

A microfluidic technology was developed and used to probe epigenomic differences between prefrontal cortex and cerebellum.
Abstract
Extensive effort is under way to survey the epigenomic landscape of primary ex vivo tissues to establish normal reference data and to discern variation associated with disease. The low abundance of some tissue types and the isolation procedure required to generate a homogenous cell population often yield a small quantity of cells for examination. This difficulty is further compounded by the need to profile a myriad of epigenetic marks. Thus, technologies that permit both ultralow input and high throughput are desired. We demonstrate a simple microfluidic technology, SurfaceChIP-seq, for profiling genome-wide histone modifications using as few as 30 to 100 cells per assay and with up to eight assays running in parallel. We applied the technology to profile epigenomes using nuclei isolated from prefrontal cortex and cerebellum of mouse brain. Our cell type–specific data revealed that neuronal and glial fractions exhibited profound epigenomic differences across the two functionally distinct brain regions.
INTRODUCTION
Histone modifications play critical roles in normal development and disease processes by dynamically tuning chromatin conformations and regulating gene expressions. The genome-wide profile of a specific histone mark is highly specific to a particular cell type. Mapping histone modifications using tissue homogenates with mixed cell types creates ambiguity and confusion in identifying molecular drivers. On the other hand, a homogeneous population of cells extracted from primary tissues is often in very small quantity because of low abundance and tedious isolation. This limitation of sample size is often further compounded by the fact that a large number of histone modifications may need to be examined. There are more than 100 distinct histone modifications, and tens of these modifications are studied routinely (1, 2). Comprehensive large-scale data sets on various histone marks permit accurate description of chromatin states using advanced computational algorithms (3). Thus, there is a pressing need for profiling histone modifications with both low input and high throughput.
Chromatin immunoprecipitation coupled with next-generation sequencing (ChIP-seq) is the gold standard for mapping in vivo genome-wide histone modifications. There have been various strategies developed in recent years to minimize the input of ChIP-seq [for example, nano-ChIP (4), LinDA (5), iChIP (6), MOWChIP (7), and Drop-ChIP (8)]. Single-cell ChIP-seq technique (that is, Drop-ChIP) offers insights into cell-to-cell variation but provides only a low number of unique reads per cell (~1000). Single-molecule imaging combined with single DNA sequencing was also applied to study combinatory histone modifications (9). Thus, techniques providing a broad coverage of genome using 10 to 100 cells per assay may be the most suitable for establishing reference epigenomes and probing disease states. In addition, the ideal platform should also allow high-throughput processing to facilitate study of various histone marks and a large number of samples. For example, we developed MOWChIP-seq (7) that permitted profiling histone modifications with as few as 100 cells. However, scaling up these devices for multiplexed operation has been complicated by the need to manipulate multiple batches of ChIP beads.
Here, we demonstrate a microfluidic technology based on ChIP on an antibody-coated channel surface, referred to as SurfaceChIP-seq, for multiplexed and ultralow-input profiling of histone modifications. Our technology does not involve manipulation of immunomagnetic beads; thus, it markedly simplifies the device design and facilitates in-parallel operation of multiple assays. We demonstrated input as low as 30 cells per assay and throughput of up to eight parallel assays on a single chip. These ChIP-seq data offered 3 million to 13 million unique reads per data set (with input of 30 to 1000 cells per assay, respectively), with data quality that rivals those of conventional assays.
We applied the technology to study NeuN+ (neuronal) and NeuN− (glial) fractions from two functionally distinct regions of a mouse brain: prefrontal cortex (PFC) and cerebellum. There have been very few reports on cell type–specific profiles of histone modifications in mammalian brains (10–12), and differences in histone modifications across various regions in these earlier studies were not studied systematically. We focused on three histone marks (H3K4me3, H3K27me3, and H3K27ac) and identified extensive differences in the epigenomes across the two brain regions.
RESULTS
We reasoned that ultralow-input ChIP would not require a large surface area for immunoprecipitation due to the tiny amount of chromatin fragments involved. Thus, we eliminated the immunomagnetic beads that had been used in all previous low-input ChIP assays (7, 13–15), and instead utilized a polydimethylsiloxane (PDMS)/glass microfluidic channel that was functionalized by an antibody. A channel with dimensions of 40 mm × 1 mm × 60 μm and a volume of 2.4 μl was sequentially treated with poly-l-lysine (PLL), a DNA oligo linker [33 nucleotides (nt)], and a DNA-conjugated antibody that hybridized with the oligo linker (Fig. 1A) (16). Previous studies showed that this protocol yielded high antibody density and activity (16, 17). During the SurfaceChIP process (Fig. 1A and fig. S1), micrococcal nuclease (MNase)–digested chromatin [170 to 550 base pairs (bp)] was loaded into the device in segments. Each segment of the chromatin solution (with a volume of 2.4 μl) filled the entire volume of the channel for 6 min before it exited the device. ChIP occurred on the glass substrate during the incubation. The loading/ChIP process took 1 hour. Oscillatory washing (that is, applying alternating pressure pulses at the two ends of the channel) was then conducted to remove nonspecific binding in a low-salt buffer and a high-salt buffer sequentially (7). Next, the channel was filled with proteinase K for 30 min to release ChIP DNA from the substrate surface. Finally, the ChIP DNA was eluted for off-chip processing (which included a step to remove the DNA linker), library preparation, and sequencing. The operational conditions were optimized by selecting high enrichment at known loci via quantitative polymerase chain reaction (qPCR) quantification of ChIP DNA.
Fig. 1. Overview of SurfaceChIP protocol and profiling of H3K4me3 and H3K27me3 marks in GM12878 cells.
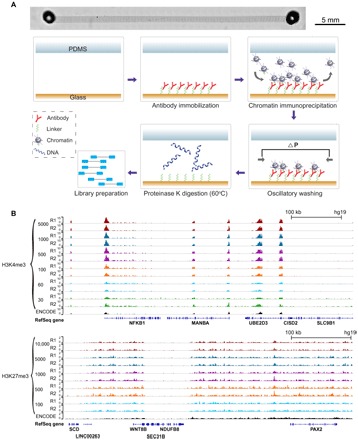
(A) Microscopic image of a single-channel device (stitched from multiple images) and steps involved in SurfaceChIP-seq. The microfluidic channel had dimensions of 40 mm × 1 mm × 60 μm. There were supporting pillars (50 μm in diameter) inside the channel to prevent collapse. (B) Normalized H3K4me3 signals generated using 30 to 5000 cells per assay and H3K27me3 signals using 100 to 10,000 cells per assay. ENCODE data (GSE29611) were included for comparison.
Using a cell line (GM12878), we demonstrated that our device generated high-quality ChIP-seq data with input as low as 30 and 100 cells per assay for H3K4me3 and H3K27me3, respectively (Fig. 1B and fig. S2). Pearson correlations between replicates were 0.99, 0.99, and 0.90 for 5000-, 100-, and 30-cell samples of H3K4me3, respectively, and 0.98, 0.97, and 0.95 for 10,000-, 1000-, and 100-cell samples of H3K27me3, respectively (fig. S2, A and B). Our data also compared favorably with those obtained with bead-based MOWChIP-seq, as quantified by receiver operating characteristic (ROC) curves (fig. S2C) (7). In SurfaceChIP processes, we obtained 700 and 25 pg of ChIP DNA from 5000 and 100 cells for H3K4me3, respectively. These yields were comparable to those in our previous study using an excess amount of ChIP beads (7). We generated 3 million to 13 million unique reads (after deducting redundancy created by PCR amplification) with 30 to 1000 cells, respectively (table S1). This performance was superior to our previous MOWChIP-seq (1.6 million reads with 100 cells) (7) and Drop-ChIP (1000 reads per cell) (8) in terms of unique reads yielded per cell.
