

Abstract
Morphologic assessment of formalin-fixed, paraffin-embedded (FFPE) tissue samples has been the gold standard for cancer diagnostics for decades due to its excellent preservation of morphology. Personalized medicine increasingly provides individually adapted and targeted therapies for characterized individual diseases enabled by combined morphological and molecular analytical technologies and diagnostics. Performance of morphologic and molecular assays from the same FFPE specimen is challenging because of the negative impact of formalin due to chemical modification and cross-linking of nucleic acids and proteins. A non-cross-linking, formalin-free tissue fixative has been recently developed to fulfil both requirements, i.e., to preserve morphology like FFPE and biomolecules like cryo-preservation. Since FISH is often required in combination with histopathology and molecular diagnostics, we tested the applicability of FISH protocols on tissues treated with this new fixative. We found that formalin post-fixation of histological sections of non-cross-linking, formalin-free and paraffin-embedded (NCFPE) breast cancer tissue generated equivalent results to those with FFPE tissue in human epidermal growth factor receptor 2 (HER2) FISH analysis. This protocol describes how a FISH assay originally developed and validated for FFPE tissue can be used for NCFPE tissues by a simple post-fixation step of histological sections.
Keywords: Cancer Research, Issue 130, Breast cancer, non-cross-linking formalin-free tissue fixative, formalin, pre-analytics, HER2 FISH analysis, molecular diagnostics
Introduction
Personalized medicine increasingly relies on multi-parameter tests involving morphological and molecular tissue analyses. Formalin fixation of tissues as the gold standard provides excellent morphologic quality1,2. However, formalin-induced chemical modification and cross-linking of proteins and nucleic acids3,4,5 negatively interferes with molecular analysis6. These molecular modifications limit the quality of nucleic acids and proteins and may result in gene sequence artefacts7 or reduced sensitivity of polymerase chain reaction (PCR)-based assays8. Although major efforts were taken to optimize molecular tests for FFPE tissue, cryo-preservation of tissues is in general superior to formalin fixation, making it necessary to split tissue samples for different preservation procedures. To avoid the need for cryo-preservation for molecular analyses, a non-cross-linking, formalin-free fixative, PAXgene Tissue was developed. This commercially available system consists of a fixation and a stabilization solution containing different alcohols, acetic acid and a soluble organic compound. Proper preservation of nucleic acids, (phospo)proteins and morphology was shown in several studies6,9,10,11,12,13.
One particular application in cancer diagnostics is FISH detecting a comprehensive range of chromosomal alterations, such as translocations, submicroscopic deletions and amplifications 14. For example, twenty percent of invasive breast cancer tumors show amplification of the human epidermal growth factor receptor 2 (HER2) gene15,16,17, which is associated with poor prognosis18,19. Determination of the HER2 overexpression and amplification status in breast cancer by FISH is needed to select patients for anti-HER2-directed therapy and is a companion diagnostic listed by the Federal Drug Association (FDA) (http://www.fda.gov/MedicalDevices/ProductsandMedicalProcedures/InVitroDiagnostics/ucm301431.htm)17. In order to allow a broad application of FISH-based companion diagnostics in health care, assays were developed and approved for FFPE tissues.
In a previous study, we showed that NCFPE tissues cannot be used for FISH assays which have been approved for FFPE tissue. However, tests of time series of different post fixation procedures of NCFPE tissues with 4 % buffered formaldehyde showed that post fixation times of 18 h or longer of NCFPE tissue sections achieved equivalent results to those with FFPE tissues14.
In this study, we provide a detailed protocol and demonstrate the impact of non-cross-linking, formalin-free tissue fixation and 24 h 4 % buffered formaldehyde post-fixation on sections used for HER2 FISH and RNA quality from the same primary tissue specimen.
