

Abstract
Pyramidal cells and astrocytes have differential susceptibility to oxygen-glucose deprivation and reperfusion (OGD-RP). It is known that excessive reactive oxygen species (ROS) in mitochondria initiates the cell death, while glutathione (GSH) is one of the major defenses against ROS. Although it is known that astrocytes contain a higher concentration of GSH than neurons, and that astrocytes can provide neurons with GSH, we are unaware of a detailed and quantitative examination of the dynamic changes in the mitochondrial GSH system in the two cell types during OGD-RP. Here, we determined mitochondrial membrane potential and the degrees of oxidation of the mitochondrially targeted roGFP-based sensors for hydrogen peroxide (OxDP) and GSH (OxDG). We also developed a method to estimate the mitochondrial GSH (mGSH) concentration in single cells in the CA1 region of organotypic hippocampal slice cultures at several time-points during OGD-RP. We find that mitochondrial membrane potential drops in pyramidal cells during OGD while it is relatively stable in astrocytes. In both types of cell, the mitochondrial membrane potential decreases during RP. During OGD-RP, mitochondrial peroxide levels are the same. Astrocytic mGSH is more than four times higher than in pyramidal cells (3.2 vs 0.7 mM). Astrocytic mGSH is drained from mitochondria during OGD, whereas in pyramidal cells it remains fairly constant. OxDGSH prior to and during OGD is lower (less oxidized) in pyramidal cells than astrocytes but the two nearly converge during RP. The larger changes of redox status in the GSH system in pyramidal cells than astrocytes is an upstream sign of the higher mortality of the pyramidal cells after facing an insult. The pattern of [mGSH] changes in the two cell types could be recognized as another mechanism by which astrocytes protect neurons from transient, extreme conditions.
Keywords: Mitochondrial GSH, mitochondrial H2O2, mitochondrial membrane potential, oxygen-glucose deprivation and reperfusion
TOC GRAPHIC
INTRODUCTION
An appropriate amount of ROS is necessary for signal transduction and the release of certain neurotransmitters.1–6 However, an excess of ROS can be harmful to neurons7–9, in stroke10–12, trauma13–15, and Alzheimer’s disease16–20. Glutathione (GSH) is an important antioxidant for removing ROS generated during oxidative metabolism in cells.21, 22 For instance, applying exogenous GSH helps to rescue neurons in primary cell culture from an ischemic insult by reducing the ROS generated.23 Interestingly, GSH in different cellular compartments has different influences on neuronal protection. Wüllner et al.24 observed that depletion of neuronal cytoplasmic GSH (cGSH) did not result in a ROS increase whereas depletion of neuronal mitochondrial GSH (mGSH) led to a significant increase in ROS and neuronal cell death in primary cultures24. Further, different cell types are known to have different GSH-mediated antioxidant capacities. Astrocytes are more resistant to OGD-RP (an in vitro ischemia model) than pyramidal cells in primary cultures.25 There is some evidence showing that the better survivability of astrocytes after OGD-RP is correlated with more efficient ROS removal by the GSH system. For example, the consumption of extracellular H2O2 is faster in astrocytes than in neurons in primary cultures26, which is ascribed to the fact that the intracellular GSH level is higher in astrocytes than in neurons in primary cultures27. Dringen et al.28–30 reported that astrocytes export GSH that cannot be directly used by neurons, and instead, GSH is hydrolyzed into GSH precursors before uptake by neurons for intracellular GSH regeneration. Such observations from primary neuron/astrocyte cultures and their co-cultures illustrate the importance of having an understanding the GSH systems in neuron and astrocytes independently. That is, cell-specific measurements provide insight that is not obtainable from whole-tissue measurements. This type of insight is quite important as Dringen et al.28–30 have shown that the GSH systems in neurons and astrocytes are interdependent. The foregoing results together provide a general understanding; however, they are based on one or a small number of time points and for the most part in cell cultures.
The goal of this work was to establish a finer-grained, more quantitative understanding of the changes that occur in astrocytes and pyramidal cells during OGD-RP31. We targeted the pyramidal cells in stratum pyramidale and protoplasmic astrocytes in stratum radiatum32 of organotypic hippocampal slice cultures33 (OHSCs). By using GFP-based probes for mitochondrial H2O2 and the GSH/GSSG couple - mito-roGFP2-Orp134 and mito-Grx1-roGFP235, respectively, the mitochondrial H2O2 and GSH redox status were measured in single astrocytes and neurons in OHSCs. We also monitored the mitochondrial membrane potential36 with the dye, TMRM. We demonstrate a new method to estimate mitochondrial and cytoplasmic GSH and GSSG concentrations at several points in time during OGD-RP37. A recently published, reversible, fluorogenic reagent for GSH holds promise for future studies38. We find that mitochondrial membrane potential drops in pyramidal cells during OGD while it is relatively stable in astrocytes. In both types of cell, the potential decreases during RP. During OGD and RP, mitochondrial peroxide levels are the same. Astrocytic [mGSH] is more than four times higher than pyramidal cells’ (3.2 vs 0.7 mM), but it decreases sigificantly during OGD, while that in pyramidal cells remains fairly constant. Basal OxDGSH and the one during OGD is lower (less oxidized) in pyramidal cells but the two nearly converge during RP.
RESULTS AND DISCUSSION
Real-time changes of the mitochondrial membrane potential during OGD-RP
OGD-RP induces changes in mitochondrial membrane potentials of hippocampal pyramidal cells and astrocytes (Figure 1). The dye, TMRM was applied at a low concentration, 10 µM, to insure that it functions in its non-quench mode in which a decrease in mitochondrial fluorescence intensity indicates mitochondrial membrane depolarization.39 The non-quench mode of TMRM benefits the recording of both fast and slow mitochondrial membrane potential (MMP) changes.40 We induce complete mitochondrial membrane depolarization by an uncoupler, FCCP, as shown in Figure 1. During OGD (20/30 min OGD-RP), pyramidal cells endure more steep and continuous mitochondrial membrane depolarization than astrocytes. During RP (20/30 min OGD-RP), depolarization continues in both cell types and reaches a similar extent at the end. A shorter 5-min, OGD period was also used with the same 30-min reperfusion (see Figure S1). The depolarization during the 5-min OGD is the same as during the first 5-min of the 20-min OGD as expected. However, mitochondrial membrane hyperpolarization follows during RP in pyramidal cells. Changes of the mitochondrial membrane potential are not apparent in astrocytes in this OGD-RP protocol.
Figure 1. Mitochondrial membrane potential performs differently between pyramidal cells and astrocytes during OGD-RP.

(a) Representative images of mitochondrial membrane potential (MMP) determinations in single cells (top, pyramidal cell; bottom, astrocyte) in OHSCs. From left to right: OHSC was stained with 10 nM TMRM solution, then imaged at Ex: 514 nm, Em: 555–585 nm; mitochondria of a single cell are visualized by GFP localized in mitochondria (Ex: 488 nm, Em: 500–530 nm); Mitochondria are outlined in yellow to create a contour; Overlay of the mito-contour of a single cell with the TMRM image. (b) Cells from OHSC CA1 region were recorded. The profiles of MMP, during 10/20/30 min Basal/OGD/RP are presented as mean ± SEM from six separate experiments. Positive control (basal condition) and negative control (50 µM FCCP treatment) are used for 100% of original MMP and 0% of original MMP, respectively. All images were acquired with 63× water-immersion lens with N.A. = 0.9. Refractive Index of OHSC is around 1.36.
