are in line with these reported changes, with the focus on the effect of aging on atrial OXPHOS activity and how it correlates with observed transcriptional changes in gene expression.
In summary, our study in a clinically well-matched group of adult and elderly patients with preserved left ventricular function undergoing open heart surgery provides evidence that aging is associated with widespread downregulation of genes regulating mitochondrial energetics and oxidative phosphorylation accompanied with decline in the OXPHOS proteins. However, at the functional level, only reduction in ETC complex I activity was predominantly observed. This could potentially explain the reduced mitochondrial reserves, and complex I of the OXPHOS could represent a target to improve energetic efficiency in the senescent human atria.
Supplementary Material
Supplementary data are available at The Journals of Gerontology, Series A: Biological Sciences and Medical Sciences online.
Funding
This work was supported by the National Institutes of Health grants (NIH R01 HL101240 and R01 HL089542).
Conflicts of Interest
None.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
Dr. Arshad Jahangir is supported by grants from the National Heart, Lung and Blood Institute (HL-089542). The authors acknowledge Jennifer Cooper for assistance in obtaining patient consents and collecting tissue samples, Dr. Yang Shi for manuscript review, Jennifer Pfaff and Susan Nord of Aurora Cardiovascular Services and Sandra Kear of Aurora Research Institute for editorial preparation of the manuscript, and Brian Miller and Brian Schurrer of Aurora Research Institute for help with the figures.
References
- 1. Emelyanova L, Ashary Z, Cosic M, et al. Selective downregulation of mitochondrial electron transport chain activity and increased oxidative stress in human atrial fibrillation. Am J Physiol Heart Circ Physiol. 2016;311:H54–H63. doi:10.1152/ajpheart.00699.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics-2016 update: A report from the American Heart Association. Circulation. 2016;133:e38–360. doi:10.1161/CIR.0000000000000350 [DOI] [PubMed] [Google Scholar]
- 3. Choksi KB, Papaconstantinou J. Age-related alterations in oxidatively damaged proteins of mouse heart mitochondrial electron transport chain complexes. Free Radic Biol Med. 2008;44:1795–1805. doi:10.1016/j.freeradbiomed.2008.01.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Preston CC, Oberlin AS, Holmuhamedov EL, et al. Aging-induced alterations in gene transcripts and functional activity of mitochondrial oxidative phosphorylation complexes in the heart. Mech Ageing Dev. 2008;129:304–312. doi:10.1016/j.mad.2008.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res. 2009;81:412–419. doi:10.1093/cvr/cvn301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frezza C, Cipolat S, Scorrano L. Organelle isolation: Functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2:287–295. doi:10.1038/nprot.2006.478 [DOI] [PubMed] [Google Scholar]
- 7. Darley-Usmar VM Rickwood D. and Wilson M. T.. Mitochondria: a practical approach. Oxford: IRL Press; 1987. [Google Scholar]
- 8. January CT, Wann LS, Alpert JS, et al. ; American College of Cardiology/American Heart Association Task Force on Practice Guidelines 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: A report of the American College of Cardiology/American Heart Association task force on practice guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2014;64:e1–76. doi:10.1016/j.jacc.2014.03.022 [DOI] [PubMed] [Google Scholar]
- 9. Tatarková Z, Kuka S, Račay P, et al. Effects of aging on activities of mitochondrial electron transport chain complexes and oxidative damage in rat heart. Physiol Res. 2011;60:281–289. [DOI] [PubMed] [Google Scholar]
- 10. Köstler H, Landschütz W, Koeppe S, et al. Age and gender dependence of human cardiac phosphorus metabolites determined by SLOOP 31P MR spectroscopy. Magn Reson Med. 2006;56:907–911. doi:10.1002/mrm.21027 [DOI] [PubMed] [Google Scholar]
- 11. Conley KE, Jubrias SA, Esselman PC. Oxidative capacity and ageing in human muscle. J Physiol. 2000;526 Pt 1:203–210. doi:10.1111/j.1469-7793.2000.t01-1-00203.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. O’Toole JF, Patel HV, Naples CJ, Fujioka H, Hoppel CL. Decreased cytochrome c mediates an age-related decline of oxidative phosphorylation in rat kidney mitochondria. Biochem J. 2010;427:105–112. doi:10.1042/BJ20091373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gómez LA, Hagen TM. Age-related decline in mitochondrial bioenergetics: Does supercomplex destabilization determine lower oxidative capacity and higher superoxide production?Semin Cell Dev Biol. 2012;23:758–767. doi:10.1016/j.semcdb.2012.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gómez LA, Monette JS, Chavez JD, Maier CS, Hagen TM. Supercomplexes of the mitochondrial electron transport chain decline in the aging rat heart. Arch Biochem Biophys. 2009;490:30–35. doi:10.1016/j.abb.2009.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Genova ML, Baracca A, Biondi A, et al. Is supercomplex organization of the respiratory chain required for optimal electron transfer activity?Biochim Biophys Acta. 2008;1777:740–746. doi:10.1016/j.bbabio.2008.04.007 [DOI] [PubMed] [Google Scholar]