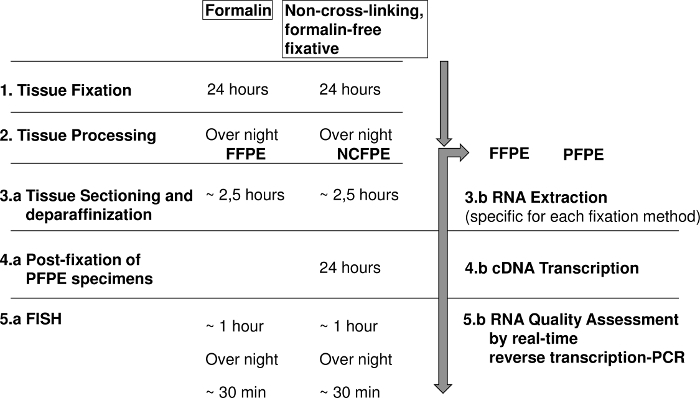
Figure 1: Diagram showing the steps for fluorescence in situ hybridization (FISH) and RNA analysis of formalin fixed paraffin embedded (FFPE) and non-cross-linking, formalin-free fixed paraffin embedded (NCFPE) tissues. Please click here to view a larger version of this figure.
Protocol
The study was approved by the Ethics Committee of the Medical University of Graz (reference number 20-066), Austria. Tissue samples were obtained from two cases of surgically resected primary invasive breast cancers after the patients had given written informed consent.
1. Tissue fixation
NOTE: Tissue fixation is performed either with standard buffered formalin solution (SBFS), defined as 10 % formalin solution containing a mass fraction of 3.7 % (corresponding to a volume fraction of 4 %) formaldehyde buffered to pH 6.8 to pH 7.2 according to CEN/TS 16827-1:2015 or the non-cross-linking, formalin-free tissue fixation system (also referred to as the alternate fixation solution below) consisting of a fixative and a stabilizer solution at room temperature (RT).
Cut the breast cancer tissue samples with a sterile scalpel into two halves each 4 mm thick to ensure adequate tissue quantity. Use a maximum size 4 x 15 x 15 mm for NCFPE or SBFS tissue fixation.
Place each half of the tumor specimen in a standard tissue cassette and submerge it in SBFS or the alternative fixation solution for 24 h with a volume per volume ratio (v/v) of 4 parts fixative and one part tissue (4 mL/1 g tissue (ideally 10 mL/1 g)).
Transfer the alternatively-fixed samples into the stabilizer solution for 2-72 h by opening the container, turning the tissue cassette by 180° and submerging it.
Place the tissue cassettes with the SBFS-fixed samples in 250 mL 70 % ethanol for 30 min at RT to remove residual SBFS. NOTE: This step is important to avoid SBFS contamination of non-cross-linking fixative-treated samples in the automated tissues processor, which would destroy the beneficial properties of non-cross-linking fixatives for preservation of biomolecules. CAUTION: SBFS is a hazardous solution, causes skin irritation, serious eye damage, may cause an allergic skin reaction, is suspected of causing genetic defects, may cause cancer, respiratory irritation, drowsiness and causes damages to organs. The alternative fixative is toxic by inhalation, in contact with skin and if swallowed, irritating to eyes and skin, danger of very serious irreversible effects through inhalation, in contact with skin and if swallowed. Inhalation and physical contact should be avoided for both fixation solutions. Wear suitable protective clothing, gloves, and eye and face protection. Work in a fume hood.
Proceed to tissue processing, embedding and tissue sectioning (sections 2 and 3).
2. Tissue processing and embedding
Perform stepwise dehydration of tissue cassettes with the fixed tissue samples in 70 % (2 x 15 min), 80 % (30 min), 90 % (60 min), and 99 % (2 x 60 min) ethanol, followed by xylene (2 x 60 min). Then infiltrate with paraffin in an automated tissue processor (4 x 45 min).
Using forceps, place the dehydrated and paraffin-infiltrated samples (alternatively and SBFS-fixed) in metal molds.
Embed the samples in 5 - 10 mL molten paraffin (solidification point 56-58 °C) using a paraffin embedding instrument.