In addition, our simple design permits multiplexed ChIP devices that run multiple parallel assays with minimal ancillary control system and a small footprint. We fabricated two-layered devices that contained four or eight parallel units for running SurfaceChIP assays with medium throughput (Fig. 2, A and B). These devices had a common inlet that connected with all channels, and each channel had one additional individual inlet and an outlet. All the individual inlets and outlets had on-chip pneumatic valves for fluid control (Fig. 2, A and B, and fig. S1F). These individual channels could be functionalized with one or multiple antibodies for either producing replicates of one histone profile or probing multiple histone marks, respectively. We used the four- and eight-channel devices (Fig. 2A) to profile two histone marks (H3K4me3 and H3K27me3) with two or four replicates for each mark in one run, respectively (table S1). Data sets generated by these devices had high reproducibility among replicates (Pearson correlation of 0.99 for H3K4me3 and 0.99 for H3K27me3 with 500 cells per assay in a four-channel device; 0.98 to 0.99 for H3K4me3 and 0.91 to 0.94 for H3K27me3 with 100 cells per assay in eight-channel device no. 1) (Fig. 2C). We also examined the reproducibility of the results by eight-channel devices made in different batches (device no. 1 in Fig. 2C and device no. 2 in fig. S3A). Excluding one low-quality data set (generated by C4 of device no. 2), the seven data sets on H3K4me3 and the eight data sets on H3K27me3 produced by two eight-channel devices (nos. 1 and 2) had Pearson correlations of 0.93 to 0.99 and 0.91 to 0.96, respectively (fig. S3B). The data showed that there was no cross-contamination among different channels. The sufficient number of replicates permitted removal of occasional low-quality data set.
Fig. 2. Multiplexed four- and eight-channel devices for rapid processing.
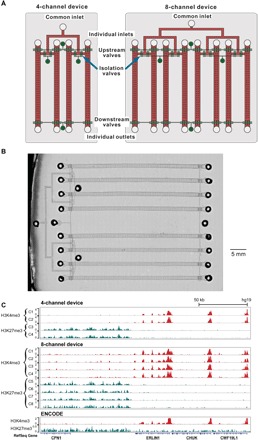
(A) Schematics of the devices. The structures in fluidic layer are in red, and the ones in control layer are in green. (B) Microscopic image of an eight-channel microfluidic device (stitched from multiple images). (C) Normalized ChIP-seq data on H3K4me3 and H3K27me3 generated using a four-channel device (500 cells per assay/channel for the four channels C1 to C4) and an eight-channel device (no. 1; 100 cells per assay/channel for the eight channels C1 to C8).
As an alternative to the PLL/oligo linker, we also tested the (3-aminopropyl)triethoxysilane/glutaraldehyde/protein A (AGP) linker system (fig. S4 and table S1) (18, 19). Using the AGP linker eliminated the need for the purification step required to remove the DNA linker before library preparation. However, SurfaceChIP-seq with the AGP linker offered less consistency between replicates than SurfaceChIP-seq with the DNA linker (fig. S4, A and B). The data quality of SurfaceChIP-seq with the AGP linker using 500 cells was similar to that of MOWChIP-seq using 600 cells (fig. S4C). However, AGP linker–based SurfaceChIP-seq produced substantially lower quality data than the PLL/oligo linker SurfaceChIP-seq or MOWChIP-seq when the input was 100 cells (fig. S4C). Thus, we used DNA linker–based SurfaceChIP-seq in all experiments, unless otherwise noted.
We applied SurfaceChIP-seq to establish epigenomes of NeuN+ and NeuN− fractions in mouse PFC and cerebellum. Although the functions of different brain regions are still being discovered (20), PFC is generally considered responsible for decision-making, personality expression, and cognitive control, whereas the cerebellum is responsible for motor control and coordination. However, there have not been studies that connect variations in epigenomic profiles with different functions of these brain regions. Epigenomes of neurons and glia in different brain regions potentially link genome-wide molecular features with specific activities and functions.
We first mapped H3K4me3, H3K27ac, and H3K27me3 using nuclei from tissue homogenates of PFC (dissected from mouse M1) and cerebellum (from mouse M2). We tested sample sizes ranging from 100 to 10,000 nuclei (table S1). There was only a slight decrease in the average Pearson correlation among replicates when fewer nuclei were used (for example, 0.99 to 0.97 for H3K4me3, 0.99 to 0.97 for H3K27ac, and 0.99 to 0.98 for H3K27me3, from 10,000 to 100 nuclei, respectively). In terms of correlations with corresponding ENCODE (Encyclopedia of DNA Elements) data on H3K4me3 and H3K27ac (GSE31039), the data using 1000 or more nuclei per assay generally outperformed the data taken with 100 nuclei per assay. However, even 100 nuclei data offered an average Pearson correlation of 0.83 on H3K4me3 and 0.80 on H3K27ac with ENCODE data.
We isolated neuronal and glial fractions from cerebellum and PFC of the same mouse (M3) using NeuN labeling and fluorescence-activated cell sorting (FACS). The four cell populations isolated are referred to as PFC NeuN+ (PNeuN+), cerebellar NeuN+ (CNeuN+), PFC NeuN− (PNeuN−), and cerebellar NeuN− (CNeuN−) (table S1). Substantial differences were observed in various histone marks across these cell populations (Fig. 3A). Previous work showed that brain tissues presented higher H3K27me3 coverage over intergenic regions (relative to introns) than other tissue types (21). Our results revealed that PNeuN+ had distinct patterns in terms of histone mark distributions between intergenic and intragenic regions. PNeuN+ has the lowest intergenic/intragenic ratio for H3K4me3 and H3K27ac, and the highest one for H3K27me3, among the four cell populations (Fig. 3B). These features are associated with restrictive chromatin environment in intergenic regions and enhanced recognition of functional elements in introns (21). For each histone mark in the same cell type, we also examined locations of the signal that was variable between the two brain regions (PNeuN+ and CNeuN+; PNeuN− and CNeuN−) (Fig. 3C). A large fraction of these differentially marked regions (identified by diffReps, P < 0.0001) for H3K27me3 were located in intergenic regions (70% for PNeuN+/CNeuN+ and 70% for PNeuN−/CNeuN−), whereas a substantial fraction of the differentially marked H3K4me3/H3K27ac regions were located in intragenic regions (genebody).
Fig. 3. Genome-wide differential histone modifications in various brain cell populations.