Our observations in the OHSCs are supported by the work of Iijima et al. in primary hippocampal neuron cultures41. Specifically, hyperpolarization was observed during RP following a short OGD (30 min) while continuing depolarization in RP followed a longer-term (60 min) OGD depolarization. A model described by Sanderson et al.42 is also consistent with these observations. According to this model, mitochondrial injury evolves post-OGD in two ways. Mitochondrial membrane hyperpolarization and excessive ROS generation occur in hyperactive mitochondria that regain oxygen after a brief OGD; mitochondrial membrane depolarization and energy failure happen in dysfunctional mitochondria after a longer OGD exposure. Abramov and Duchen43 found that a ten-minute glutamate-induced Ca2+ increase and mitochondrial membrane depolarization can be rescued by scavenging mitochondrial Ca2+ and applying NADH- or ATP-generating substrates. The same treatment did not prevent mitochondrial membrane potential collapse after 20-min exposure to glutamate. The time frame of our OGD-RP experiment is similar to theirs. Like glutamate, OGD-RP triggers escalation of mitochondrial Ca2+44 and NADH shortage45. It is reasonable to suggest that the opposing trajectories of the mitochondrial membrane potential between 5 and 20 min OGD we found here can be ascribed to the existence of mitochondrial permeability transition pores at the longer time. In Figure 1 and S1, a general observation is that when mitochondrial membrane depolarization takes place, it changes at a smaller magnitude and slower speed in astrocytes than in pyramidal cells under our experimental conditions. Similar observations were reported by others working with brain cells (i.e. neuroblastoma cell cultures46, primary astrocyte cultures47, different regions in acute hippocampal slices48). The differences might be due to the uncoupling proteins (UCP) that favor mitochondrial membrane depolarization and reduce the ROS-induced injury.49, 50 The most abundant UCP isoforms 2, 4, and 5 are distributed differently in brain cells. UCP 4 and 5 are both expressed transcriptionally approximately twice as high in neurons as in astrocytes. Astrocytes have higher UCP2 (by less than two-fold) compared to pyramidal cells.49 It appears that UCP4 and 5 weigh heavier than UCP 2 in controlling the mitochondrial membrane potential.49, 50 The overall higher contents of UCPs in neurons can lead to a greater tendency towards mitochondrial membrane depolarization than in astrocytes. The protein inhibitor factor 1 (IF1) also can be considered to affect mitochondrial membrane potential.51 IF1 is an inhibitor of ATPase and facilitates mitochondrial membrane depolarization.51 The higher level of IF1 in neurons than in astrocytes may also contribute to the greater mitochondrial membrane potential susceptibility to OGD in neurons than astrocytes.51 In contrast, during reperfusion, astrocyte mitochondrial membrane potential decreases more rapidly than in neurons.
Real-time oxidation/reduction in mitochondria reflected by hydrogen peroxide- and glutathione-sensitive GFP probes during OGD-RP
We used redox-sensitive green fluorescent protein-based probes to investigate mitochondrial H2O2 levels and GSH system oxidation status. Probes based on roGFP have many merits such as photo-stability, being ratiometric, pH insensitivity, cellular compartment selectivity and a reversible response to redox change34, 52, 53 Grx1-roGFP2 (both mitochondrial and cytosolic versions) are unique probes for determining redox changes of the GSH/GSSG couple. They are capable of achieving a time resolution of about 10 s. Mito-roGFP2-Orp134 detects mitochondrial H2O2. It is preferred over MitoSox, the commercial small-molecule dye in common use, for many reasons. MitoSox is the mitochondrial targeted analog of hydroethidine (HEt), often used in the determination of ROS in brain cells47, 54, 55 But HEt lacks selectivity and is involved in non-catalyzed and enzymatic reactions with a broad series of reactive oxygen/nitrogen species. MitoSox’s fluorescence is influenced by many confounding factors, in fact, may be affected by processes other than ROS generation56–58 Importantly, HEt cannot record redox changes reversibly and can experience photo-bleaching and export from cells. Cell swelling can also lead to the misinterpretation of oxidative changes54, 55
When using these probes, the measured quantity is the oxidation degree which we will refer to as OxDP for the peroxide probe and OxDG for the GSH probe (see Supporting Information for more details)37. It is important to note that OxDP and OxDG do not directly show the absolute concentrations of H2O2 and GSH, respectively. Also, the roGFP-based sensor property, OxDG, does not equal the degree of oxidation of the GSH system, OxDGSH, but they are related (Figure S2). For example, when OxDG is 0.6, then OxDGSH is about 0.0002 or 0.002 when total [GSH] is 1 mM or 10 mM respectively. Nonetheless, OxDP and OxDG demonstrate degree of oxidation/reduction occurring inside cellular compartments as well as dynamic changes. A larger OxDP represents a higher H2O2 level. Similarly, a larger OxDG represents the GSH system in a more oxidized state whereas a lower OxDG indicates that the GSH system is in a less oxidized state.
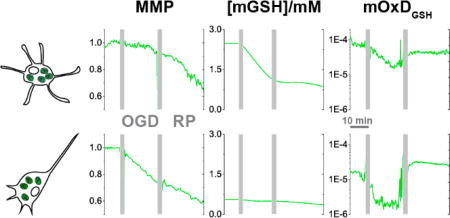
Figure 2 demonstrates that the pattern of changes of mitochondrial OxDP and OxDG are similar throughout OGD. OxDP and OxDG decrease during OGD, then increase during RP compared to their basal values. This phenomenon indicates a less oxidizing situation with lower mH2O2 and less oxidized mGSH during OGD and the opposite during RP. During OGD, ROS generation is limited due to lack of the precursor, O2. Low OxDG is observed primarily for the same reason. Also, the decreasing pH in the mitochondrial matrix accompanying mitochondrial membrane depolarization favors (thermodynamically) the reduction of GSSG by NADPH as well37. It is important to note that no time-dependent change of cytosolic OxDG was found during OGD-RP (see Figure S3). As discussed in our previous work37, the lack of GSH oxidation in cytosol is due to the paucity of cytosolic ROS during a short OGD59 and the abundant GSH in cytosol60.
Figure 2. Pyramidal and astrocyte mitochondrial H2O2 respond similarly and GSH systems respond differently to OGD-RP.

(a) Demonstration of the expression of tdTomato (Ex: 561 nm, Em: 580–600 nm) fluorescent protein in a pyramidal cell (top) and an astrocyte (bottom) from OHSCs (gene gun). (Left) bright field (BF) image of the OHSC and (middle) fluorescence image of tdTomato are taken with a 5× objective lens. The dotted line indicates approximately the Cornu Ammonis (CA). (Right) Enlarged images of single cells (indicated by arrows in the middle image) are taken with a the 63× objective lens (see Figure 1). Gold particles (see blue arrow) carrying plasmids were introduced to the cell by gene gun. (b) Representative images of a pyramidal cell (top) and an astrocyte (bottom) expressing mitochondrially-targeted fluorescent protein. (Left) overlay of BF image and fluorescence image of and Mito-Red (Ex: 561 nm, Em: 580–600 nm). (Middle/right) ratiometric images of the mitochondrial GSH probe (Mito-GP, Ex: 405/488 nm, Em: 500–530 nm) at basal, H2O2, and DTT treatment. (c–d) (Top) the OxD derived from the fluorescence measurements during OGD-RP. OHSCs were sequentially treated with 10 min basal/20 min OGD/30 min RP followed by H2O2 and DTT sequentially for calibration. Data are represented as mean ± SEM from six separate experiments (see Eq. S6 – S9 in Supporting Information). (Bottom) Comparisons of OxD values taken from the last five minutes at each condition (prior to the visible transients in the data trace above). Student’s t-test and one-way ANOVA were applied (no symbol if p > 0.05, *p < 0.05, **p < 0.01, ***p<0.001, n = 6 for each case). Symbols above the horizontal lines represent the comparisons between different cell types. Symbols in the legend below the bar chart represent the ANOVA comparisons among the OGD-RP periods.