Place the molds onto a cooling plate at -5 °C for 30 min.
Recover the paraffin-embedded tissue blocks from the molds.
Store the resulting blocks (alternatively-fixed, paraffin-embedded (NCFPE) and SBFS-fixed paraffin-embedded (FFPE)) in the dark at 4 °C until use.
3. Tissue sectioning and slide preparation for FISH
NOTE: FFPE sections are directly used for FISH, while slides with NCFPE sections are post-fixed in SBFS for 24 h before FISH procedure.
Insert a single-use blade into the microtome and adjust the microtome to 10 µm.
Take the first block from the cooling plate and insert it into the instrument.
Trim the block until an even surface is achieved. Clean the microtome using a brush and adjust it to 3 µm. Discard the first 2-3 sections.
Cut 3 µm sections of NCFPE and FFPE blocks and place them in a cold water bath (ultrapure water). NOTE: To obtain better photographs without overlapping nuclei, here, images of 2 µm sections instead of 3-5 µm as recommended in the manufacturer´s instructions are shown.
Pick up each floating section with a fine paint brush and a needle onto a coated microscope slide for adhesion of the tissue sections.
Plunge this slide into a warm (40 °C) water bath and let the section stretch completely.
Put the section back onto the slide by carefully pulling the slide out of the water and avoid wrinkles.
Let all sections dry at at room temperature for 1 h.
Heat the slides at 70 °C for 30 min in an incubator to melt the paraffin.
Put the slides horizontally in a glass staining jar.
Rehydrate the sections stepwise at RT in the staining jars filled with 250 mL each of the following liquids: 100 % xylene 2 x 15 min, 96 % ethanol 2 x 15 min, 90 % ethanol 2 min, 80 % ethanol 2 min, 70 % ethanol 2 min, 50 % ethanol 2 min, and distilled water 2 x 10 min. CAUTION: Perform deparaffinization and rehydration steps in a fume hood, do not inhale toxic volatilized solvents. NOTE: Depending on the sections and tissue, it may be necessary to test different incubation times in advance.
4. Post-fixation of NCFPE specimens
Perform post-fixation of NCFPE slides by putting them into a new staining jar filled with SBFS for 24 h at RT. Place the jar in a fume hood during the 24 h post-fixation step.
Remove excess SBFS by washing with phosphate buffered saline (3 x 10 min) and distilled water (2 x 10 min).
5. Fluorescence in situ hybridization (FISH)
NOTE: Here FISH is performed using the commercially available HER2/CEN17 dual color probe Kit according to the manufacturer's instructions. Do not allow the tissue sections to dry during the hybridization and washing steps. Do not allow the DNA probe and 4', 6-diamidino-2-phenylindole (DAPI) DNA counterstain solutions to be exposed to light for a longer period. Perform these steps in the dark using lightproof containers. Depending on the age and the fixation step of the sample material, increase or decrease the denaturing and wash temperatures to obtain better hybridization results.
Place a staining jar containing 250 mL of heat pre-treatment solution citric in a 98 °C water bath.
Place the rehydrated FFPE and post-fixed NCFPE slides into the jar and incubate for 15 min. Do not use more than six slides per staining jar.
Wash the slides in distilled water for 3 min at RT in a new staining jar.
- For proteolysis, place the slides into a temperature controlled hybridization system at 37 °C.
- Immediately, apply the-ready-to-use Pepsin solution (2mg/ml) dropwise (~ 30 µl per drop) to the tissue sections until it is completely covered. Incubate for 9 min at 37 °C in the hybridization system. NOTE: An incubation time of 5-15 min is recommended. The optimum time for proteolysis should be ascertained in advance.
- Place the slides into a jar containing wash buffer SSC and incubate for 5 min.
- Rinse the slides in distilled water for 1 min at RT.
- Perform dehydration of the sections in the following graded alcohols: 50 %, 70 %, 90 % and 100 % each for 1 min. Air dry the sections.