- 16. Smeitink J, van den Heuvel L, DiMauro S. The genetics and pathology of oxidative phosphorylation. Nat Rev Genet. 2001;2:342–352. doi:10.1038/35072063 [DOI] [PubMed] [Google Scholar]
- 17. Tatarkova Z, Kovalska M, Timkova V, Racay P, Lehotsky J, Kaplan P. The effect of aging on mitochondrial complex I and the extent of oxidative stress in the rat brain cortex. Neurochem Res. 2016;41:2160–2172. doi:10.1007/s11064-016-1931-z [DOI] [PubMed] [Google Scholar]
- 18. Nickel AG, von Hardenberg A, Hohl M, et al. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. 2015;22:472–484. doi:10.1016/j.cmet.2015.07.008 [DOI] [PubMed] [Google Scholar]
- 19. Stroud DA, Formosa LE, Wijeyeratne XW, Nguyen TN, Ryan MT. Gene knockout using transcription activator-like effector nucleases (TALENs) reveals that human NDUFA9 protein is essential for stabilizing the junction between membrane and matrix arms of complex I. J Biol Chem. 2013;288:1685–1690. doi:10.1074/jbc.C112.436766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Babot M, Birch A, Labarbuta P, Galkin A. Characterisation of the active/de-active transition of mitochondrial complex I. Biochim Biophys Acta. 2014;1837:1083–1092. doi:10.1016/j.bbabio.2014.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Babot M, Labarbuta P, Birch A, et al. ND3, ND1 and 39kDa subunits are more exposed in the de-active form of bovine mitochondrial complex I. Biochim Biophys Acta. 2014;1837:929–939. doi:10.1016/j.bbabio.2014.02.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Loeffen JL, Triepels RH, van den Heuvel LP, et al. cDNA of eight nuclear encoded subunits of NADH:ubiquinone oxidoreductase: Human complex I cDNA characterization completed. Biochem Biophys Res Commun. 1998;253:415–422. doi:10.1006/bbrc.1998.9786 [DOI] [PubMed] [Google Scholar]
- 23. Davis CW, Hawkins BJ, Ramasamy S, et al. Nitration of the mitochondrial complex I subunit NDUFB8 elicits RIP1- and RIP3-mediated necrosis. Free Radic Biol Med. 2010;48:306–317. doi:10.1016/j.freeradbiomed.2009.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Emahazion T, Jobs M, Howell WM, et al. Identification of 167 polymorphisms in 88 genes from candidate neurodegeneration pathways. Gene. 1999;238:315–324. doi:10.1016/S0378-1119(99)00330-3 [DOI] [PubMed] [Google Scholar]
- 25. Jarr KU, Eschricht S, Burkly LC, et al. TNF-like weak inducer of apoptosis aggravates left ventricular dysfunction after myocardial infarction in mice. Mediators Inflamm. 2014;2014:131950. doi:10.1155/2014/131950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ngu LH, Nijtmans LG, Distelmaier F, et al. A catalytic defect in mitochondrial respiratory chain complex I due to a mutation in NDUFS2 in a patient with Leigh syndrome. Biochim Biophys Acta. 2012;1822:168–175. doi:10.1016/j.bbadis.2011.10.012 [DOI] [PubMed] [Google Scholar]
- 27. Sánchez-Caballero L, Guerrero-Castillo S, Nijtmans L. Unraveling the complexity of mitochondrial complex I assembly: A dynamic process. Biochim Biophys Acta. 2016;1857:980–990. doi:10.1016/j.bbabio.2016.03.031 [DOI] [PubMed] [Google Scholar]
- 28. Guarani V, Paulo J, Zhai B, Huttlin EL, Gygi SP, Harper JW. TIMMDC1/C3orf1 functions as a membrane-embedded mitochondrial complex I assembly factor through association with the MCIA complex. Mol Cell Biol. 2014;34:847–861. doi:10.1128/MCB.01551-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jahangir A, Ozcan C, Holmuhamedov EL, Terzic A. Increased calcium vulnerability of senescent cardiac mitochondria: Protective role for a mitochondrial potassium channel opener. Mech Ageing Dev. 2001;122:1073–1086. doi:10.1016/S0047-6374(01)00242-1 [DOI] [PubMed] [Google Scholar]
- 30. Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007;292:C670–C686. doi:10.1152/ajpcell.00213.2006 [DOI] [PubMed] [Google Scholar]
- 31. Miyake N, Yano S, Sakai C, et al. Mitochondrial complex III deficiency caused by a homozygous UQCRC2 mutation presenting with neonatal-onset recurrent metabolic decompensation. Hum Mutat. 2013;34:446–452. doi:10.1002/humu.22257 [DOI] [PubMed] [Google Scholar]
