

Abstract
Rationale:
The life-threatening drug rash with eosinophilia and systemic symptoms (DRESS) syndrome occurs most commonly after exposure to drugs, clinical features mimic those found with other serious systemic disorders. It is rarely associated with thrombotic microangiopathy.
Patient concerns:
We describe the unique case of a 44-year-old man who simultaneously experienced DRESS syndrome with thrombotic microangiopathy (TMA) after a 5 days treatment with fluindione.
Diagnoses:
Clinical evaluation leads to the discovery of an underlying lymphangiomatosis, due to a Noonan syndrome.
Intervetions:
The anticoagulant was withdrawn, and corticosteroids (1 mg/kg/day) and acenocoumarol were started.
Outcomes:
Clinical improvement ensued. At follow-up the patient is well.
Lessons:
The association of DRESS with TMA is a rare condition; we believe that the presence of the underlying Noonan syndrome could have been the trigger. Moreover, we speculate about the potential interrelations between these entities.
Keywords: fluindione, lymphangiomatosis, thrombotic thrombocytopenic purpura
1. Introduction
Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome is a rare and life-threatening idiosyncratic drug reaction occurring 2 to 8 weeks after exposure to an offending agent.[1] Among the most commonly implicated medications are allopurinol, sulfonamides, and aromatic anticonvulsants such as phenytoin, phenobarbital, and carbamazepine. The association between thrombotic microangiopathy is exceedingly rare.[2,3]
2. Case report
A 44-year-old man, with a history of asthma and Charcot–Marie–Tooth disease, was initially admitted to another hospital for the sudden onset of fever, erythematous rash and 5 days after receiving the oral anticoagulant fluindione after ankle sprain repair. The patient was 1.84 m tall and presented mild dysmorphic features (thick lips with prominent nasolabial folds, epicanthal folds, low-set posteriorly rotated ears, short neck, high anterior and low posterior hairline, pectus carinatum, and arachnodactyly; Fig. 1A–C) besides skin rash, physical examination revealed diffuse lymphadenopathy, hyperlaxity of the hands and wrists, and foot deformities (high arch, or pes cavus, and hammer toes) typical of Charcot–Marie–Tooth disease (Fig. 1D). The patient reported that his grandfather had similar dysmorphic features.
Figure 1.
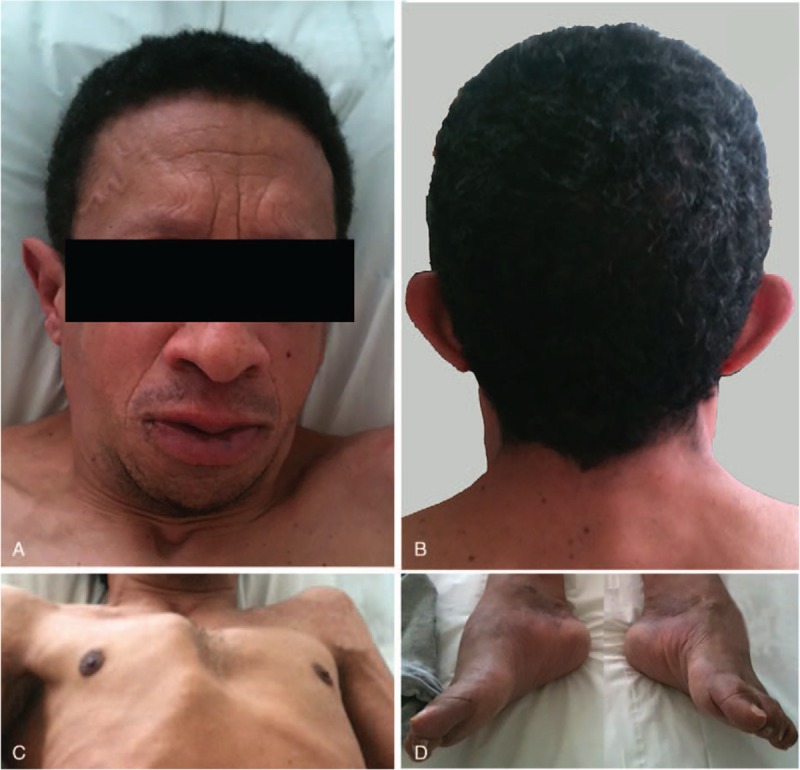
Dysmorphic features. (A) Thick lips with prominent nasolabial folds, high anterior airline, short neck. (B) Low-set posteriorly rotated ears, low posterior hairline. (C) Pectus carinatum. (D) Foot deformities typical of Charcot–Marie–Tooth disease.