- For hybridization, pipette 10 µL of HER2/CEN17 probe onto the dried tissue sections, cover each sample with a coverslip and seal it with rubber cement. NOTE: 10 µL of probe is recommended per 22 x 22 mm tissue area. A gentle warming of the probe makes the pipetting process easier. Avoid bubbles and long exposure of the probe to the light.
- Co-denature probe and specimen DNA in the hybridization system at 75 °C for 10 min.
- Set the hybridization system to 37 °C and hybridize overnight.
- Carefully remove the rubber cement with forceps for post-hybridization and detection.
- Incubate the slides in a staining jar containing 37 °C pre-warmed 1 x wash buffer A for 5 min. Remove the coverslips.
- Wash the slides using pre-warmed 1 x wash buffer A twice for 5 min at 37 °C.
- Dehydrate the slides in 70 %, 90 % and 100 % ethanol, 1 min each.
- Dry the samples in air and protect from light.
- For staining, apply 30 µL DAPI-solution to the hybridized area avoiding bubbles. Cover the sections with a coverslip.
- Incubate for 15 min protected from light.
- Carefully remove excess DAPI-solution by gently pressing the slide between filter papers.
- Store the slides at 2 - 8 °C in the dark for up to 2 weeks until evaluation by confocal microscopy.
6. Confocal imaging and scanning of FISH slides
Perform confocal imaging and scanning of the slides on a confocal digital scanner equipped with a 40x/1.2 NA objective, quad band filters (DAPI/FITC/TRITC/Cy5) and a 5.5Mpx, 16 bit, cooled scientific complementary metal oxide semiconductor (CMOS) camera.
Acquire the images with filters for green (excitation: 503 nm, emission: 528 nm) and orange (excitation: 547 nm, emission: 572 nm) channels.
7. Interpretation
NOTE: The HER2/CEN17 probe is a mixture of a red fluorochrome-labelled CEN17 probe specific for the alpha satellite centromeric region of chromosome 17 (D17Z1) and a green fluorochrome-labelled HER2 probe specific for the HER2 gene (probe location 17q12). Tumor specimens with a HER2:CEN17 ratio ≤2.0 per nucleus are scored as normal, whereas those with a HER2:CEN17 ratio ≥2.0 are scored as amplified17.
- Perform quality analysis of the images with the open source CaseViewer 2.120.
- Evaluate at least 20 cells that are located in two different regions of the invasive component of the carcinoma as defined by a pathologist. Include clearly distinguishable well distributed nuclei in the counting area. Exclude from counting tissue regions at the boundary and retracted or squeezed areas.
8. RNA Quality
- RNA Extraction
- Ensure that all instruments and tools are RNAse free. Use a RNAse-removing solution.
- Cut 10 to 20 sections (5 µm thick) of NCFPE or FFPE samples using a microtome following the protocol until step 3.8.
- Extract RNA from NCFPE samples using the RNA isolation kit (see the Table of Materials).Extract RNA from FFPE samples by using an FFPE RNA isolation kit. NOTE: Extraction of RNA from non-cross-linking fixatives requires an isolation method adapted to the fixation method. Do not use any other RNA isolation method (e.g. Trizol, RNeasy FFPE, etc. for NCFPE specimen).
- Determine the RNA concentration and purity using a spectrophotometer. The ratio of the absorbance at 260 nm and 280 nm (A260/280) should be ~ 2.0.
- Store the isolated RNA at -70 °C until further analysis.
- Perform a glyceraldehyde 3-phosphate dehydrogenase (GAPDH) real-time reverse transcription PCR using primers to generate amplicons of different lengths8,9.
- Use a cDNA reverse transcription kit according to manufacturer´s instructions for cDNA synthesis. Use 500 ng RNA of each sample. Store cDNA at -20 °C until further use.
- Prepare the PCR master mix according to manufacturer´s instructions.