- 32. Pérez-Pérez R, Lobo-Jarne T, Milenkovic D, et al. COX7A2L is a mitochondrial complex III binding protein that stabilizes the III2+IV supercomplex without affecting respirasome formation. Cell Rep. 2016;16:2387–2398. doi:10.1016/j.celrep.2016.07.081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fannin SW, Lesnefsky EJ, Slabe TJ, Hassan MO, Hoppel CL. Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Arch Biochem Biophys. 1999;372:399–407. doi:10.1006/abbi.1999.1508 [DOI] [PubMed] [Google Scholar]
- 34. Huang TT, Naeemuddin M, Elchuri S, et al. Genetic modifiers of the phenotype of mice deficient in mitochondrial superoxide dismutase. Hum Mol Genet. 2006;15:1187–1194. doi:10.1093/hmg/ddl034 [DOI] [PubMed] [Google Scholar]
- 35. Rydström J. Mitochondrial NADPH, transhydrogenase and disease. Biochim Biophys Acta. 2006;1757:721–726. doi:10.1016/j.bbabio.2006.03.010 [DOI] [PubMed] [Google Scholar]
- 36. Aon MA, Stanley BA, Sivakumaran V, et al. Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: An experimental-computational study. J Gen Physiol. 2012;139:479–491. doi:10.1085/jgp.201210772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim SH, Ma X, Klupa T, et al. Genetic modifiers of the age at diagnosis of diabetes (MODY3) in carriers of hepatocyte nuclear factor-1alpha mutations map to chromosomes 5p15, 9q22, and 14q24. Diabetes. 2003;52:2182–2186. doi:10.2337/diabetes.52.8.2182 [DOI] [PubMed] [Google Scholar]
- 38. Kates AM, Herrero P, Dence C, et al. Impact of aging on substrate metabolism by the human heart. J Am Coll Cardiol. 2003;41:293–299. doi:10.1016/S0735-1097(02)02714-6 [DOI] [PubMed] [Google Scholar]
- 39. Hyyti OM, Ledee D, Ning XH, Ge M, Portman MA. Aging impairs myocardial fatty acid and ketone oxidation and modifies cardiac functional and metabolic responses to insulin in mice. Am J Physiol Heart Circ Physiol. 2010;299:H868–H875. doi:10.1152/ajpheart.00931.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chiao YA, Kolwicz SC, Basisty N, et al. Rapamycin transiently induces mitochondrial remodeling to reprogram energy metabolism in old hearts. Aging (Albany NY). 2016;8:314–327. doi:10.18632/aging.100881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Southworth LK, Owen AB, Kim SK. Aging mice show a decreasing correlation of gene expression within genetic modules. PLoS Genet. 2009;5:e1000776. doi:10.1371/journal.pgen.1000776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zahn JM, Poosala S, Owen AB, et al. AGEMAP: A gene expression database for aging in mice. PLoS Genet. 2007;3:e201. doi:10.1371/journal.pgen.0030201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ma L, Zhu J, Gao Q, Rebecchi MJ, Wang Q, Liu L. Restoring pharmacologic preconditioning in the aging heart: Role of mitophagy/autophagy. J Gerontol A Biol Sci Med Sci. 2017;72:489–498. doi:10.1093/gerona/glw168 [DOI] [PubMed] [Google Scholar]
- 44. Gaborit N, Steenman M, Lamirault G, et al. Human atrial ion channel and transporter subunit gene-expression remodeling associated with valvular heart disease and atrial fibrillation. Circulation. 2005;112:471–481. doi:10.1161/CIRCULATIONAHA.104.506857 [DOI] [PubMed] [Google Scholar]
- 45. Nattel S. Changes in the atrial transcriptome and atrial fibrillation: Susceptibility, persistence, causes, and consequences. Circ Arrhythm Electrophysiol. 2015;8:5–7. doi:10.1161/CIRCEP.115.002673 [DOI] [PubMed] [Google Scholar]
- 46. Ellinghaus P, Scheubel RJ, Dobrev D, et al. Comparing the global mRNA expression profile of human atrial and ventricular myocardium with high-density oligonucleotide arrays. J Thorac Cardiovasc Surg. 2005;129:1383–1390. doi:10.1016/j.jtcvs.2004.08.031 [DOI] [PubMed] [Google Scholar]
- 47. Hassouna A, Loubani M, Matata BM, Fowler A, Standen NB, Galiñanes M. Mitochondrial dysfunction as the cause of the failure to precondition the diabetic human myocardium. Cardiovasc Res. 2006;69:450–458. doi:10.1016/j.cardiores.2005.11.004 [DOI] [PubMed] [Google Scholar]
- 48. Guibert EE, Petrenko AY, Balaban CL, Somov AY, Rodriguez JV, Fuller BJ. Organ preservation: Current concepts and new strategies for the next decade. Transfus Med Hemother. 2011;38:125–142. doi:10.1159/000327033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Klebanov L, Qiu X, Welle S, Yakovlev A. Statistical methods and microarray data. Nat Biotechnol. 2007;25:25–6; author reply 26. doi:10.1038/nbt0107-25 [DOI] [PubMed] [Google Scholar]
- 50. Yang J, Huang T, Petralia F, et al. ; GTEx Consortium Synchronized age-related gene expression changes across multiple tissues in human and the link to complex diseases. Sci Rep. 2015;5:15145. doi:10.1038/srep15145 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.