Surprisingly, pyramidal cell and astrocyte mitochondrial OxDP are virtually indistinguishable (Figure 2c). Earlier, it was found that ROS is higher in pyramidal cells in stratum pyramidal than astrocytes in stratum radiatum under similar OGD-RP conditions32, 55. However, the measurement reported here is quite specific as it is confined to H2O2 only in mitochondria whereas the cited work made whole-cell measurements using a probe with less chemical specificity. Intriguingly, mitochondrial OxDG is different in pyramidal cells and astrocytes (Figure 2d). Pyramidal cells have more extreme changes in reduction/oxidation of the mGSH system during OGD-RP compared to astrocytes. Because there are similar mH2O2 changes in the two types of cells, we hypothesize that the differences in OxDG can be attributed to differences in the mGSH systems. This observation led us to try to determine how the concentration of GSH and the oxidation degree of GSH change in mitochondria of astrocytes and pyramidal cells in the organotypic tissue cultures over the course of OGD-RP.
Quantitative measurement of mitochondrial GSH concentration and its changes during OGD-RP
Determining the relationship of OxDG and OxDGSH35 in mitochondria. quantitatively requires knowing the mGSH concentration. Ideally, we could determine the basal mGSH concentration in the different cell types, and monitor the changes of mGSH during OGD-RP. Dissociating and separating the cells from cultures could provide a route to cell-specific measurements. However, the internal GSH concentration could be altered during the process.30 There is a similar problem when isolating mitochondria from cells.61 Also, because of the intense communication between astrocytes and neurons62, we cannot expect that measurements on separated cells reflect the status in intact tissue.
Thus, we developed a cell-specific mGSH concentration determination built on several measurements, and observations from the literature (see Figure 3, Table 1, and SI): 1) the total GSH concentration, [GSH] + 2[GSSG], in OHSCs was measured in extracts of tissue cultures by using an enzyme-based colorimetric method.63, 64 2) Separately, intact tissue cultures were exposed to a fluorogenic, thiol-specific reagent, Thiol Probe IV. As the major thiol is GSH and the majority of GSH exists in its reduced form (a statement that we will confirm below based on experimental evidence), the relative fluorescence intensities, I, from the two cell types indicate approximately their relative total GSH concentrations. Protein thiols will contribute to fluorescence, but these should not contribute significantly65 despite their presence.66 From these fluorescence measurements, we obtained FA/P, the astrocyte-to-pyramidal cell ratio of the fluorescence from the GSH adducts. We take this to be the ratio of the total GSH concentration in astrocytes to that in pyramidal cells. 3) After exposing cultures to a mitochondrially-directed fluorescent protein and the thiol-specific reagent; we create two-color images, then use the fluorescent protein’s fluorescence to create a mitochondrial mask for individual cells. This mask delineates the region containing fluorescently labeled mitochondrial thiols. An analogous procedure using a mask based on tdTomato to delineate single cells provides the region containing fluorescently labeled thiols in the whole cell. Thus, we determine the relative concentration of GSH in mitochondria vs. the whole cell in each cell type (see the derivation and discussion in the SI). We discuss separately below an analysis of the magnitude of the error induced by making certain assumptions.
Figure 3. Procedures to obtain the total mitochondrial GSH concentration of pyramidal cells or astrocytes in an intact OHSC.

(1) The average total concentration of GSH from OHSC homogenate is measured with Ellman’s reagent following Rahman’s protocol64. (2) The GSH (and other thiols) reacts with thiol-specific fluorogenic reagent superfused over cultures rapidly forming the fluorescent adduct. FA/P, the ratio of total GSH concentration of astrocytes over pyramidal cells, equals the ratio of their respective fluorescence intensities, I. (3) Mitochondrial and whole-cell masks are created with mitochondria and cytosol targeting fluorescent proteins. Fluorescence intensity from the GSH adduct is measured within each masked region separately. FM/C, the ratio of GSH concentration in mitochondria to that of the whole cell, equals the ratio of I from mitochondria to that of the whole cell.
Table 1.
Summary of parameters measured in Figure 3
| Measurement | Method | Notation |
|---|---|---|
| [GSH] + 2[GSSG] total | Enzymatic on tissue homogenate | Total [GSH] |
| Free thiol fluorescence ratio in astrocytes to pyramidal cells in OHSC | Thiol Probe IV and fluorescence microscopy of single cells in OHSCs | FA/P |
| Free thiol fluorescence ratio in mitochondria to whole cells in OHSC for each cell type | Thiol Probe IV/mitochondrial mask with fluorescence microscopy of single cells in OHSCs | FM/C |
The next step is to estimate the concentrations in each cell type. Following Rice and Russo-Menna67, given the average tissue culture concentration from the first measurement and the ratio of the fluorescence intensities from the fluorescent thiol adducts from the second measurement, we can obtain the GSH concentration in each cell type. In an analogous fashion, the mitochondrial-to-cytosol ratio of GSH is estimated by the relative intensities inside the mitochondrial mask and the whole cell mask; then the mitochondrial concentrations in each cell type can be deduced from the whole-cell concentrations of the respective cell types. In the foregoing, we have assumed that the fluorescence measurements of reduced GSH adequately represents the total GSH. In many circumstances, the reduced GSH and total GSH concentrations are very similar. Considering that the range of our experimental OxDG is between 0 to 0.6 and the GSH concentration is about 1 mM, the portion of the total attributable to reduced GSH is greater than 99.99% indicating that reduced GSH is an adequate surrogate for the total GSH (see Figure S2).
There are two potential sources of error that affect the accuracy of the total mitochondrial GSH concentration in each cell type. One is at the step to obtain GSH concentration (Figure 3-2, Figure S4, Eq. S2 and S3) where the ratio of the volume of astrocytes to the volume of pyramidal cells, VA/P, and the fraction of the extracellular space to the whole culture volume (its porosity), fEC, are used. We have used values of 1.0 and 0.22, respectively, here. According to the literature, fEC can vary between 0.12 and 0.468–70 but is 0.21–0.22 in CA1 in vivo.71, 72. Rice and Russo-Menna67 found that VA/P is about 2 for postnatal day 3 (p3) rats and 0.31 for adult rats (> p63). Our animals are sacrificed at age p7 and used after culturing for 5 – 7 days. VA/P is the product of two ratios for the two cell types: the ratio of single cell volumes and the ratio of cell numbers. By setting each ratio to 1 and letting each ratio vary up or down by a factor of 1.5, values of VA/P in a range of 0.44 to 2.25 were considered for the error analysis. This range of VA/P overlaps the reported range by Rice and Russo-Menna67 for our age range. Importantly, estimates for mGSH in astrocytes and pyramidal cells both depend to the same degree on the numerical values of these two parameters. Thus, the relative concentrations of mGSH in the two cell types is measured fairly robustly.
The other potential contribution to error is the selection of the mitochondrial mask. This process is subjective. We determined the best mask-defining conditions for mitochondria based on the observations made by changing the low threshold of the mitochondrially directed GFP-labeled image as shown in Figure S5. There are no abrupt changes of the GFP intensity (Figure S5b) or area magnitude (Figure S5c) that could serve as an indicator of a proper choice of mitochondrial mask. Fortunately, the average intensity of the fluorescent adducts of thiols from the selection does not vary much as the low threshold changes. In the example (Figure S5), the average intensities of GSH from different selections vary merely −1% to 2% when the low threshold of GFP-labeled mitochondrial changes by ± 30% from our chosen low threshold. With the same manipulations, the GFP intensities change by ± 20% and the mask areas change from −43% to 81% compared to the chosen threshold. Thus, the choice of threshold does not have a significant impact on the outcome.