- Pipette the PCR master mix in triplicates in a 384-well reaction plate. Add 4 µL of 1:16 diluted cDNA (PCR template). Perform the PCR according to manufacturer´s instructions. NOTE: For human GAPDH, the following primers were used: GAPDH forward: 5´-CCACATCGCTCAGACACCAT-3´; GAPDH reverse: 71 bp 5´-ACCAGGCGCCCAATACG-3´, 153 bp 5´-GTAAACCATGTAGTTGAGGTC-3´, 200 bp 5´-TTGACGGTGCCATGGAATTT-3´, 277 bp 5´-ACTTGATTTTGGAGGGATCT-3´, 323 bp 5´-AAGACGCCAGTGGACTCCA-3´, and 530 bp 5´-ACGATACCAAAGTTGTCATG-3´.
- Analyze the results using a software applicable to the PCR machine. Nonspecific products must be excluded based on melting curve analysis21.
Representative Results
FISH HER2 determination:
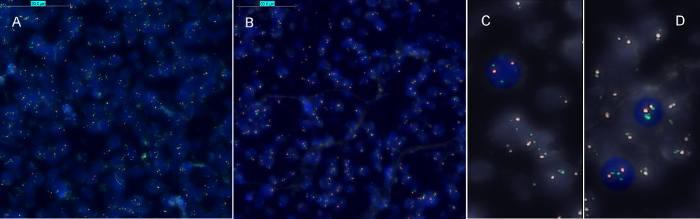
The HER2-FISH signal intensities from FFPE and NCFPE are similar (Figure 2). Both cases in most cells show a clear excess of the green signal (indicative of the amplified HER2 gene locus) as compared to the red signal (CEN17 reference gene). Therefore, the case shows an amplified HER2 gene, which means that this patient is expected to respond to trastuzumab, an anti-HER2 targeted therapy. The FFPE specimen served as positive control. The signals observed in non-neoplastic cells (both in FFPE and NCFPE) serve as internal reference and negative control for HER2 gene amplification. For every analysis series, at least one section with known gene amplification status should be used as positive control for the hybridization reaction.
RNA Quality:
Corresponding FFPE and NCFPE samples from two different cases show differences in RNA quality by employing real-time reverse transcription PCR. Results obtained show different amplification efficiencies for different amplicon lengths (i.e., 71 bp, 153 bp, 200 bp, 275 bp, 323 bp) of the GAPDH mRNA. All Ct (cycle threshold) values obtained from FFPE samples were higher than those from NCFPE. This difference demonstrates the lower PCR amplificability of mRNA isolated from FFPE tissues, suggesting a more pronounced chemical modification. Furthermore, the higher Ct values for long compared to short amplicons are an indicator for mRNA fragmentation. In comparison, the Ct value slopes of the different amplicon lengths shows more pronounced mRNA fragmentation in FFPE than NCFPE tissues (Figure 3).
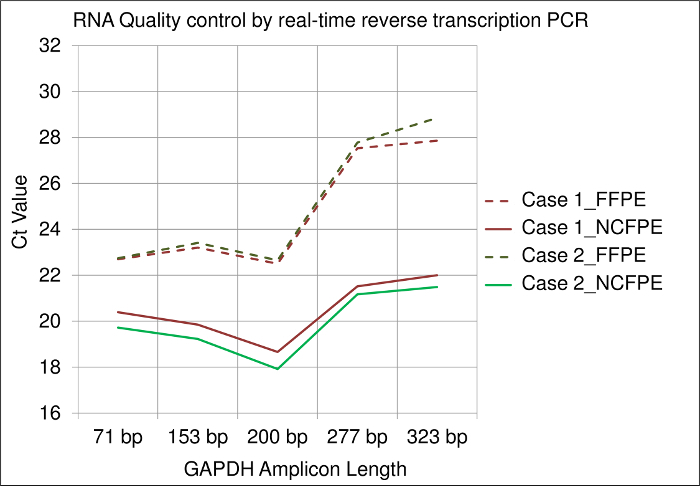
Figure 2: Representative results of HER2 FISH for corresponding formalin fixed paraffin embedded (FFPE) and non-cross-linking, formalin-free fixed paraffin embedded (NCFPE) tissue sections. Corresponding samples from the same FFPE (A)and NCFPE (B) tissues are shown. In contrast to normal interphase cells (C), where two green (HER2) and two red (CEN17) signals are expected, cells with amplified HER2 gene locus represent multiple copies of green signals (HER2) (D). HER2 amplification status was identical in both FFPE and the post-fixed NCFPE sections (A, B). Please click here to view a larger version of this figure.