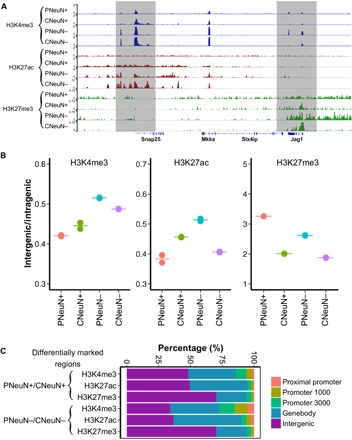
The data under analysis were generated with 1000 nuclei per assay using samples from mouse M3. (A) SurfaceChIP-seq signals around Snap25 gene for all four cell populations. (B) H3K4me3, H3K27ac, and H3K27me3 coverage in intergenic regions relative to intragenic regions (genebody). The pair-wise comparisons of H3K4me3, H3K27ac, and H3K27me3 for four cell populations show significant difference (P < 0.01), except PNeuN+/CNeuN+ and CNeuN+/CNeuN− on H3K4me3 mark and PNeuN+/CNeuN+ on H3K27ac mark. (C) Distributions of differentially marked regions in various genomic features.
Histone modifications in promoter regions critically affect gene activities. Our data sets showed a high correlation between replicates for all cell populations and histone marks (Pearson correlations in the range of 0.91 to 0.99). We found that the H3K4me3 level at gene promoters was fairly consistent across the four cell populations (average Pearson correlation, 0.91 among the four cell populations) (Fig. 4A). In comparison, H3K27ac and H3K27me3 levels at these promoters were much more variable (average Pearson correlation coefficients, 0.80 and 0.72, respectively) (Fig. 4A). Furthermore, being of the same cell type did not necessarily guarantee high level of consistency (for example, in terms of H3K27me3 level, the average Pearson correlation was 0.61 for PNeuN+ and CNeuN+, whereas it was 0.85 for CNeuN− and CNeuN+; Fig. 4A). We identified 1518, 4184, and 5588 promoter regions with significantly different levels (false discovery rate < 0.05) in H3K4me3, H3K27ac, and H3K27me3, respectively, when PNeuN+ and CNeuN+ data were compared (Fig. 4B and tables S2 to S4). Among them, 1512 promoters showed differences on two histone marks, and 431 promoters showed significant variability on all three histone marks. We also obtained mRNA-sequencing (mRNA-seq) data (table S1) and found that a substantial fraction of differentially expressed genes between PNeuN+ and CNeuN+ (1238 of 2672) involved differential histone marking in the promoter region. Consistent with previous reports (4, 21), we observed positive correlations between promoter H3K4me3 and H3K27ac levels and gene expression (fig. S5) and a negative correlation between promoter H3K27me3 level and gene expression (fig. S5) in both populations.
Fig. 4. Differential histone modifications at promoters in various brain cell populations.
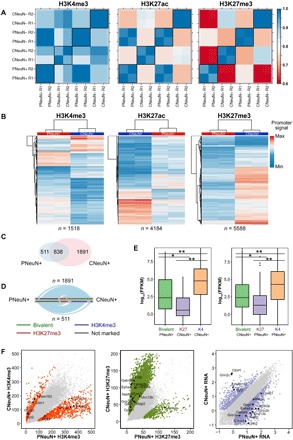
The data under analysis were generated with 1000 nuclei per assay using samples from mouse M3. (A) Pearson correlations of SurfaceChIP-seq signals in promoter regions across various cell populations. Each set of data has two replicates (R1 and R2). (B) Heatmap of all differentially marked promoters identified by DESeq2 (adjusted P < 0.05) between PNeuN+ and CNeuN+. (C) Venn diagram of bivalent promoters in PNeuN+ and CNeuN+. (D) Hive plots showing the proportions of bivalent promoters specific to PNeuN+ (or CNeuN+) being H3K4me3-marked, H3K27me3-marked, or not marked in the other neuronal population. Ribbons are proportional to the number of promoters changing marking. (E) The change in histone modification state is associated with gene expression change. *P < 10−4; **P < 10−15. (F) Pair-wise comparisons of H3K4me3 level, H3K27me3 level, and gene expression [log10(FPKM), measured by mRNA-seq] at various genes across PNeuN+ and CNeuN+ populations. The differential histone modifications (adjusted P < 0.05) and differentially expressed (fold change > 4) genes are shown as colored points in these. Selected genes associated with synapse GO term are labeled.
We also examined bivalent promoters that were marked by both activating H3K4me3 and repressive H3K27me3. Bivalent domains play critical roles in pluripotency by keeping developmental genes silent but poised for subsequent expression during differentiation (22). We discovered that CNeuN+ had roughly twice as many bivalent promoters (2729) as PNeuN+ (1349), with 838 bivalent promoters in both cell populations (Fig. 4C). Among the bivalent promoters that were uniquely present in PNeuN+ (n = 511), the vast majority (67%) was marked by only H3K4me3 in CNeuN+, whereas the rest was marked by H3K27me3 (28%) or unmarked (5%) (Fig. 4D). Similarly, among the bivalent promoters that were unique to CNeuN+ (n = 1891), a substantial fraction of them were marked by H3K4me3 (72%) (together with 22% marked by H3K27me3 and 6% unmarked) in PNeuN+. The change in the bivalent state of promoters was closely associated with variation in the gene expression level. In general, increased transcript levels were observed with loss of H3K27me3 mark, whereas decreased transcription was associated with loss of H3K4me3 at the bivalent promoters (Fig. 4E).
We further discovered 1074 genes with differential histone modifications at promoters that were correlated with changes in gene expression, when PNeuN+ and CNeuN+ were compared (table S5). In these cases, the increase or decrease in transcription level (based on mRNA-seq data) matched the trend predicted by differential H3K4me3 or H3K27me3 level at promoters (for example, increase in expression was associated with either an H3K4me3 increase or an H3K27me3 decrease at the gene promoter). Among these genes, 63% (673) of them were associated with only one differential histone mark (that is, change in either H3K4me3 or H3K27me3) across PNeuN+ and CNeuN+, with the rest associated with both differential histone marks. Of the 401 genes associated with two differential marks, there were only four genes, with the two differential histone marks predicting opposite trends for gene expression change. We also conducted gene ontology (GO) enrichment analysis of these 1074 genes, whose expression appeared to be dictated by promoter histone modifications using DAVID (23). We found that these genes were highly enriched in terms associated with neuronal functions and pathways, such as synapse (Fig. 4F) (P < 10−18), neuronal cell body (P < 10−10), neurogenesis (P < 10−9), and neuron projection (P < 10−8). These findings suggest that epigenetic variance plays a significant role in differentiating molecular profiles of NeuN+ fractions between the two brain regions.
We investigated active enhancers (marked by H3K4me3low + H3K27achigh) in the four cell populations. We predicted 8534, 8152, 11,151, and 10,533 enhancers in PNeuN+, CNeuN+, PNeuN−, and CNeuN−, respectively. Enhancers of the same cell type largely overlapped (Fig. 5A). In contrast, super-enhancers [enhancers stitched together when they are not within ±2 kb from annotated transcription start sites (TSSs), and the distance between them is within 15 kb] showed high specificity to different brain regions. PNeuN+ super-enhancers (80%) were not present in CNeuN+, and the common super-enhancers between CNeuN− and PNeuN− only accounted for 23 and 18% of the total in CNeuN− and PNeuN−, respectively, (Fig. 5A). The target genes of PNeuN+-specific super-enhancers (including a number of important neuronal function regulators, such as Nrxn1, Nedd4l, Nlgn1, and Ank3) were enriched in neuronal functions, such as synapse (P < 10−17, hypergeometric test) and neuronal cell body (P < 10−11, hypergeometric test). An unsupervised clustering analysis of genome-wide regions marked by H3K27ac revealed that the four cell populations could be separated from each other, whereas PNeuN+, CNeuN+, and PNeuN− bore more similarity among themselves than with CNeuN− (Fig. 5B). Finally, we searched for sequence motifs that differentiated PNeuN+ and CNeuN+ (Fig. 5C). The corresponding transcription factors included BRN1, TBET, and PAX3 that were specifically enriched in PNeuN+, and RUNX2, FOXH1, and STAT6 that were specifically enriched in CNeuN+. These transcription factors are well known to be critical for neuronal development and neuron differentiation (24).