Laboratory tests showed hemolytic anemia with schistocytes on a peripheral smear, thrombocytopenia, eosinophilia (11 × 109/L), elevated lactate dehydrogenase level and renal failure with proteinuria and hematuria. His condition rapidly worsened with jaundice, oliguria, and hemodynamic instability. A renal biopsy showed microangiopathy characterized by wrinkling of glomerular capillaries, parietal arteriolar edema, focal glomerular ischemia, widening of the subendothelial space, and focal glomerular ischemia (Fig. 2A). Trans-thoracic echocardiography demonstrated left ventricular thrombus, endocardial infiltration of both ventricles compatible with an eosinophilic cardiomyopathy and moderate left ventricular dysfunction (left ventricular ejection fraction, LVEF: 45%) without chamber dilatation (Fig. 2B). The patient was transferred to our hospital for investigations. Echocardiography-guided endomyocardial biopsy was performed upon arrival; histology showed focal thrombotic microangiopathy and scattered foci of myocardial necrosis.
Figure 2.
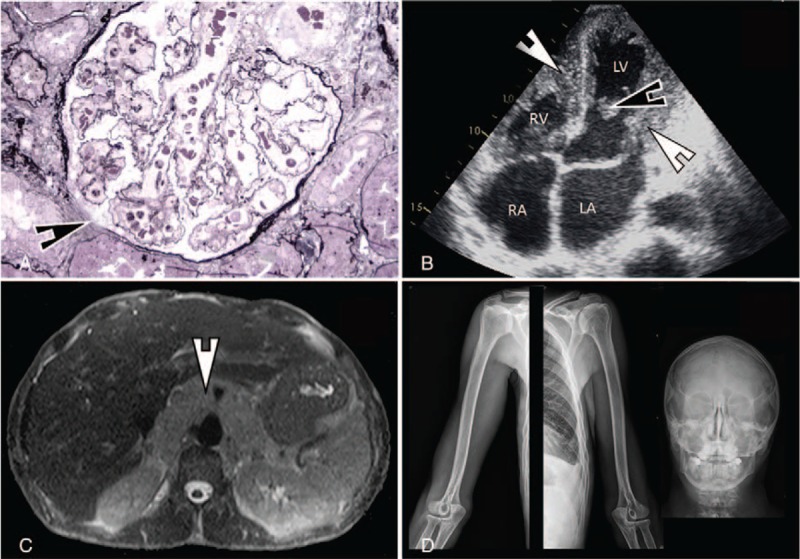
(A) Glomerulus showing wrinkling of capillary walls, parietal arteriolar edema, focal glomerular ischemia, and widening of the subendothelial space (arrows) (Jones’ basement membrane stain, ×400). (B) Trans-thoracic echocardiography showing a nondilated left ventricle with infiltration of both left and right ventricles endocardium (thick arrows) and a left ventricular thrombus (thin arrow). (C) MRI (T2 weighted images) showing a diffuse infiltration of the retroperitoneal space surrounding the aorta and the caval vein with no mass effect. (D) Representative x-ray images (from left to right: right and left humerus and shoulder girdle, skull) showing normal bone structure without pathologic features. LA = left atrium, LV = left ventricle, RA = right atrium, RV = right ventricle.
Clinical (fever, exanthema, and lymphadenopathy) and laboratory (eosinophilia) findings raised the suspicion of a drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome associated with thrombotic microangiopathy (TMA), as suggested by thrombocytopenia, anemia, and ischemic injury of kidney and heart, as well as kidney histology. The offending agent was suspected to be the vitamin K-antagonist anticoagulant fluindione. The anticoagulant was immediately withdrawn, and corticosteroids (1 mg/kg/day) and acenocoumarol were started with rapid clinical improvement.
Search for the FIP1L1–PDGFRA rearrangement was negative, thus excluding a primary hypereosinophilic syndrome. Mild deficiency of ADAMTS13 (43% of plasma levels) and high anti-ADAMTS13 titers were found (40 UI; normal <15 UI) suggesting thrombotic thrombocytopenic purpura (TTP). Diffuse lymphadenopathy prompted us to perform whole body MRI. The exam showed mediastinal and retroperitoneal lymphangiomatosis (Fig. 2C). There was no bone involvement, as confirmed by plain radiographs of the humerus, forearm, shoulder girdle, femur, leg, foot, and skull that proved normal (Fig. 2D). The extensive lymphatic involvement as well as morphological features evoked an underlying genetic abnormality: Noonan syndrome (NS) was considered despite his height. Plasmatic vascular endothelial growth factor-C (VEGF-C) levels were normal (41 pg/mL; normal < 115 pg/mL). Genetic analysis showed a mutation in the SOS2 gene but a normal karyotype, confirming the clinical suspicion of NS.