Figure 3: Results of a real-time reverse transcription PCR assay used for RNA quality control. Y-Axis: Ct (cycle threshold) values of the GAPDH gene are shown for different amplicon lengths (71 bp, 153 bp, 200 bp, 275 bp, 323 bp) on the X-axis. mRNA was isolated from FFPE and NCFPE of two different breast cancer tissue samples processed in parallel and transcribed to cDNA for PCR. Please click here to view a larger version of this figure.
Discussion
The results demonstrate two key findings. First, a simple 24 h SBFS post-fixation step of sections cut from NCFPE tissues is sufficient to obtain equivalent results to those with FFPE in FISH analyses using FISH assays approved for FFPE tissues (see also reference14). This protocol has the advantage of using FISH assays originally developed and approved for FFPE without the need for comprehensive revalidation (e.g., by optimizing the pre-hybridization conditions for non-cross-linked tissue). The FISH protocol can be used exactly as described in the manufacturer's instructions.
Minor modifications from the manufacturer´s instructions described in this protocol (e.g. tissue section diameter and incubation periods at the deparaffinization steps) result from previous adaptations, which is explicitly recommended by the manufacturer due to the use of different tissue types and fixation methods.
The post-fixation step is critical because it requires a chemical reaction time of 18-24 h, which extends the analysis time by one day but allows the same daily workflow as developed for FFPE tissues (Figure 1). SBFS is reported to penetrate tissue at an average rate of 1 mm per hour22 to 5 mm in 2 h depending on the tissue type23,24. The observation that only prolonged post-fixation periods of more than 18 h could change the properties of NCFPE to FFPE sections, indicates that not the penetration of the fixative but the chemical reaction time is critical for achieving the desired results1,14.
Second, the remaining NCFPE tissue that is not used for post-fixation can be used for further molecular analyses as the biomolecules are well preserved. This was demonstrated by real-time reverse transcription PCR (Figure 3). The assay is more sensitive and specific than the electropherogram-derived RIN value (RNA integrity number) with regard to RNA quality (i.e., chemical modification and fragmentation) for mRNA isolated from paraffin-embedded tissues8. Quality control was restricted in this study to real-time PCR since independent groups have shown that in addition to RNA, the quality of DNA and proteins isolated from NCFPE is better than that from FFPE tissues 8,9,13.
The protocol presented follows, where applicable (e.g., SBFS recipe, fixation conditions, RNA quality control, validation), the requirements of the CEN technical specifications for pre-analytical procedures for in-vitro diagnostics (IVD), which have been recently released by the European Committee of Standardization (CEN) (e.g. CEN/TS 16827-1:2015 for RNA-Isolation from FFPE tissues which refers to the ISO 15189 standard).
The method is limited to this specific fixative. Although the good preservation of biomolecules and morphology using the non-cross-linking, formalin-free fixative has been presented in several studies, it is mainly used in research but not in routine medical applications. One reason is that replacing the gold standard SBFS fixation by another fixative would require a variety of validation studies as not all protocols optimized for FFPE material can be used for materials fixed with non-cross-linking fixatives. Other tissue fixation methods were not tested for this FISH protocol.
The simple post-fixation step described in this manuscript has the advantage of using IVD approved assays both for FFPE and NCFPE tissues.