Fig. 5. Enhancers and super-enhancers identified in various brain cell populations.
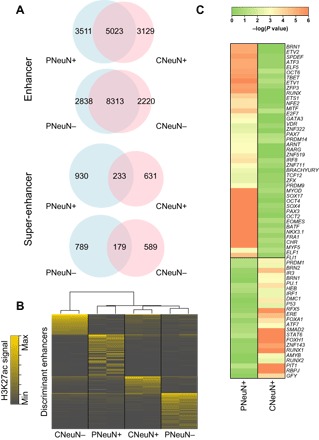
The data under analysis were generated with 1000 nuclei per assay using samples from mouse M3. (A) Venn diagrams of enhancers and super-enhancers identified in various cell populations. (B) Unsupervised hierarchical clustering of H3K27ac signals for the top 25% most-variable peaks (enhancers) located outside promoters (±2 kb from TSSs). (C) Heatmap of P values for transcription factor binding motifs enriched at enhancers.
DISCUSSION
Our SurfaceChIP-seq technology offers a couple of unique characteristics that facilitate high-throughput profiling of histone marks using primary tissues. First, our technology marks a balance between low input and high data quality. While allowing histone modification profiling using a small and highly purified cell population (down to 30 to 100 cells per assay), SurfaceChIP-seq offers data quality rivaling that of standard reference epigenomes (for example, those generated by the ENCODE consortium). Second, the extremely simple structure of SurfaceChIP-seq device (that is, one channel per assay) permits facile implementation of rapid and parallel operations. This is especially important for analyzing a number of histone marks and a large quantity of patient samples in a precision medicine setting.
We applied SurfaceChIP-seq to analyze different neuronal and glial epigenomic landscapes across mouse PFC and cerebellum. We applied NeuN labeling in this study to select neuronal nuclei (NeuN+ fraction) with the possible exception of cerebellar Purkinje and Golgi cells (25). Nevertheless, the majority of neurons in cerebellum are granule cells, and cerebellar Purkinje and Golgi cells only account for a tiny fraction of all neurons in cerebellum (each <0.1% of the cerebellar cell population) (26). Thus, cerebellar epigenomes profiled in this study should still be largely representative of the neuronal and glial populations. Our data show that the neuronal and glial fractions from the two brain regions exhibit extensive differences in histone modifications. These molecular differences are likely to be linked with the markedly different functions and activities for PFC and cerebellum. There are thousands of subtypes for neurons (27), and there is early evidence that their epigenomic profiles may have substantial variability (28). Thus, the different epigenomic features observed across PFC and cerebellum may reflect different sets of cell subtypes involved in each region of the brain.
MATERIALS AND METHODS
Fabrication of microfluidic devices
PDMS/glass microfluidic chips were fabricated using soft lithography. Photomasks with microscale patterns were designed using LayoutEditor (juspertor GmbH) and printed on high-resolution (10,000 dots per inch) transparencies. For making single-channel devices, a single master was made by replicating the features in the photomask on a 76-mm silicon wafer (University Wafers) with spun-on SU-8 2025 photoresist of 60-μm thickness (MicroChem) using photolithography. PDMS (RTV615, Momentive) with a mass ratio of A/B = 10:1 was poured onto the master in a petri dish to yield ~4-mm thickness. PDMS was baked at 75°C for 1 hour to cure. The cured PDMS was peeled off from the master, and access holes were punched. Four- or eight-channel devices were made using multilayer soft lithography (7, 29). Briefly, a control layer master was fabricated on a silicon wafer with 24-μm-thick spun-on SU-8 2025. A fluidic layer master was fabricated on a silicon wafer with 60-μm-thick SU-8 2025 and 21-μm-thick AZ 9260 (Clariant) features. The fluidic layer master was baked at 130°C for 30 s to round AZ 9260 features so that the resulting fluidic channels could be fully closed by microscale pneumatic valves. The PDMS control layer (~4 mm thick) was fabricated by pouring PDMS prepolymer with a mass ratio of A/B = 5:1 onto the control layer master in a petri dish. The fluidic layer (~80 μm thick) was fabricated by spinning PDMS (A/B = 20:1) at 500 rpm for 10 s and then at 1500 rpm for 30 s on the fluidic layer master. Both layers were baked at 75°C for 15 min. The control layer was then peeled off from the master, and access holes to the control layer channels were punched. The control layer was laid on top of the fluidic layer for alignment and contact. The entire structure was baked at 75°C for 1 hour to achieve thermal boning. The PDMS structure was then peeled off from the fluidic layer master, and access holes to the fluidic layer were punched. The PDMS structure (either single- or multi-layered) was bonded to a glass slide that was precleaned in 27% NH4OH/30% H2O2/H2O = 1:1:5 (volumetric ratio) at 80°C for 30 min. Both PDMS surface and glass surface were oxidized in plasma (PDC-32G, Harrick Plasma) for 1 min and then immediately brought into contact. The bonded device was immediately used for antibody coating.
Antibody coating on the channel substrate
PLL/oligo linker
Our antibody coating procedure was similar to previous works with minor modifications (16, 17). The microfluidic channel (with freshly oxidized glass surface) was filled with 0.1% PLL (P2636, Sigma-Aldrich) and sealed with Microseal ’B’ adhesive seals (MSB1001, Bio-Rad). Channel filling or washing was conducted by loading solution into a channel reservoir (or a 200-μl pipette tip plugged into the reservoir in the case of liquid volumes >15 μl) on one end followed by aspiration using a pipettor on the other end (fig. S1B). The chip was incubated at 37°C for 15 min in an incubator. The channel was then washed by 100 μl of H2O and incubated at 75°C for 1 hour in an oven. The microfluidic channel was filled with 4 μl of 200 μM coating DNA (5′-AAAAAAAAAAAAAATCCTGGAGCTAAGTCCGTA-3′) dissolved in dimethyl sulfoxide/H2O = 1:2 (volumetric ratio) and kept in a desiccator at room temperature for 3 days until all solvent evaporated. The chip was then baked at 75°C for 3 hours. The microfluidic channel was washed with 100 μl of blocking buffer [3% bovine serum albumin (BSA) and 0.05% Tween 20 in phosphate-buffered saline (PBS) buffer] before incubation with the blocking buffer in the channel at 37°C for 1 hour. An antibody-oligo conjugate was generated by conjugating an antibody (anti-H3K4me3, EMD Millipore 07-473; anti-H3K27ac, Abcam ab4729; or anti-H3K27me3, EMD Millipore 07-449) and a DNA oligo (5′-NH3-AAAAAAAAAATACGGACTTAGCTCCAGGAT-3′) using the Protein-Oligo Conjugation Kit (Solulink), following the manufacturer’s instructions. The antibody-oligo conjugate had a concentration of 250 to 500 μg/ml in 100 μl of PBS buffer after the conjugation. We used specific concentrations for the antibody-oligo conjugate solution used for coating (generated by diluting the produced conjugate using the blocking buffer), depending on the histone mark and the number of cells/nuclei used in a ChIP assay. For H3K4me3 and H3K27ac marks, conjugate concentrations equivalent to 50, 25, and 10 μg of antibody per milliliter were used for input amounts of 5000, 500 to 1000, and less than 500 cells/nuclei, respectively. For H3K27me3, conjugate concentrations equivalent to 100, 50, and 25 μg of antibody per milliliter were used for 5000 to 10,000, 500 to 1000, and 100 cells/nuclei, respectively. The coating step was conducted by filling a channel with 8 to 10 μl of the conjugate solution and by incubating at 37°C for 1 hour.