Four years after diagnosis the patient is well and laboratory findings are normal with no eosinophilia; echocardiography shows no signs of thrombosis with normal LVEF.
3. Discussion
Difficult to diagnose and a major mimicker, DRESS is characterized by the presence of at least 3 of the following findings: fever, exanthema, eosinophilia, atypical circulating lymphocytes, lymphadenopathy, and hepatitis.[1] Multiple organ involvement can occur, as in our patient, including myocarditis, pericarditis, interstitial nephritis, necrotizing granulomatous vasculitis of the kidney, encephalitis, meningitis, colitis, thyroiditis, pancreatitis, and myositis.[4] Fluindione is the most prescribed oral anticoagulant in France (it represents nearly 80% of all prescribed anticoagulants).[5] Since 1987, hepatic and renal adverse effects through immunoallergic mechanisms have been well documented with fluindione.[5] Association of DRESS syndrome to fluindione was first desbribed by Sparsa et al.[6] Since then, 36 cases of DRESS syndrome associated to fluindione were reported in a 10 years period.[7] The mean time-to-onset of DRESS syndrome after fluindione treatment was 28.4 days; occurrence was more rapid in patients with previous hypersensitivity syndrome with a chemically related drug. Kidneys and liver were the most frequently involved organs, but no cases of cardiac involvement were observed. Moreover, the co-occurrence of TMA was never reported.
TMAs represent a heterogeneous group of syndromes sharing the classic triad of thrombocytopenia, microangiopathic hemolytic anemia, and organ injury (due to microvascular occlusion and ischemia). Frequently triggered by infections, drug-induced TMA has also been described.[8] Up to now, only 2 cases reporting a potential association between DRESS and thrombotic thrombocytopenic purpura, a type of TMA, have been published.[2,3] ADAMTS13 testing can be useful in differentiating TTP from others TMA, as in our case.[9]
Lymphangiomatosis is a rare disease characterized by lymphatics proliferation in different organs. When occurring in bones (preferentially the humerus, shoulder girdle, pelvis, skull and mandible) it is called Gorham–Stout or “vanishing bone” disease; typical features are massive osteolysis followed by fibrotic replacement. When proliferation occurs in soft tissues (mainly the lung, spleen, mediastinum, and retroperitoneum), the disease is referred to as disseminated (also called generalized or diffuse) lymphangiomatosis.[10] Often considered as 2 separate entities, recent data suggest that they could represent 2 forms of the same process sharing a common pathogenetic mechanism: altered lymphangiogenesis.[11] Circulating levels of lymphangiogenic factors, including the VEGF, seem to be relevant markers of disease activity.[12] Disseminated lymphangiomatosis can be associated with NS, a genetically heterogeneous autosomal dominant disorder characterized by facial dysmorphism, short stature, heart defects and lymphatic vascular disease;[13] mutations in SOS2 are associated with normal stature, as in our patient.[14] Although association between abnormally low VEGF-C levels and NS has been reported,[15] we detected normal VEGF-C levels in our patient.
We hypothesize that NS could have been the backbone of the development of both DRESS and TMA with fluindione as trigger. Immune dysregulation has been described in NS[16] and could have been playing a facilitating role in the development of DRESS syndrome, whose pathogenesis is immune mediated. Coagulation defects, and among them platelet dysfunction, is a typical feature of NS; a conceivable role for such defect in TMA can therefore be claimed.[17] A single case of TTP associated with NS has been previously described.[18] Instead, although the patient was also suffering from Charcot–Marie–Tooth disease, we could not find a link between this disorder with the development of DRESS and TMA.
4. Conclusion
We report the unique presentation of a DRESS syndrome associated with TMA, most likely induced by fluindione, occurring in a patient with a serendipitously revealed NS. This case suggests an intricate interplay between both acquired and congenital defects in the immune and coagulation system contributing to the final clinical picture.
Ethical approval was not necessary since it is the retrospectively description of case of an anonymized patient; moreover, the patient in the study has been treated according to guidelines and no experimental procedures or medications have been used. Patient gave his informed consent.
Author contributions
Conceptualization: Gilbert Habib, Jacques Serratrice, Matteo Coen.
Data curation: Jacques Serratrice, Mickaël Bobot.
Formal analysis: Jacques Serratrice.
Investigation: Clémentine Simon, Jacques Serratrice, Laurent Daniel, Mickaël Bobot.
Supervision: Jacques Serratrice, Laurent Daniel, Matteo Coen.
Validation: Jacques Serratrice, Laurent Daniel, Matteo Coen.
Visualization: Clémentine Simon, Jacques Serratrice, Matteo Coen.
Writing – original draft: Gilbert Habib, Jacques Serratrice, Mickaël Bobot.