Disclosures
D.G. is employed by Qiagen and has stock options or bond holdings in the company's pension plan. The data presented in this manuscript refer to the PAXgene Tissue System, which has been developed in a joint effort by Qiagen and PreAnalytiX. Qiagen is industrial partner of the publicly funded Christian Doppler Laboratory for Biospecimen Research and Biobanking Technologies. All authors have no conflict of interest and no competing financial interests.
Acknowledgments
We thank the team of the laboratory for molecular pathology at the Institute of Pathology of the Medical University Graz for their expertise and support. Furthermore, we thank Iris Kufferath and Daniela Pabst for technical assistance, Bernadette Rieger and Sylvia Eidenhammer for processing the HER2 cases as well as Kinga Szurian (pathologist) and Tamas Regényi (3DHISTEC). We thank Penelope Kungl for proofreading the manuscript. This work has been financially supported by the Christian Doppler Research Fund, the Austrian Federal Ministry of Science, Research and Economy and the National Foundation for Research, Technology and Development.
References
- Fox CH, Johnson FB, Whiting J, Roller RP. Formaldehyde fixation. J. Histochem.Cytochem. 1985;33(8):845–853. doi: 10.1177/33.8.3894502. [DOI] [PubMed] [Google Scholar]
- Oosterhuis JW, Coebergh JW, van Veen EB. Tumour banks: well-guarded treasures in the interest of patients. Nat. Rev. Cancer. 2003;3(1):73–77. doi: 10.1038/nrc973. [DOI] [PubMed] [Google Scholar]
- Masuda N, Ohnishi T, Kawamoto S, Monden M, Okubo K. Analysis of chemical modification of RNA from formalin-fixed samples and optimization of molecular biology applications for such samples. Nucleic Acids Res. 1999;27(22):4436–4443. doi: 10.1093/nar/27.22.4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz B, Kersten GF, Hoogerhout P, Brugghe HF, Timmermans HA, de Jong A, et al. Identification of formaldehyde-induced modifications in proteins: reactionswith model peptides. J. Biol. Chem. 2003;279(8):6235–6243. doi: 10.1074/jbc.M310752200. [DOI] [PubMed] [Google Scholar]
- Evers DL, Fowler CB, Cunningham BR, Mason JT, O'Leary TJ. The effect of formaldehyde fixation on RNA: optimization of formaldehyde adduct removal. J.Mol. Diagn. 2011;13(3):282–288. doi: 10.1016/j.jmoldx.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groelz D, Sobin L, Branton P, Compton C, Wyrich R, Rainen L. Non-formalin fixative versus formalin-fixed tissue: A comparison of histology and RNA quality. Exp. Mol. Pathol. 2013;94(1):188–194. doi: 10.1016/j.yexmp.2012.07.002. [DOI] [PubMed] [Google Scholar]
- Do H, Dobrovic A. Sequence artifacts in DNA from formalin-fixed tissues: causes and strategies for minimization. Clin. Chem. 2015;61:64–71. doi: 10.1373/clinchem.2014.223040. [DOI] [PubMed] [Google Scholar]
- Kashofer K, Viertler C, Pichler M, Zatloukal K. Quality Control of RNA Preservation and Extraction from Paraffin-Embedded Tissue: Implications for RT-PCR and Microarray Analysis. PloS One. 2013;8(7):e70714. doi: 10.1371/journal.pone.0070714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viertler C, Groelz D, Gündisch S, Kashofer K, Reischauer B, Riegman PH, et al. A new technology for stabilization of biomolecules in tissues for combined histological and molecular analyses. J. Mol. Diagn. 2012;14(5):458–466. doi: 10.1016/j.jmoldx.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Gundisch S, Slotta-Huspenina J, Verderio P, Maura C, Ciniselli SP, Schott S, et al. Evaluation of colon cancer histomorphology: A comparison between formalin and PAXgene tissue fixation by an international ring trial. Virchows Arch. 2014;465(5):509–519. doi: 10.1007/s00428-014-1624-4. [DOI] [PubMed] [Google Scholar]