AGP linker
In the experiments summarized in fig. S4, we used SurfaceChIP devices that were produced using the AGP linker. Twenty-five microliters of freshly prepared 10% 3-aminopropyltriethoxysilane (440140, Sigma-Aldrich) in ethanol was filled into the microfluidic channel, with the two reservoirs fully filled with the solution and sealed with Microseal ’B’ adhesive seals to avoid contact with moisture in the air. The chip was incubated at room temperature for 30 min for surface silanization. The channel was then washed with 100 μl of 95% ethanol (containing 5% water). The channel was then dried by pushing air at 10 psi (68.95 kPa) through. The chip was then baked at 125°C for 30 min to remove residual ethanol and promote silanization. The microfluidic channel was filled with 8% glutaraldehyde (G7651, Sigma-Aldrich), sealed by Microseal ’B’ adhesive seals at the reservoirs, and then incubated at room temperature for 30 min. The channel was washed by 100 μl of water and dried by air at 10 psi. Eight to 10 μl of protein A (2 mg/ml; 21184, Life Technologies) in PBS were then filled into the channel followed by incubation at room temperature for 60 min and washing by 100 μl of 0.5% BSA in PBS. Finally, 10 μl of antibody (200 μg/ml) in PBS (containing 0.5% BSA) was loaded into the channel, with the reservoirs sealed and incubated at 4°C overnight.
After antibody coating (with either linker), the channel was washed by 100 μl of 1× lysis buffer [2% Triton X-100, 50 mM tris (pH 7.5), 50 mM NaCl, and 15 mM MgCl2] with freshly added 1 μl of protease inhibitor cocktail (PIC; P8340, Sigma-Aldrich) and 1 μl of 100 mM phenylmethylsulfonyl fluoride (PMSF; Sigma-Aldrich). The channel was washed again with 100 μl of PBS (containing 0.5% BSA) before ChIP. Extra care was taken to avoid introducing bubbles into channels.
Cell culture
GM12878 cells were purchased from Coriell Institute for Medical Research. The cells were cultured in RPMI 1640 medium (11875-093, Gibco) with 15% fetal bovine serum (26140-079, Gibco) and penicillin-streptomycin (100 U/ml) (15140-122, Gibco) at 37°C in a humidified incubator with 5% CO2. Cells were maintained in exponential growth phase by subculturing every 2 to 3 days.
Mouse strain and brain dissection
Eight-week-old C57BL/6NHsd male mice were purchased from Envigo and allowed a week of acclimation in the animal facility with 12-hour light/12-hour dark cycles and food/water ad libitum before sacrificing. The mice were sacrificed by the use of compressed CO2 followed by cervical dislocation and decapitation. The cerebellum and PFC were rapidly dissected and frozen on dry ice. The tissues were stored at −80°C until use for nuclei isolation. The Institutional Animal Care and Use Committee (IACUC) at Virginia Tech approved this study, and IACUC guidelines were closely followed.
Nuclei isolation from brain tissues
We conducted nuclei isolation from brain tissues using a published protocol (30). All procedures were performed on ice. All centrifugation was performed at 4°C. One PFC or cerebellum was placed in 5 ml of ice-cold nuclei extraction buffer [0.32 M sucrose, 5 mM CaCl2, 3 mM Mg(Ac)2, 0.1 mM EDTA, 10 mM tris-HCl, and 0.1% Triton X-100] with freshly added 50 μl of PIC (P8340, Sigma-Aldrich), 5 μl of 100 mM PMSF, and 5 μl of 1 M dithiothreitol (DTT). The ribonuclease (RNase) inhibitor (7.5 μl of 40 U/μl; N2611, Promega) was mixed with nuclei extraction buffer when mRNA-seq was conducted. Once thawed, the tissue was homogenized by slowly douncing 15 times with a loose pestle (D9063, Sigma-Aldrich) and then 25 times with a tight pestle (D9063, Sigma-Aldrich). The homogenate was filtered with a 40-μm nylon mesh cell strainer (22363547, Thermo Fisher Scientific) and transferred into a 15-ml centrifugation tube. The sample was centrifuged at 1000g for 10 min. The supernatant was removed, and pellet was gently resuspended in 1 ml of cold nuclei extraction buffer with freshly added 10 μl of PIC, 1 μl of 100 mM PMSF, and 1 μl of 1 M DTT. The RNase inhibitor (1.5 μl of 40 U/μl) was also added when mRNA-seq was conducted. The 1-ml sample suspension was evenly split into two 1.5-ml microcentrifuge tubes. Each 0.5-ml sample was mixed with 0.75 ml of 50% iodixanol that was prepared by adding 0.8 ml of diluent [150 mM KCl, 30 mM MgCl2, and 120 mM tris-HCl (pH 7.8)] to 4 ml of 60% iodixanol (D1556, Sigma-Aldrich). The solution was then centrifuged at 10,000g for 20 min. After centrifugation, the supernatant was removed and 0.5 ml of 2% normal goat serum (50062Z, Life Technologies) in Dulbecco’s PBS (DPBS; 14190144, Life Technologies) was added in each tube followed by incubation on ice for 10 min. Nuclei were then suspended by pipetting, and the two tubes were pooled together to generate ~1 ml in total. The RNase inhibitor (1.5 μl of 40 U/μl) was added when mRNA-seq was conducted. For labeling and separation of NeuN+ and NeuN− fractions, anti-NeuN antibody conjugated with Alexa 488 (MAB377X, EMD Millipore) was diluted to 2 ng/μl in DPBS. Anti-NeuN antibody (16 μl of 2 ng/μl) was added to the nuclei suspension. Then, the 1-ml nuclei suspension was evenly split into five 1.5-ml centrifugation tubes (with 200 μl in each) and incubated at 4°C for 1 hour on an end-to-end rotator. The samples were finally pooled (to generate approximately 1 ml in total) and then sorted using FACS (BD FACSAria, BD Biosciences). Nonlabeled nuclei (50,000 to 100,000) were used as unstained control.
Chromatin fragmentation
PIC (0.1 μl) and PMSF (0.1 μl of 100 mM) were freshly added in 10 μl of suspension (containing cells/nuclei ranging from 30 to 10,000). The sample was then mixed with 10 μl of 2× lysis buffer [4% Triton X-100, 100 mM tris (pH 7.5), 100 mM NaCl, and 30 mM MgCl2] and incubated at room temperature for 10 min. CaCl2 (1 μl of 0.1 M) and MNase (2.5 μl of 10 U/μl; 88216, Thermo Fisher Scientific) were rapidly mixed with the sample and incubated at room temperature for 10 min. EDTA (2.22 μl of 0.5 M) (pH 8) was added and incubated on ice for 10 min. The sample was centrifuged at 16,100g at 4°C for 5 min. The supernatant (~24 μl) was transferred to a new microcentrifuge tube and stored on ice.