Figure 4 shows results obtained by following the protocol in Figure 3 at five time-points spanning the 10/20/30 min basal/ OGD-RP conditions. Figure 4a, shows that [GSH] of OHSC decreases continuously in OHSCs with the major change occurring during OGD. Our results indicate a net GSH efflux from the OHSCs. Similar observations were also reported in previous work73. Figure 4b–c shows that the ratio of total GSH concentration in astrocytes to pyramidal cells falls during OGD-RP from about four in the basal condition to about two during reperfusion. Thus, while the whole cell concentration stays higher in astrocytes than pyramidal cells, the gap between the two cell types decreases dramatically during OGD and stabilizes during RP. It is noteworthy that the pyramidal cell [GSH] is relatively stable during OGD-RP. In both cell types, the mitochondria-to-cytosol ratio of [GSH] decreases subtly but with statistical significance (Figure 4f) during OGD-RP. Finally, the steep decrease in astrocytic [mGSH] and relative constancy in pyramidal cell [mGSH] seen in Figure 4g reflect the same pattern as seen in the whole cell. Quantitatively, basal [mGSH] for pyramidal cells is approximately 0.7 mM whereas for astrocytes it is approximately 3.2 mM. However, [mGSH] in astrocytes drops after OGD-RP but is still higher than in pyramidal cells.
Figure 4. Mitochondrial GSH concentration changes significantly in astrocytes but not in pyramidal cells in OHSCs during OGD-RP.

(a) The volumetric concentration of total GSH (mM) in OHSCs was sampled at five time points of 10/20/30 min basal/OGD/RP. Data are shown as mean ± SEM, n = 9 repeats for each case. Data in (c), (f), and (g) are acquired at the same time points as (a). (b–g) GSH in cells is labeled with the membrane-permeable, maleimide-based, fluorogenic Thiol probe IV (Ex: 405, Em: 450–480 nm). (b) OHSC exposed to 100 µM thiol probe IV for 8 min before imaging. (left) Nine pyramidal cells are outlined in white dash lines, while nine astrocytes are outlined in yellow solid lines. (right) The distribution of fluorescence intensity is shown in a false color pattern referencing to the color bar. The nine pyramidal cells and nine astrocytes are noted with (Δ) and (*), respectively. In (c, f, and g), the OGD period is indicated by a cyan box, and the RP period is right after it. (c) (left) The fluorescence intensity ratio of labelled GSH in astrocytes over pyramidal cells (FA/P) changes during OGD-RP. Data are shown as mean ± SEM (n = 38 cells for each case) with t test results (vs. the first data point (basal), p*** < 0.001, no label shown when p>0.05). (right) the GSH concentration in the two cell types. The GSH level is significantly higher in astrocytes than pyramidal cells. (d) Examples of a pyramidal cell in OHSC. Mitochondria (top) and whole cell (bottom) are visualized by Mito-GFP (Ex: 488, Em: 500–530 nm) and tdTomato (Ex: 561, Em: 580–600 nm), respectively. Column headings from left to right, FL: fluorescence image of fluorescent protein; Contour: the contour of the ratio of fluorescence from mitochondria/whole cell; Thiol Stain: overlay of the contour with the OHSC image (Before) and (After) GSH-staining. (e) Examples of an astrocyte are displayed in a similar way as in (d). (f) The fluorescence intensity ratios of labelled GSH in mitochondria over whole cell (FM/C) were record, and represented as mean ± SEM (n = 19 cells for each case). Each data point excluding the first one is compared with the first data point at the basal (**p<0.01, *p<0.05, no label shown when p>0.05) via t test. Red and black are used for pyramidal cells and astrocytes, respectively. (g) Volumetric concentrations of mitochondrial GSH are calculated from data in (a), (c), and (f) and shown as mean ± SEM (n = 9 repeats for each case). All images were acquired with 63× water-immersion lens with N.A. =0.9. Refractive Index of OHSC is around 1.36.
The degree of oxidation of mitochondrial GSH during OGD-RP
With an estimate of [mGSH], we can deduce the fraction of oxidized GSH out of the total concentration of GSH (Eq. S10). In principle, this fraction could be determined from measurements of the concentrations of GSH and GSSG.This is a daunting task when [GSSG] is small compared to [GSH] even under well-controlled, in vitro conditions, and more difficult with a tissue culture preparation. In addition, OxDGSH is sensitive to small (µM) changes in GSSG, but the same change in GSH hardly changes OxDGSH at all because [GSSG] is typically very small compared to [GSH]. We attempted to circumvent the direct measurement of [GSSG], instead deriving OxDGSH from OxDG and [GSH]35.
The measurements of OxDG have a higher data density than the measurements of [mGSH]. In order to deduce mitochondrial OxDGSH over the whole time course of the OGD-RP experiment we must estimate [mGSH] from the experimental data at times between the measured points. [mGSH] is well-approximated by simple linear fits (see Figure S6). Mitochondrial OxDGSH values derived from these linear fits and OxDG are shown in Figure 5. We note that basal mitochondrial OxDGSH is higher in astrocytes (~1×10−4) than in pyramidal cells (~2×10−5) despite the fact that basal mitochondrial OxDG is similar in the two cell types. The difference relates to the fact that the probe’s OxDG depends only on its ratio of oxidized and reduced forms while the mitochondrial OxDGSH depends on that ratio and also the concentration of GSH35, 53. (For clarification, see Figure S2 which shows that OxDGSH increases when [GSH] increases at constant OxDG.) Analogously, astrocytes have higher basal mitochondrial OxDGSH and [mGSH] than pyramidal cells. OxDGSH during OGD is ten-fold lower in pyramidal cell mitochondria than astrocytic mitochondria. Interestingly, mitochondrial OxDGSH during RP becomes similar in pyramidal cells and astrocytes. This is largely driven by the approach of [mGSH] in the two cell types. The decrease in the difference of total GSH concentration and oxidation degree of GSH after OGD-RP reflects the observation that astrocytes are a source of GSH for neurons.62
Figure 5. In mitochondria, the oxidation degree of glutathione (OxDGSH) of the two cell types converge during RP.

The OxDGSH data was derived from the OxDG data in Figure 2d (top) and the mitochondrial GSH concentration equations in Figure S-6. The mitochondrial GSH concentration equations were fitted from the mitochondrial GSH concentration data in Figure 4g. Eq. S10 is used in this calculation. All other related details are in Supporting Information. All data are represented as mean ± SEM, n = 6 repeats.
We recently reported that CA1/CA3 differences in the mitochondrial thioredoxin system’s activity during OGD-RP is a significant factor in reducing neuronal injury seen 18 hours later.45 Here, we have focused on changes in the GSH system during OGD-RP and in particular trying to understand quantitatively the GSH system’s status in mitochondria (the source of ROS at this short timescale of OGD59) of both astrocytes and neurons in the OHSC. The OHSC is a well-documented31 preparation that permits investigations that would be impossible or complex in vivo while maintaining the observed cells in a somewhat natural environment. Astrocytes assist neurons to survive under anoxic and hypoglycemic stress. They provide neurons with energy substrates such as lactate.74 In contrast to neurons, oxidative stress is managed in astrocytes by having stable Nrf2 levels that promote antioxidant genes75, 76. The relatively high astrocytic GSH concentration is ascribed to the effect of Nrf2 which enables astrocytes to release GSH for the de novo synthesis of GSH in neurons.75 GSH can be oxidized enzymatically and non-enzymatically by ROS such as hydrogen peroxide.77 We saw no difference in hydrogen peroxide during OGD-RP. At this time scale, differences in GSH oxidation status between pyramidal cells and astrocytes are due to the differences in GSH concentration - a larger GSH pool is accompanied by a smaller change in the degree of oxidation of GSH itself.