- Ergin B, Meding S, Langer R, Kap M, Viertler C, Schott C, et al. Proteomic analysis of PAXgene-fixed tissues. J. Proteome Res. 2010;9(10):5188–5196. doi: 10.1021/pr100664e. [DOI] [PubMed] [Google Scholar]
- Kap M, Smedts F, Oosterhuis W, Winther R, Christensen N, Reischauer B, et al. Histological Assessment of PAXgene Tissue Fixation and Stabilization Reagents. PLoS ONE. 2011;6(11):e27704. doi: 10.1371/journal.pone.0027704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieson W, Marcon N, Antunes L, Ashford DA, Betsou F, Frasquilho SG, et al. A Critical Evaluation of the PAXgene Tissue Fixation System: Morphology, Immunohistochemistry, Molecular Biology, and Proteomics. Am J Clin Pathol. 2016;146:25–40. doi: 10.1093/ajcp/aqw023. [DOI] [PubMed] [Google Scholar]
- Oberauner-Wappis L, Loibner M, Viertler C, Groelz D, Wyrich R, Zatloukal K. Protocol for HER2 FISH determination on PAXgene-fixed and paraffin-embedded tissue in breast cancer. Int J. Exp. Path. 2016;97(2):202–206. doi: 10.1111/iep.12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauletti G, Godolphin W, Press MF, Slamon DJ. Detection and quantitation ofHER-2/neu gene amplification in human breast cancer archival material using fluorescence in situ hybridization. Oncogene. 1996;13(1):63–72. [PubMed] [Google Scholar]
- Owens MA, Horten BC, Da Silva MM. HER2 amplification ratios by fluorescence in situ hybridization and correlation with immunohistochemistry in a cohort of 6556 breast cancer tissues. Clin.Breast Cancer. 2004;5(1):63–69. doi: 10.3816/cbc.2004.n.011. [DOI] [PubMed] [Google Scholar]
- Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch Pathol Lab Med. 2007;131(1):18–43. doi: 10.5858/2007-131-18-ASOCCO. [DOI] [PubMed] [Google Scholar]
- Press MF, Bernstein L, Thomas PA, Meisner LF, Zhou JY, Ma Y, et al. HER-2/neu gene amplification characterized by fluorescence in situ hybridization: poor prognosis in node-negative breast carcinomas. J. Clin. Oncol. 1997;15(8):2894–2904. doi: 10.1200/JCO.1997.15.8.2894. [DOI] [PubMed] [Google Scholar]
- Yamauchi H, Stearns V, Hayes DF. When is a tumor marker ready for prime time? A case study of c-erbB-2 as a predictive factor in breast cancer. J. Clin. Oncol. 2001;19(8):2334–2356. doi: 10.1200/JCO.2001.19.8.2334. [DOI] [PubMed] [Google Scholar]
- Paulik R, Micsik T, Kiszler G, Kaszál P, Székely J, Paulik N, Várhalmi E, Prémusz V, Krenács T, Molnár B. An optimized image analysis algorithm for detecting nuclear signals in digital whole slides for histopathology. Cytometry A. 2017. [DOI] [PubMed]
- Applied Biosystems. QuantStudio 6 and 7 Flex Real-Time PCR System Software. 2013. Publication Number 4489822.
- NCCLS. Quality Assurance for Immunocytochemistry. Wayne: 1999. [Google Scholar]
- Baker JR. Principles of Biological Microtechnique. London: Methuen: 1958. Reprinted 1970, with corrections. [Google Scholar]
- Hewitt SM, Lewis F, Cao Y, Conrad RC, Cronin M, Danenberg KD, et al. Tissue handling and specimen preparation in surgical pathology: issues concerning the recovery of nucleic acids from formalin-fixed, paraffin-embedded tissue. Arch Pathol. Lab Med. 2008;132(12):1929–1935. doi: 10.5858/132.12.1929. [DOI] [PubMed] [Google Scholar]