Operation of the microfluidic device
Single-channel device
We used a tubing system that had three connected parts (fig. S1, D and E): 1.5-m perfluoroalkoxyalkane (PFA) high-purity tubing [ID (inner diameter), 0.02 in. (0.51 mm) and OD (outer diameter), 0.0625 in. (1.59 mm); 1622L, IDEX Health & Science], a 5-cm Clear C-Flex tubing (ID, 0.0313 in. (0.795 mm) and OD, 0.0938 in. (2.383 mm); EW-06422-01, IDEX Health & Science), and 1-cm PFA tubing. The flexible C-Flex tubing connected the two pieces of PFA tubing and allowed the short PFA tubing to plug into the microchannel reservoir without tension. Freshly prepared chromatin solution (24 μl) was loaded into the tubing system from the short PFA tubing end. The short PFA tubing was then plugged into one end of the microfluidic channel. The free end of the long PFA tubing was plugged into a solenoid valve (18801003-12V, ASCO Scientific) so that pressure pulses could be applied to push the chromatin solution into the microfluidic channel. The chromatin solution (24 μl) was flowed through the microfluidic channel (with a volume of 2.4 μl) in 10 segments. A pressure pulse of 5 psi and 0.7-s duration was applied to push roughly 2.4 μl of the chromatin solution into the channel each time. Each segment of the chromatin solution was incubated in the channel for 6 min before the next pressure pulse filled the channel with fresh chromatin solution. This entire ChIP process took 1 hour. The channel was then washed with 20 μl of low-salt washing buffer [10 mM tris-HCl (pH 7.4), 50 mM NaCl, 1 mM EDTA (pH 8.0) with freshly added 1 mM PMSF, and 1% PIC]. Oscillatory washing was conducted by filling 20 μl of low-salt washing buffer in each of two tubing systems and then plugging them into both ends of the channel (fig. S1E). The other ends of the two pieces of tubing were connected to pressure source via two solenoid valves. Pressure pulses (at 5 psi with 1-s duration) were applied alternatingly at either end of the microfluidic channel without intervals for 4 min via control of the two solenoid valves by a LabVIEW program. The oscillatory washing effectively flowed the washing buffer over the channel surface back and forth to remove nonspecifically adsorbed chromatin fragments. Another round of oscillatory washing was performed under the same parameters with high-salt washing buffer [10 mM tris-HCl (pH 7.4), 100 mM NaCl, 1 mM EDTA (pH 8.0) with freshly added 1 mM PMSF, and 1% PIC].
After washing, 15 μl of freshly made elution buffer [proteinase K (1 mg/ml), 10 mM tris-HCl (pH 7.4), 50 mM NaCl, 10 mM EDTA (pH 8.0), and 0.03% SDS] was flowed into the channel (with the majority hold in the reservoirs that were sealed). The chip was placed on a hot plate and incubated at 60°C for 30 min to allow digestion of histones and antibody to release DNA. After proteinase K digestion, the sample was eluted by flushing the channel with 100 μl of water.
Four- or eight-channel devices
The setup for four-channel device involved the use of a syringe pump and control system for on-chip pneumatic valves and application of pressure pulses via the channel inlet/outlet (fig. S1F). The eight-channel device was set up in a similar fashion. These devices were two-layer devices with a control layer and a fluidic layer (29). There were two sets of solenoid valves (18801003-12V, ASCO Scientific) that regulated oscillatory washing (at 5 psi) and on-chip pneumatic valves (at 25 psi) separately due to difference in pressure. The operation of the solenoid valves was carried out via a LabVIEW program and a data acquisition card (PCI-6509, National Instruments). The control layer channels were filled with water before experiments.
In general, four- or eight-channel devices followed the same steps for antibody coating and ChIP as single-channel devices, except for reagent delivery and dispensing. For example, the early steps in antibody coating (before immobilization of antibody or antibody-oligo conjugate) of four-channel device were conducted by delivering 4× amount of reagents from the common inlet using the syringe pump so that all four channels were treated simultaneously. The upstream pneumatic valves were closed, and the downstream and isolation ones open during these steps (various valves are shown in Fig. 2A). When we needed to coat the four channels with different antibodies, we closed the isolation valves and kept the upstream and downstream valves open so that different antibodies were delivered into the channels separately from the individual inlets without cross-contamination. During ChIP, the chromatin solution (4×) was dispensed into all four channels from the common inlet. The oscillatory washing and subsequent elution were also conducted with the channels in isolation from each other.
Purification of ChIP DNA
The 100-μl elute (per assay) was purified by phenol extraction and ethanol precipitation. DNA pellet was resuspended in 20 μl of EB buffer (19086, Qiagen). When the PLL/oligo linker was used, the DNA was further purified by adding 32 μl of SPRIselect beads (Beckman Coulter) following the manufacturer’s instructions. The DNA was resuspended in 20 μl (10 μl when a ThruPLEX DNA-seq kit was used) of EB buffer after the step. The purified ChIP DNA was stored at −80°C until use. DNA concentrations were measured using a NanoDrop 3300 fluorospectrometer with the Quant-iT PicoGreen dsDNA Assay Kit (P11496, Life Technologies).
Input DNA
Input samples (10 μl containing DNA from 1000 cells/nuclei) were mixed with 94 μl of elution buffer [containing proteinase K (0.4 mg/ml)] and incubated at 65°C for 1 hour. The sample was then purified by phenol extraction and ethanol precipitation. DNA pellet was resuspended in 20 μl of EB buffer and stored at −80°C until use.
Construction of ChIP-seq libraries
All ChIP-seq libraries were constructed using an Accel-NGS 2S Plus DNA Library kit (Swift Biosciences), with the exception of GM12878 H3K4me3 samples with ≥100 cells (prepared using a ThruPLEX DNA-seq kit of Rubicon Genomics).
Accel-NGS 2S Plus DNA Library kit
ChIP DNA in 20 μl of EB buffer was used for library preparation. We followed the manufacturer’s instructions with minor modifications as detailed below. EvaGreen dye (1×; Biotium) was added for monitoring and quantifying PCR amplification. After PCR amplification (with DNA library in a 50-μl volume), a double-size selection using SPRIselect beads was performed to collect DNA of 300 to 700 bp. Briefly, large DNA fragments were removed by adding 25-μl beads and incubating for 5 min. The beads that had large DNA bound were discarded, and the supernatant was preserved. SPRIselect beads (12.5 μl) were then added into the supernatant and incubated for 5 min. This time, the supernatant was discarded and the DNA was eluted from the beads using 7 μl of EB buffer.
ThruPLEX DNA-seq kit
ChIP DNA in 10 μl of EB buffer was processed following the manufacturer’s instructions. The sequencing library (in a volume of 50 μl) was purified with 50 μl of SPRIselect beads and eluted using 7 μl of EB buffer.
Library quality control and quantification
Library fragment size was examined using a High Sensitivity DNA Analysis kit (Agilent) on a TapeStation (Agilent). The libraries were quantified by a KAPA Library Quantification kit (Kapa Biosystems) and pooled at 10 nM for sequencing by Illumina HiSeq 4000 with single-end 50-nt read.