Our observations are consistent with the well-established fact that astrocytes are more resilient to OGD-RP than pyramidal cells77. Their stability in the face of the OGD insult is reflected in the slower decrease in mitochondrial membrane potential seen here during OGD. Pyramidal cell mitochondrial membranes depolarize faster during OGD than those of astrocytes. However, this may also reflect the fact that uncoupling proteins can act to depolarize the mitochondrial inner membrane in neurons as a protective mechanism.50 The larger changes of redox status in the GSH system in pyramidal cells than astrocytes is an upstream sign of the higher mortality of the pyramidal cells after facing an insult. The pattern of [mGSH] changes in the two cell types could be recognized as another mechanism by which astrocytes protect neurons from transient, extreme conditions.
In the course of OGD-RP, [GSH] in both cytoplasm and mitochondria of astrocytes remain higher than that in neurons. As mentioned above, higher Nrf2 and enzymes related to GSH synthesis in astrocytes contribute to higher astrocytic [GSH]76, but astrocytes are also enriched in other enzymes related to redox control including glutathione peroxidase and glutathione reductase.78 Along with the higher astrocyte [GSH] compared to pyramidal cells, these differences in enzyme activity may account for the less extreme changes in the OxDs seen in the two cell types during OGD-RP.
It is intriguing and unexpected to find that astrocytic mitochondria lose GSH much faster than neuronal mitochondria during OGD. The important role of astrocytes in providing the components of GSH to neurons through GSH release, extracellular hydrolysis and neuronal uptake is well established.29, 62, 79, 80 Our data show that the pattern of the change of [GSH] in mitochondria is very similar to that in cytoplasm both for neurons and astrocytes. GSH is not produced in mitochondria, but imported from cytoplasm instead,81, 82 although not as previously assumed by the mitochondrial dicarboxylate and 2-oxoglutarate carriers.83 Thus, in the case of OGD, astrocytes’ cytoplasmic GSH is apparently exported in preference to being transported to mitochondria. This export of cytoplasmic GSH from astrocytes weakens the support of their own mitochondrial GSH pool. Our observation is consistent with the carrier of GSH in the inner mitochondrial membrane being reversible. Related to this is the observation that, while astrocytes maintain mitochondrial membrane potential early in the OGD-RP protocol better than pyramidal cells, at the end of the 50-min protocol, mitochondrial membrane potentials are similar in the two cell types, and low. Further work may reveal that astrocytic support for neurons wanes during more extended OGD insults.
The protocol described here makes the real-time tracking of the mGSH system’s redox changes accessible. In conjunction with measurements of mitochondrial membrane potential, we have obtained a more quantitative picture of the events in these cells during OGD-RP. We find the mitochondria in pyramidal cells are more sensitive to OGD-RP than in astrocytes as indicated by mitochondrial membrane potential. OxDP is remarkably similar in the mitochondria of pyramidal cells and astrocytes. It is tempting to suggest that this reflects the facile permeation of hydrogen peroxide across membranes in conjunction with the proximity of the cells to each other. However, we do not see a rise in cytosolic hydrogen peroxide during the same time period. This observation is not consistent with the idea that peroxide’s facile diffusion is the cause of the observed similarity. On the other hand, the oxidation status of the two cell type’s mitochondria as reflected by the OxDGSH are quite different. The astrocytic mGSH system is always more oxidized than that of pyramidal cells, while astrocytic [mGSH] changes more dramatically during OGD than pyramidal cell [mGSH]. These patterns reveal more insight about the chemical events involved in OGD-RP. We anticipate that the protocols presented here can be applied to other studies that require thorough understanding of the redox changes in the mGSH system.
METHODS
Imaging, data processing and statistical analysis
We used a Leica TCS SP5II broadband confocal microscope equipped with an HCX PL FLUOTAR 5× objective lens with N.A. = 0.15 and an HCX APO-L U-V-I water immersion 63× objective lens with N.A. = 0.90. The z-direction shift was minimized by the “autofocus” function of this microscope during imaging. Rapid sequential excitations were applied when more than one source of fluorescence was imaged. Images were acquired approximately one frame per 10 s and processed by ImageJ (http://imagej.nih.gov/ij/). Slices in the image series were realigned to remove the x, y-plane shift of the target of interest by using the plug-in “Template Matching” in ImageJ84, then the fluorescence intensities were extracted. Numerical data were processed in Matlab (version R2015b, MathWorks, Inc.) and OrginPro (version 2015, OriginLab Corp.). Statistical analysis was done in Matlab (version R2015b, MathWorks, Inc.) and R (www.r-project.org).
Measurement of the mitochondrial membrane potential
OHSCs were incubated with 10 nM TMRM (ThermoFisher, Ex: 514 nm, Em: 555–585 nm) in HBSS solution for 45 min at 37°C before imaging36. The mitochondrial mask was created by labeling the mitochondria with GFP (Ex: 488 nm, Em: 500–530 nm, Table S1). The intensity within the mitochondrial mask of single cells was recorded (n = 6 cultures, Figure 1a). OHSCs were treated with OGD-RP conditions followed by 20 min 50 µM FCCP (Sigma-Aldrich), a mitochondrial uncoupler36. The fluorescence intensity of TMRM was normalized to its initial value and the value after the FCCP treatment.
Measurements of the H2O2 and GSH systems
The roGFP2 based probes for hydrogen peroxide and GSH (Ex: 405/488 nm, Em: 500–530 nm) were expressed in OHSCs following insertion into cells with gene gun85. Mito-Red (Ex: 561 nm, Em: 580–600 nm) was introduced as an internal standard for the probes37. OHSCs were exposed to the OGD-RP conditions then they were exposed to H2O2 and DTT for calibration. More information is in Supporting Information.
Measurement of the GSH concentration of cells in tissue culture
The total concentration of GSH in the tissue culture extract was measured with Ellman’s reagent by following Rahman’s protocol.64 Proteins are precipitated in this method, minimizing interference from protein thiols. The concentration of total protein of the OHSC homogenate was measured by using the Pierce BCA protein assay kit (Thermo Scientific, USA) following the manufacturer’s instructions89. Four OHSCs were lysed in 1 mL potassium phosphate-EDTA buffer64 for the GSH measurement, while four others were lysed in 1 mL RIPA buffer (Cell Signaling Technology) for the total protein analysis. To achieve an effective extraction, the lysed samples were sonicated while on ice with ten rounds of pulses (12 s pulse on and 20 s pulse off) at 10% power with a 550 Sonic Dismembrator (Fisher Scientific). Then samples were centrifuged at 4500g for 10 min to obtain the supernatant. The GSH concentration was first calculated in the units of nmol/mg protein, then converted to the volumetric concentration in the unit of mM. More details are described in Supporting Information.
To compare the GSH levels between different cell types in OHSC and between mitochondria and cytosol in one cell without isolating the cells and cellular organelles, GSH was measured via fluorescence imaging (Exi/Em: 405 nmol/450–480 nm) after reaction with Thiol Probe IV (EMD Millipore)86. OHSCs were stained with 100 µM Thiol Probe IV in HBSS solution for 5 min. The thiol-stained pyramidal cells and astrocytes were distinguished based on their difference in fluorescence intensity and morphology. Mito-GFP and tdTomato were used to visualize the mitochondria and cytosol respectively and to create masks for defining an ROI and thus the GSH-dependent fluorescence in a particular ROI.
Supplementary Material
Table 2.