Writing – review & editing: Jacques Serratrice, Matteo Coen.
Footnotes
Abbreviations: DRESS = drug reaction with eosinophilia and systemic symptoms, LVEF = left ventricular ejection fraction, NS = Noonan syndrome, TMA = thrombotic microangiopathy, TTP = thrombotic thrombocytopenic purpura, VEGF-C = vascular endothelial growth factor-C.
MB and MC equally contributed to this work.
The authors have no conflicts of interest to disclose.
References
- [1].Spriet S, Banks TA. Drug reaction with eosinophilia and systemic symptoms syndrome. Allergy Asthma Proc 2015;36:501–5. [DOI] [PubMed] [Google Scholar]
- [2].Fleming P, Marik PE. The DRESS syndrome: the great clinical mimicker. Pharmacotherapy 2011;31:332. [DOI] [PubMed] [Google Scholar]
- [3].Sandouk Z, Alirhayim Z, Khoulani D, et al. DRESS syndrome and thrombotic thrombocytopaenic purpura: are they related? BMJ Case Rep 2012;2012:bcr2012007558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mahé I, Bal dit Sollier C, Duru G, et al. Use and monitoring of vitamin K antagonists in everyday medical practice. French results of the international ISAM study of patients with nonvalvular atrial fibrillation. Presse Med 2006;35:1797–803. [DOI] [PubMed] [Google Scholar]
- [5].Cadranel JF, Demontis R, Seddik M, et al. Hepatic and renal toxicity to fluindione (Previscan). Gastroenterol Clin Biol 2008;32:816–8. [DOI] [PubMed] [Google Scholar]
- [6].Sparsa A, Bedane C, Benazahary H, et al. Drug hypersensitive syndrome caused by fluindione. Ann Dermatol Venereol 2001;128:1014–8. [PubMed] [Google Scholar]
- [7].Daveluy A, Milpied B, Barbaud A, et al. Fluindione and drug reaction with eosinophilia and systemic symptoms: an unrecognised adverse effect? Eur J Clin Pharmacol 2012;68:101–5. [DOI] [PubMed] [Google Scholar]
- [8].George JN, Nester CM. Syndromes of thrombotic microangiopathy. N Engl J Med 2014;371:654–66. [DOI] [PubMed] [Google Scholar]
- [9].Shah N, Rutherford C, Matevosyan K, et al. Role of ADAMTS13 in the management of thrombotic microangiopathies including thrombotic thrombocytopenic purpura (TTP). Br J Haematol 2013;163:514–9. [DOI] [PubMed] [Google Scholar]
- [10].Trenor CC, 3rd, Chaudry G. Complex lymphatic anomalies. Semin Pediatr Surg 2014;23:186–90. [DOI] [PubMed] [Google Scholar]
- [11].Aviv RI, McHugh K, Hunt J. Angiomatosis of bone and soft tissue: a spectrum of disease from diffuse lymphangiomatosis to vanishing bone disease in young patients. Clin Radiol 2001;56:184–90. [DOI] [PubMed] [Google Scholar]
- [12].Dellinger MT, Garg N, Olsen BR. Viewpoints on vessels and vanishing bones in Gorham-Stout disease. Bone 2014;63:47–52. [DOI] [PubMed] [Google Scholar]
- [13].Roberts AE, Allanson JE, Tartaglia M, et al. Noonan syndrome. Lancet 2013;26;381:333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Cordeddu V, Yin JC, Gunnarsson C, et al. Activating mutations affecting the Dbl homology domain of SOS2 cause Noonan syndrome. Hum Mutat 2015;36:1080–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fuchs S, Gat-Yablonski G, Shtaif B, et al. Vascular endothelial growth factor (VEGF) levels in short, GH treated children: a distinct pattern of VEGF-C in Noonan syndrome. J Endocrinol Invest 2015;38:399–406. [DOI] [PubMed] [Google Scholar]
- [16].Loddo I, Romano C, Cutrupi MC, et al. Autoimmune liver disease in Noonan syndrome. Eur J Med Genet 2015;58:188–90. [DOI] [PubMed] [Google Scholar]
- [17].Artoni A, Selicorni A, Passamonti SM, et al. Hemostatic abnormalities in Noonan syndrome. Pediatrics 2014;133:e1299–304. [DOI] [PubMed] [Google Scholar]
- [18].Flick JT, Singh AK, Kizer J, et al. Platelet dysfunction in Noonan's syndrome. A case with a platelet cyclooxygenase-like deficiency and chronic idiopathic thrombocytopenic purpura. Am J Clin Pathol 1991;95:739–42. [DOI] [PubMed] [Google Scholar]