Construction of mRNA-seq libraries
Nuclei from brain tissues (either homogenate or sorted; 100,000 nuclei per assay) were used for total RNA extraction by the RNeasy Mini Kit (Qiagen) with deoxyribonuclease (DNase) treatment step to remove genomic DNA contamination, following the manufacturer’s instructions. mRNA-seq libraries were prepared using a SMART-Seq v4 Ultra Low Input RNA kit (Clontech) and a Nextera XT DNA Library Prep kit (Illumina) following the manufacturer’s instructions. We used ~1 ng of RNA (in 9.5 μl of water) and generated complementary DNA (cDNA) after amplification by 14 cycles of PCR. Purified cDNA (100 to 150 pg) was used for Nextera library preparation. The fragment size of the library was determined by a TapeStation and quantified by a KAPA qPCR Library Quantification kit. Each library was pooled at 10 nM for sequencing by Illumina HiSeq 4000 with single-end 50-nt read.
ChIP-seq read mapping and normalization
Sequencing reads were trimmed by Trim Galore! with default settings. The trimmed reads were aligned to hg19 or mm9 genome using Bowtie (31). Mapped reads were used for narrow peak calling with MACS2 (q < 0.05) (32) and broad peak calling with SICERpy (33) (-rt 0 --windowSize 1000 --gapSize 3). Uniquely mapped reads from both ChIP and input samples were extended to 250 bp and used to compute a normalized signal for each 100-nt bin across the genome. Normalized signal was defined as follows
Construction of ROC curves
We compared the performance of SurfaceChIP-seq to that of MOWChIP-seq (7) using ROC curves. We focused on promoter regions (defined as 2 kb upstream and 500 bp downstream of a TSS based on RefSeq genes). The ROC curves were constructed using previously described methods (7). We used data generated by ENCODE consortium [Gene Expression Omnibus (GEO): GSM733708 and GSM945188] as the gold standard.
Analysis of promoter signals
ChIP-seq read counts in promoter regions were extracted. Promoter regions were defined as ±2 kb around TSSs. Signals across the promoter regions were used for computing Pearson correlation coefficients. The read counts were analyzed by DESeq2 (adjusted P < 0.05) (34) to identify differentially marked promoters. Bivalent promoters were defined as promoter regions that overlapped with both H3K4me3 and H3K27me3 peaks for at least 400 bp. The genes with differential histone marks were extracted as targets for GO term enrichment analysis using DAVID with default settings (23).
Genome-wide differentially marked regions
Genome-wide regions that were differentially marked by histone modifications were identified and annotated to RefSeq genes by diffReps (35) with default settings.
Prediction of enhancers and super-enhancers
Enhancers were defined as H3K4me3low + H3K27achigh and predicted using CSI-ANN (36) with normalized H3K4me3 and H3K27ac signals as inputs. The predicted enhancers and H3K27ac signal were then used as inputs to predict super-enhancers using ROSE (http://bitbucket.org/young_computation/rose). ROSE stitches enhancers within 15 kb and excludes enhancers within ±2 kb from annotated TSSs. The coordinates of the super-enhancers were extracted as targets for GO term enrichment analysis using GREAT with default settings (37).
Transcription factor motif enrichment analysis
We used coordinates of the predicted enhancers as input for motif analysis. We used Homer (-size 200 -mask -nomotif) (38) to find enrichment (P value) of known transcription factors in the default database.
mRNA-seq data analysis
Sequencing reads were trimmed by Trim Galore! with default settings. The trimmed reads were aligned by TopHat (39) and further analyzed by cufflinks (40). Gene expression levels were plotted by CummeRbund (http://compbio.mit.edu/cummeRbund/), SeqMonk, and custom R scripts. Differentially expressed genes were identified by CummeRbund.
Supplementary Material
Acknowledgments
We thank P. W. Faber of University of Chicago Genomics Facility for facilitating sequencing of the samples. Funding: This work was supported by U.S. NIH grants EB017235 (C.L.), HG009256 (C.L.), HG008623 (C.L.), CA214176 (C.L.), and HG007352 (J.M.) and a seed grant from Center for Engineered Health of Virginia Tech Institute for Critical Technology and Applied Science (C.L.). Author contributions: C.L. and S.M. designed the microfluidic device. S.M. and C.L. developed the SurfaceChIP-seq protocol. Y.-P.H. conducted mouse brain dissection. S.M. generated SurfaceChIP-seq and mRNA-seq data. S.M., C.L., and J.M. conducted the data analysis. C.L. supervised the research. C.L. and S.M. wrote the manuscript. All authors proofread the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. GEO: SurfaceChIP-seq and mRNA-seq data are deposited under accession number GSE99757 (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE99757).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/4/eaar8187/DC1
fig. S1. Fabrication, operation, and setup of the SurfaceChIP microfluidic device.
fig. S2. Data quality of SurfaceChIP-seq.
fig. S3. Reproducibility of SurfaceChIP-seq data by eight-channel devices produced in different batches.
fig. S4. SurfaceChIP-seq with AGP linker.
fig. S5. Rank of H3K4me3, H3K27ac, and H3K27me3 signals at promoters plotted against mRNA rank (generated by mRNA-seq).
table S1. Metadata of SurfaceChIP-seq and mRNA-seq.
table S2. A list of genes with differential H3K4me3 mark at promoters across PNeuN+ and CNeuN+ populations.
table S3. A list of genes with differential H3K27ac mark at promoters across PNeuN+ and CNeuN+ populations.
table S4. A list of genes with differential H3K27me3 mark at promoters across PNeuN+ and CNeuN+ populations.
table S5. A list of differentially expressed genes (fold change > 4; P < 0.05, t test) with correlated differential histone modification in H3K4me3 or H3K27me3 at promoters across PNeuN+ and CNeuN+ populations.