Summary of relationships of the terms used in error analysis
| Variables affecting derived results |
Definition | Application |
|---|---|---|
| fEC, fP, fA | Volume fractions of extracellular space, pyramidal cells, and astrocytes resp. ; fEC + fP + fA = 1 | Determination of [GSH] in pyramidal cells and astrocytes (Eqs. S1 – S3) |
| FA/P, FM/C | Ratios of fluorescence intensity of astrocytes to pyramidal cells (A/P) and mitochondria to whole cell (M/C) following exposure to Thiol Probe IV | Determination of [GSH] in pyramidal cells and astrocytes (Eqs. S1 – S3). Determination of [mGSH] in each cell type (Eq. S4) |
Acknowledgments
Funding Sources
NIH funding: Grants R01 GM066018 and R01 GM044842
We thank Jihe Liu (University of Pittsburgh) who made the plasmid for coding mito-tdTomato. Tom Harper (University of Pittsburgh) provided technical support for imaging on the confocal microscope.
ABBREVIATIONS
- cGSH
cytoplasmic GSH
- GSH
glutathione
- IF1
inhibitor factor 1
- mGSH
mitochondrial GSH
- OGD-RP
oxygen-glucose deprivation and reperfusion
- OHSC
organotypic hippocampal slice cultures
- OxD
oxidation degree of the probe
- OxDG
oxidation degree of the GSH probe
- OxDGSH
oxidation degree of the GSH/GSSG couple
- mPTP
mitochondrial permeability transition pore
- UCP
uncoupling protein
Footnotes
ASSOCIATED CONTENT
Supplemental methods contain tissue preparation and plasmid transfection, the OGD-RP experiment, derivation of the mitochondrial GSH concentration in pyramidal cells and astrocytes in OHSCs, the oxidation degree of the GFP based probes, and determination of OxDGSH for the GSH system. Six Supplemental Figures are also included. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
B.Y and S.G.W. designed the experiments. B.Y. conducted the experiments and collected the data. B.Y., G. B., and S.G.W. carried out data analysis, interpretation and construction of the manuscript.
Conflict of Interest
The authors claim no competing financial interest.
References
- 1.Finkel T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel JC, Rice ME. Classification of H2O2 as a neuromodulator that regulates striatal dopamine release on a subsecond time scale. ACS Chem. Neurosci. 2012;3:991–1001. doi: 10.1021/cn300130b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rice ME. H2O2: a dynamic neuromodulator. Neuroscientist. 2011;17:389–406. doi: 10.1177/1073858411404531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bao L, Avshalumov MV, Patel JC, Lee CR, Miller EW, Chang CJ, Rice ME. Mitochondria are the source of hydrogen peroxide for dynamic brain-cell signaling. J. Neurosci. 2009;29:9002–9010. doi: 10.1523/JNEUROSCI.1706-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waypa GB, Marks JD, Guzy RD, Mungai PT, Schriewer JM, Dokic D, Ball MK, Schumacker PT. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am. J. Respir. Crit. Care Med. 2013;187:424–432. doi: 10.1164/rccm.201207-1294OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK, Schumacker PT. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol. Cell. Biol. 2011;31:3531–3545. doi: 10.1128/MCB.05124-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herdegen T, Leah JD. Inducible and constitutive transcription factors in the mammalian nervous system: control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res. Rev. 1998;28:370–490. doi: 10.1016/s0165-0173(98)00018-6. [DOI] [PubMed] [Google Scholar]
- 8.Cheng Y, Deshmukh M, D'Costa A, Demaro JA, Gidday JM, Shah A, Sun Y, Jacquin MF, Johnson EM, Jr, Holtzman DM. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. J. Clin. Invest. 1998;101:1992–1999. doi: 10.1172/JCI2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banasiak KJ, Xia Y, Haddad GG. Mechanisms underlying hypoxia-induced neuronal apoptosis. Prog. Neurobiol. (Oxford) 2000;62:215–249. doi: 10.1016/s0301-0082(00)00011-3. [DOI] [PubMed] [Google Scholar]
- 10.McCann SK, Roulston CL. NADPH oxidase as a therapeutic target for neuroprotection against ischaemic stroke: future perspectives. Brain Sci. 2013;3:561–598. doi: 10.3390/brainsci3020561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim JY, Kawabori M, Yenari MA. Innate inflammatory responses in stroke: mechanisms and potential therapeutic targets. Curr. Med. Chem. 2014;21:2076–2097. doi: 10.2174/0929867321666131228205146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duval D, Foll ID-L, Vimard F, Gauberti M. Neuroprotective effects of N-acetylcysteine: a review. Brain Res. J. 2013;6:309–337. [Google Scholar]
- 13.Cornelius C, Crupi R, Calabrese V, Graziano A, Milone P, Pennisi G, Radak Z, Calabrese EJ, Cuzzocrea S. Traumatic brain injury: oxidative stress and neuroprotection. Antioxid. Redox Signaling. 2013;19:836–853. doi: 10.1089/ars.2012.4981. [DOI] [PubMed] [Google Scholar]
- 14.Rodriguez-Rodriguez A, Egea-Guerrero JJ, Murillo-Cabezas F, Carrillo-Vico A. Oxidative stress in traumatic brain injury. Curr. Med. Chem. 2014;21:1201–1211. doi: 10.2174/0929867321666131217153310. [DOI] [PubMed] [Google Scholar]
- 15.Abdul-Muneer PM, Chandra N, Haorah J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol. Neurobiol. 2014;51:966–979. doi: 10.1007/s12035-014-8752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lasierra-Cirujeda J, Coronel P, Aza MJ, Gimeno M. Beta-amyloidolysis and glutathione in Alzheimer's disease. J. Blood Med. 2013;4:31–38. doi: 10.2147/JBM.S35496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aliev G, Priyadarshini M, Reddy VP, Grieg NH, Kaminsky Y, Cacabelos R, Ashraf GM, Jabir NR, Kamal MA, Nikolenko VN, Jr, A AZ, Benberin VV, Bachurin SO. Oxidative stress mediated mitochondrial and vascular lesions as markers in the pathogenesis of Alzheimer disease. Curr. Med. Chem. 2014;21:2208–2217. doi: 10.2174/0929867321666131227161303. [DOI] [PubMed] [Google Scholar]
- 18.Butterfield DA, Di Domenico F, Barone E. Elevated risk of type 2 diabetes for development of Alzheimer disease: a key role for oxidative stress in brain. Biochim. Biophys. Acta, Mol. Basis Dis. 2014;1842:1693–1706. doi: 10.1016/j.bbadis.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schapira AHV. Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol. 2008;7:97–109. doi: 10.1016/S1474-4422(07)70327-7. [DOI] [PubMed] [Google Scholar]
- 20.Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700. doi: 10.1038/nature09536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radical Biol. Med. 2010;48:749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu G, Fang Y-Z, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J. Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 23.Furuichi T, Liu W, Shi H, Miyake M, Liu KJ. Generation of hydrogen peroxide during brief oxygen-glucose deprivation induces preconditioning neuronal protection in primary cultured neurons. J. Neurosci. Res. 2005;79:816–824. doi: 10.1002/jnr.20402. [DOI] [PubMed] [Google Scholar]
- 24.Wüllner U, Seyfried J, Groscurth P, Beinroth S, Winter S, Gleichmann M, Heneka M, Löschmann PA, Schulz JB, Weller M, Klockgether T. Glutathione depletion and neuronal cell death: the role of reactive oxygen intermediates and mitochondrial function. Brain Res. 1999;826:53–62. doi: 10.1016/s0006-8993(99)01228-7. [DOI] [PubMed] [Google Scholar]
- 25.Almeida A, Delgado-Esteban M, Bolaños JP, Medina JM. Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J. Neurochem. 2002;81:207–217. doi: 10.1046/j.1471-4159.2002.00827.x. [DOI] [PubMed] [Google Scholar]
- 26.Dringen R, Kussmaul L, Gutterer JM, Hirrlinger J, Hamprecht B. The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. J. Neurochem. 1999;72:2523–2530. doi: 10.1046/j.1471-4159.1999.0722523.x. [DOI] [PubMed] [Google Scholar]
- 27.Dringen R, Pawlowski PG, Hirrlinger J. Peroxide detoxification by brain cells. J. Neurosci. Res. 2005;79:157–165. doi: 10.1002/jnr.20280. [DOI] [PubMed] [Google Scholar]
- 28.Dringen R, Kranich O, Hamprecht B. The gamma-glutamyl transpeptidase inhibitor acivicin preserves glutathione released by astroglial cells in culture. Neurochem. Res. 1997;22:727–733. doi: 10.1023/a:1027310328310. [DOI] [PubMed] [Google Scholar]
- 29.Dringen R. Metabolism and functions of glutathione in brain. Prog. Neurobiol. 2000;62:649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- 30.Dringen R, Pfeiffer B, Hamprecht B. Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 1999;19:562–569. doi: 10.1523/JNEUROSCI.19-02-00562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahlgren H, Henjum K, Ottersen O, Rundén-Pran E. Validation of organotypical hippocampal slice cultures as an ex vivo model of brain ischemia: different roles of NMDA receptors in cell death signalling after exposure to NMDA or oxygen and glucose deprivation. Cell Tissue Res. 2011;345:329–341. doi: 10.1007/s00441-011-1218-2. [DOI] [PubMed] [Google Scholar]
- 32.Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic Astrocytes in CA1 Stratum Radiatum Occupy Separate Anatomical Domains. The Journal of Neuroscience. 2002;22:183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gogolla N, Galimberti I, DePaola V, Caroni P. Preparation of organotypic hippocampal slice cultures for long-term live imaging. Nat. Protocols. 2006;1:1165–1171. doi: 10.1038/nprot.2006.168. [DOI] [PubMed] [Google Scholar]
- 34.Gutscher M, Sobotta MC, Wabnitz GH, Ballikaya S, Meyer AJ, Samstag Y, Dick TP. Proximity-based Protein Thiol Oxidation by H2O2-scavenging Peroxidases. J. Biol. Chem. 2009;284:31532–31540. doi: 10.1074/jbc.M109.059246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gutscher M, Pauleau A-L, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, Dick TP. Real-time imaging of the intracellular glutathione redox potential. Nat. Methods. 2008;5:553–559. doi: 10.1038/nmeth.1212. [DOI] [PubMed] [Google Scholar]
- 36.Joshi DC, Bakowska JC. Determination of mitochondrial membrane potential and reactive oxygen species in live rat cortical neurons. J Vis. Exp. 2011:e2704. doi: 10.3791/2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yin B, Barrionuevo G, Weber SG. Optimized real-time monitoring of glutathione redox status in single pyramidal neurons in organotypic hippocampal slices during oxygen–glucose deprivation and reperfusion. ACS Chem. Neurosci. 2015;6:1838–1848. doi: 10.1021/acschemneuro.5b00186. [DOI] [PubMed] [Google Scholar]
- 38.Jiang X, Chen J, Bajić A, Zhang C, Song X, Carroll SL, Cai Z-L, Tang M, Xue M, Cheng N, Schaaf CP, Li F, MacKenzie KR, Ferreon ACM, Xia F, Wang MC, Maletić-Savatić M, Wang J. Quantitative real-time imaging of glutathione. 2017;8:16087. doi: 10.1038/ncomms16087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perry SW, Norman JP, Barbieri J, Brown EB, Gelbard HA. Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. BioTechniques. 2011;50:98–115. doi: 10.2144/000113610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nicholls DG. Simultaneous Monitoring of Ionophore- and Inhibitor-mediated Plasma and Mitochondrial Membrane Potential Changes in Cultured Neurons. J. Biol. Chem. 2006;281:14864–14874. doi: 10.1074/jbc.M510916200. [DOI] [PubMed] [Google Scholar]
- 41.Iijima T, Mishima T, Akagawa K, Iwao Y. Mitochondrial hyperpolarization after transient oxygen-glucose deprivation and subsequent apoptosis in cultured rat hippocampal neurons. Brain Res. 2003;993:140–145. doi: 10.1016/j.brainres.2003.09.041. [DOI] [PubMed] [Google Scholar]
- 42.Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Hüttemann M. Molecular Mechanisms of Ischemia–Reperfusion Injury in Brain: Pivotal Role of the Mitochondrial Membrane Potential in Reactive Oxygen Species Generation. Mol. Neurobiol. 2013;47:9–23. doi: 10.1007/s12035-012-8344-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abramov AY, Duchen MR. Mechanisms underlying the loss of mitochondrial membrane potential in glutamate excitotoxicity. Biochim. Biophys. Acta. 2008;1777:953–964. doi: 10.1016/j.bbabio.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 44.Schäfer MKE, Pfeiffer A, Jaeckel M, Pouya A, Dolga AM, Methner A. Regulators of mitochondrial Ca2+ homeostasis in cerebral ischemia. Cell Tissue Res. 2014;357:395–405. doi: 10.1007/s00441-014-1807-y. [DOI] [PubMed] [Google Scholar]
- 45.Yin B, Barrionuevo G, Batinic-Haberle I, Sandberg M, Weber SG. Differences in Reperfusion-Induced Mitochondrial Oxidative Stress and Cell Death Between Hippocampal CA1 and CA3 Subfields Are Due to the Mitochondrial Thioredoxin System. Antioxidants & Redox Signaling. 2017;27:534–549. doi: 10.1089/ars.2016.6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Agudo-López A, Miguel BG, Fernández I, Martínez AM. Involvement of mitochondria on neuroprotective effect of sphingosine-1-phosphate in cell death in an in vitro model of brain ischemia. Neurosci. Lett. 2010;470:130–133. doi: 10.1016/j.neulet.2009.12.070. [DOI] [PubMed] [Google Scholar]
- 47.Ouyang Y-B, Voloboueva LA, Xu L-J, Giffard RG. Selective Dysfunction of Hippocampal CA1 Astrocytes Contributes to Delayed Neuronal Damage after Transient Forebrain Ischemia. The Journal of Neuroscience. 2007;27:4253–4260. doi: 10.1523/JNEUROSCI.0211-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bahar S, Fayuk D, Somjen GG, Aitken PG, Turner DA. Mitochondrial and intrinsic optical signals imaged during hypoxia and spreading depression in rat hippocampal slices. J. Neurophysiol. 2000;84:311–324. doi: 10.1152/jn.2000.84.1.311. [DOI] [PubMed] [Google Scholar]
- 49.Lambert HP, Zenger M, Azarias G, Chatton J-Y, Magistretti PJ, Lengacher S. Control of Mitochondrial pH by Uncoupling Protein 4 in Astrocytes Promotes Neuronal Survival. J. Biol. Chem. 2014;289:31014–31028. doi: 10.1074/jbc.M114.570879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, Warden CH, Castilho RF, Melcher T, Gonzalez-Zulueta M, Nikolich K, Wieloch T. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. Nat. Med. 2003;9:1062–1068. doi: 10.1038/nm903. [DOI] [PubMed] [Google Scholar]
- 51.Campanella M, Casswell E, Chong S, Farah Z, Wieckowski MR, Abramov AY, Tinker A, Duchen MR. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab. 2008;8:13–25. doi: 10.1016/j.cmet.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 52.Dooley CT, Dore TM, Hanson GT, Jackson WC, Remington SJ, Tsien RY. Imaging Dynamic Redox Changes in Mammalian Cells with Green Fluorescent Protein Indicators. J. Biol. Chem. 2004;279:22284–22293. doi: 10.1074/jbc.M312847200. [DOI] [PubMed] [Google Scholar]
- 53.Meyer AJ, Dick TP. Fluorescent protein-based redox probes. Antioxid. Redox Signaling. 2010;13:621–650. doi: 10.1089/ars.2009.2948. [DOI] [PubMed] [Google Scholar]