REFERENCES AND NOTES
- 1.Strahl B. D., Allis C. D., The language of covalent histone modifications. Nature 403, 41–45 (2000). [DOI] [PubMed] [Google Scholar]
- 2.Barski A., Cuddapah S., Cui K., Roh T.-Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K., High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Ernst J., Kellis M., ChromHMM: Automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adli M., Zhu J., Bernstein B. E., Genome-wide chromatin maps derived from limited numbers of hematopoietic progenitors. Nat. Methods 7, 615–618 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shankaranarayanan P., Mendoza-Parra M.-A., Walia M., Wang L., Li N., Trindade L. M., Gronemeyer H., Single-tube linear DNA amplification (LinDA) for robust ChIP-seq. Nat. Methods 8, 565–567 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Lara-Astiaso D., Weiner A., Lorenzo-Vivas E., Zaretsky I., Jaitin D. A., David E., Keren-Shaul H., Mildner A., Winter D., Jung S., Friedman N., Amit I., Immunogenetics. Chromatin state dynamics during blood formation. Science 345, 943–949 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao Z., Chen C., He B., Tan K., Lu C., A microfluidic device for epigenomic profiling using 100 cells. Nat. Methods 12, 959–962 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rotem A., Ram O., Shoresh N., Sperling R. A., Goren A., Weitz D. A., Bernstein B. E., Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 33, 1165–1172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shema E., Jones D., Shoresh N., Donohue L., Ram O., Bernstein B. E., Single-molecule decoding of combinatorially modified nucleosomes. Science 352, 717–721 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dincer A., Gavin D. P., Xu K., Zhang B., Dudley J. T., Schadt E. E., Akbarian S., Deciphering H3K4me3 broad domains associated with gene-regulatory networks and conserved epigenomic landscapes in the human brain. Transl. Psychiatry 5, e679 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shulha H. P., Crisci J. L., Reshetov D., Tushir J. S., Cheung I., Bharadwaj R., Chou H.-J., Houston I. B., Peter C. J., Mitchell A. C., Yao W.-D., Myers R. H., Chen J.-f., Preuss T. M., Rogaev E. I., Jensen J. D., Weng Z., Akbarian S., Human-specific histone methylation signatures at transcription start sites in prefrontal neurons. PLOS Biol. 10, e1001427 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheung I., Shulha H. P., Jiang Y., Matevossian A., Wang J., Weng Z., Akbarian S., Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc. Natl. Acad. Sci. U.S.A. 107, 8824–8829 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geng T., Bao N., Litt M. D., Glaros T. G., Li L., Lu C., Histone modification analysis by chromatin immunoprecipitation from a low number of cells on a microfluidic platform. Lab Chip 11, 2842–2848 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Cao Z., Lu C., A microfluidic device with integrated sonication and immunoprecipitation for sensitive epigenetic assays. Anal. Chem. 88, 1965–1972 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou P., Gu F., Zhang L., Akerberg B. N., Ma Q., Li K., He A., Lin Z., Stevens S. M., Zhou B., Pu W. T., Mapping cell type-specific transcriptional enhancers using high affinity, lineage-specific Ep300 bioChIP-seq. eLife 6, e22039 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan R., Vermesh O., Srivastava A., Yen B. K. H., Qin L., Ahmad H., Kwong G. A., Liu C.-C., Gould J., Hood L., Heath J. R., Integrated barcode chips for rapid, multiplexed analysis of proteins in microliter quantities of blood. Nat. Biotechnol. 26, 1373–1378 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma C., Fan R., Ahmad H., Shi Q., Comin-Anduix B., Chodon T., Koya R. C., Liu C.-C., Kwong G. A., Radu C. G., Ribas A., Heath J. R., A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat. Med. 17, 738–743 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiong L., Regnier F. E., Channel-specific coatings on microfabricated chips. J. Chromatogr. A 924, 165–176 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Yu L., Chen Q., Tian Y. L., Gao A. X., Li Y., Li M., Li C. M., One-post patterning of multiple protein gradients using a low-cost flash foam stamp. Chem. Commun. 51, 17588–17591 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Wagner M. J., Kim T. H., Savall J., Schnitzer M. J., Luo L., Cerebellar granule cells encode the expectation of reward. Nature 544, 96–100 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu J., Adli M., Zou J. Y., Verstappen G., Coyne M., Zhang X., Durham T., Miri M., Deshpande V., De Jager P. L., Bennett D. A., Houmard J. A., Muoio D. M., Onder T. T., Camahort R., Cowan C. A., Meissner A., Epstein C. B., Shoresh N., Bernstein B. E., Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 152, 642–654 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bernstein B. E., Mikkelsen T. S., Xie X., Kamal M., Huebert D. J., Cuff J., Fry B., Meissner A., Wernig M., Plath K., Jaenisch R., Wagschal A., Feil R., Schreiber S. L., Lander E. S., A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Huang D. W., Sherman B. T., Lempicki R. A., Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pang Z. P., Yang N., Vierbuchen T., Ostermeier A., Fuentes D. R., Yang T. Q., Citri A., Sebastiano V., Marro S., Südhof T. C., Wernig M., Induction of human neuronal cells by defined transcription factors. Nature 476, 220–223 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mullen R. J., Buck C. R., Smith A. M., NeuN, a neuronal specific nuclear protein in vertebrates. Development 116, 201–211 (1992). [DOI] [PubMed] [Google Scholar]
- 26.Tomomura M., Rice D. S., Morgan J. I., Yuzaki M., Purification of Purkinje cells by fluorescence-activated cell sorting from transgenic mice that express green fluorescent protein. Eur. J. Neurosci. 14, 57–63 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Molyneaux B. J., Arlotta P., Menezes J. R. L., Macklis J. D., Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci. 8, 427–437 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Mo A., Mukamel E. A., Davis F. P., Luo C., Henry G. L., Picard S., Urich M. A., Nery J. R., Sejnowski T. J., Lister R., Eddy S. R., Ecker J. R., Nathans J., Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 86, 1369–1384 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Unger M. A., Chou H.-P., Thorsen T., Scherer A., Quake S. R., Monolithic microfabricated valves and pumps by multilayer soft lithography. Science 288, 113–116 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Lake B. B., Ai R., Kaeser G. E., Salathia N. S., Yung Y. C., Liu R., Wildberg A., Gao D., Fung H.-L., Chen S., Vijayaraghavan R., Wong J., Chen A., Sheng X., Kaper F., Shen R., Ronaghi M., Fan J.-B., Wang W., Chun J., Zhang K., Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352, 1586–1590 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langmead B., Trapnell C., Pop M., Salzberg S. L., Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nusbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu S., Grullon S., Ge K., Peng W., Spatial clustering for identification of ChIP-enriched regions (SICER) to map regions of histone methylation patterns in embryonic stem cells. Methods Mol. Biol. 1150, 97–111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen L., Shao N.-Y., Liu X., Maze I., Feng J., Nestler E. J., diffReps: Detecting differential chromatin modification sites from ChIP-seq data with biological replicates. PLOS ONE 8, e65598 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Firpi H. A., Ucar D., Tan K., Discover regulatory DNA elements using chromatin signatures and artificial neural network. Bioinformatics 26, 1579–1586 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McLean C. Y., Bristor D., Hiller M., Clarke S. L., Schaar B. T., Lowe C. B., Wenger A. M., Bejerano G., GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28, 495–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K., Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trapnell C., Pachter L., Salzberg S. L., TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trapnell C., Williams B. A., Pertea G., Mortazavi A., Kwan G., van Baren M. J., Salzberg S. L., Wold B. J., Pachter L., Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/4/eaar8187/DC1
fig. S1. Fabrication, operation, and setup of the SurfaceChIP microfluidic device.
fig. S2. Data quality of SurfaceChIP-seq.
fig. S3. Reproducibility of SurfaceChIP-seq data by eight-channel devices produced in different batches.
fig. S4. SurfaceChIP-seq with AGP linker.
fig. S5. Rank of H3K4me3, H3K27ac, and H3K27me3 signals at promoters plotted against mRNA rank (generated by mRNA-seq).
table S1. Metadata of SurfaceChIP-seq and mRNA-seq.
table S2. A list of genes with differential H3K4me3 mark at promoters across PNeuN+ and CNeuN+ populations.
table S3. A list of genes with differential H3K27ac mark at promoters across PNeuN+ and CNeuN+ populations.
table S4. A list of genes with differential H3K27me3 mark at promoters across PNeuN+ and CNeuN+ populations.
table S5. A list of differentially expressed genes (fold change > 4; P < 0.05, t test) with correlated differential histone modification in H3K4me3 or H3K27me3 at promoters across PNeuN+ and CNeuN+ populations.