- 54.Funke F, Gerich FJ, Müller M. Dynamic, semi-quantitative imaging of intracellular ROS levels and redox status in rat hippocampal neurons. NeuroImage. 2011;54:2590–2602. doi: 10.1016/j.neuroimage.2010.11.031. [DOI] [PubMed] [Google Scholar]
- 55.Fekete A, Vizi ES, Kovacs KJ, Lendvai B, Zelles T. Layer-specific differences in reactive oxygen species levels after oxygen-glucose deprivation in acute hippocampal slices. Free Radical Biol. Med. 2008;44:1010–1022. doi: 10.1016/j.freeradbiomed.2007.11.022. [DOI] [PubMed] [Google Scholar]
- 56.Karlsson M, Kurz T, Brunk UT, Nilsson SE, Frennesson CI. What does the commonly used DCF test for oxidative stress really show? Biochem. J. 2010;428:183–190. doi: 10.1042/BJ20100208. [DOI] [PubMed] [Google Scholar]
- 57.Kalyanaraman B, Darley-Usmar V, Davies KJA, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ, II, Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radical Biol. Med. 2012;52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zielonka J, Kalyanaraman B. Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: Another inconvenient truth. Free Radical Biol. Med. 2010;48:983–1001. doi: 10.1016/j.freeradbiomed.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J. Neurosci. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.García-Ruiz C, Morales A, Ballesta A, Rodés J, Kaplowitz N, Fernández-Checa JC. Effect of chronic ethanol feeding on glutathione and functional integrity of mitochondria in periportal and perivenous rat hepatocytes. J. Clin. Invest. 1994;94:193–201. doi: 10.1172/JCI117306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat. Protocols. 2009;4:1582–1590. doi: 10.1038/nprot.2009.151. [DOI] [PubMed] [Google Scholar]
- 62.Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain. Eur. J. Biochem. 2000;267:4912–4916. doi: 10.1046/j.1432-1327.2000.01597.x. [DOI] [PubMed] [Google Scholar]
- 63.Olson BJSC, Markwell J. Current Protocols in Protein Science. John Wiley & Sons, Inc; 2001. Assays for determination of protein concentration. [Google Scholar]
- 64.Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protocols. 2007;1:3159–3165. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- 65.Curbo S, Reiser K, Rundloef A-K, Karlsson A, Lundberg M. Is Trichloroacetic Acid an Insufficient Sample Quencher of Redox Reactions? Antioxid. Redox Signaling. 2013;18:795–799. doi: 10.1089/ars.2012.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hansen RE, Roth D, Winther JR. Quantifying the global cellular thiol-disulfide status. Proc. Natl. Acad. Sci. U. S. A. 2009;106:422–427. doi: 10.1073/pnas.0812149106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rice ME, Russo-Menna I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience. 1997;82:1213–1223. doi: 10.1016/s0306-4522(97)00347-3. [DOI] [PubMed] [Google Scholar]
- 68.McBain CJ, Traynelis SF, Dingledine R. Regional variation of extracellular space in the hippocampus. Science. 1990;249:674. doi: 10.1126/science.2382142. [DOI] [PubMed] [Google Scholar]
- 69.van der Toorn A, Syková E, Dijkhuizen RM, Vořišek I, Vargová L, Škobisová E, van Lookeren Campagne M, Reese T, Nicolay K. Dynamic changes in water ADC, energy metabolism, extracellular space volume, and tortuosity in neonatal rat brain during global ischemia. Magn. Reson. Med. 1996;36:52–60. doi: 10.1002/mrm.1910360110. [DOI] [PubMed] [Google Scholar]
- 70.Syková E, Nicholson C. Diffusion in Brain Extracellular Space. Physiol. Rev. 2008;88:1277–1340. doi: 10.1152/physrev.00027.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mazel T, Simonova Z, Sykova E. Diffusion heterogeneity and anisotropy in rat hippocampus. Neuroreport. 1998;9:1299–1304. doi: 10.1097/00001756-199805110-00008. [DOI] [PubMed] [Google Scholar]
- 72.Sykova E, Mazel T, Simonova Z. Diffusion constraints and neuron-glia interaction during aging. Exp Gerontol. 1998;33:837–851. doi: 10.1016/s0531-5565(98)00038-2. [DOI] [PubMed] [Google Scholar]
- 73.Li X, Wallin C, Weber SG, Sandberg M. Net efflux of cysteine, glutathione and related metabolites from rat hippocampal slices during oxygen/glucose deprivation: dependence on γ-glutamyl transpeptidase. Brain Res. 1999;815:81–88. doi: 10.1016/s0006-8993(98)01097-x. [DOI] [PubMed] [Google Scholar]
- 74.Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proceedings of the National Academy of Sciences. 1994;91:10625–10629. doi: 10.1073/pnas.91.22.10625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH. Coordinate Regulation of Glutathione Biosynthesis and Release by Nrf2-Expressing Glia Potently Protects Neurons from Oxidative Stress. The Journal of Neuroscience. 2003;23:3394–3406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bolaños JP. Bioenergetics and redox adaptations of astrocytes to neuronal activity. J. Neurochem. 2016;139:115–125. doi: 10.1111/jnc.13486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rossi DJ, Brady JD, Mohr C. Astrocyte metabolism and signaling during brain ischemia. Nat. Neurosci. 2007;10:1377–1386. doi: 10.1038/nn2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang J, Philbert MA. Distribution of glutathione and glutathione-related enzyme systems in mitochondria and cytosol of cultured cerebellar astrocytes and granule cells. Brain Res. 1995;680:16–22. doi: 10.1016/0006-8993(95)00209-9. [DOI] [PubMed] [Google Scholar]
- 79.Dringen R, Pfeiffer B, Hamprecht B. Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 1999;19:562–569. doi: 10.1523/JNEUROSCI.19-02-00562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rana S, Dringen R. Gap junction hemichannel-mediated release of glutathione from cultured rat astrocytes. Neurosci. Lett. 2007;415:45–48. doi: 10.1016/j.neulet.2006.12.043. [DOI] [PubMed] [Google Scholar]
- 81.Griffith OW, Meister A. Origin and turnover of mitochondrial glutathione. Proceedings of the National Academy of Sciences. 1985;82:4668–4672. doi: 10.1073/pnas.82.14.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marí M, Morales A, Colell A, García-Ruiz C, Fernández-Checa JC. Mitochondrial Glutathione, a Key Survival Antioxidant. Antioxidants & Redox Signaling. 2009;11:2685–2700. doi: 10.1089/ars.2009.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Booty LM, King MS, Thangaratnarajah C, Majd H, James AM, Kunji ERS, Murphy MP. The mitochondrial dicarboxylate and 2-oxoglutarate carriers do not transport glutathione. FEBS Lett. 2015;589:621–628. doi: 10.1016/j.febslet.2015.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tseng Q, Duchemin-Pelletier E, Deshiere A, Balland M, Guillou H, Filhol O, Théry M. Spatial organization of the extracellular matrix regulates cell–cell junction positioning. Proc. Natl. Acad. Sci. USA. 2012;109:1506–1511. doi: 10.1073/pnas.1106377109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Woods G, Zito K. Preparation of Gene Gun Bullets and Biolistic Transfection of Neurons in Slice Culture. J Vis. Exp. 2008:675. doi: 10.3791/675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yi L, Li H, Sun L, Liu L, Zhang C, Xi Z. A Highly Sensitive Fluorescence Probe for Fast Thiol-Quantification Assay of Glutathione Reductase. Angew. Chem. 2009;121:4094–4097. doi: 10.1002/anie.200805693. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.