

Abstract
Tetracarboxylate pyrroline nitroxides undergo very fast reduction with ascorbate/GSH, with second order rate constants that are five orders of magnitude greater than those for gem-diethyl pyrroline nitroxides. For tetracarboxylate nitroxides, the electrochemical reduction potentials, measured by square wave voltammetry, are much less negative (by about 0.8 V), compared to the corresponding gem-diethyl nitroxides, while the oxidation potentials become more positive (by about 0.7 V). Electrochemical potentials correlate well via simple regressions with field/inductive parameters such as Swain/Lupton F-parameters (and/or Charton σI-parameters). Rates of reduction with ascorbate/GSH similarly correlate well for four pyrroline nitroxides, except for the slowest reducing gem-diethyl nitroxide. These results suggest that the electron withdrawing groups adjacent to the nitroxide moiety have a strong accelerating impact on the reduction rates, and thus they are not suitable for the design of hydrophilic nitroxides for in vivo applications.
Keywords: nitroxide, electron paramagnetic resonance, electrochemistry, voltammetry, Hammett correlation
Introduction
Nitroxides are among the very few stable organic radicals that have a wide range of applications in chemistry, biochemistry, biophysics and biomedicine [1-6]. In recent years, a significant effort has been made in the development of biocompatible nitroxides for in vivo biophysical and biomedical applications [7-13]. Nitroxides that are highly water soluble (hydrophilic) and resistant to in vivo reduction are of particular interest for in vivo spin-labeling study of biomolecules and the development of contrast agents for magnetic resonance imaging (MRI).
Most of the typical nitroxides undergo fast in vivo reduction to diamagnetic hydroxylamines by antioxidants, such as ascorbate (vitamin C) or enzymatic systems, which hinder their applications in biomedicine and biophysics [14-16]. 5-Membered ring nitroxides with gem-diethyl substituent groups, such as pyrrolidine nitroxide 1 (Figure 1), were found to undergo reduction with ascorbate at rates that are at least one order of magnitude slower than the corresponding gem-dimethyl nitroxides such as 2 [9,14]; similar findings were reported for 1 in cell extracts and in live oocyte cells [11]. Notably, gem-diethyl pyrroline nitroxides, such as 3, are reduced by ascorbate at only slightly faster (up to 20%) rates than 1 [10]. Similar relationships between the rate of reduction and the structure of the gem-diethyl vs. gem-dimethyl structure motifs were found in other nitroxides, though their rates of reduction were considerably faster than that of 1 [7,12,17,18].
Figure 1.
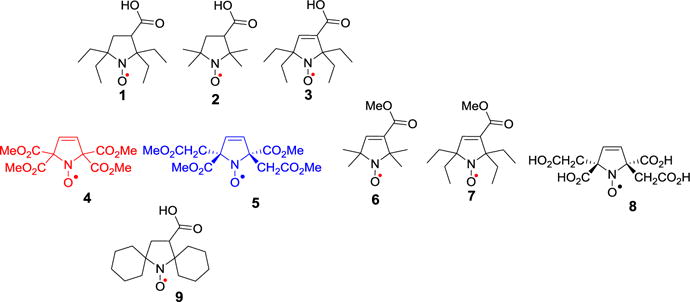
Pyrrolidine nitroxide 1, 2, and 9, and pyrroline nitroxides 3 – 8.
We recently synthesized hydrophilic pyrroline nitroxides 4 and 5. We found that, when immobilized in trehalose/sucrose glass, these nitroxides possessed relatively long electron spin phase memory times (Tm) and short spin–lattice relaxation times (T1) at room temperature [18]. Therefore, spin labels derived from 4 and 5 should provide highly promising tools for determination of distances by DEER (or DQC) in doubly labelled biomolecules at room temperature [19-21].
Our research team also successfully prepared the i.v. injectable organic radical contrast agents (ORCA) for MRI. The agents possess relatively long in vivo lifetimes, high 1H water relaxivities (r1 or r2), and provide contrast-enhanced high resolution MRI in mice for over 1 – 24 hours [5,6,22]. The molecular design of ORCA was based on spirocyclohexyl nitroxide radicals 9 and polyethylene glycol (PEG) chains conjugated to a generation 4 polypropylenimine (PPI) dendrimer scaffold, in which the strategic PEGylation provided both immobilization and increased water access to the paramagnetic nitroxides [5]. The branched-bottlebrush macromolecular structures have also successfully been implemented in the design of ORCA [6,22]. Hydrophilic nitroxides such as 4 and 5 would provide a promising avenue to further increase of r1 (and r2) in the ORCA. However, the crucial prerequisite for their usefulness as MRI contrast agents for clinical imaging is the slow rate of reduction with ascorbate.
Relative rates of reduction of nitroxides have been long considered to correlate with steric factors [7-12]. State-of-the-art quantitative structure activity relationships (QSAR) approaches that account for steric factors rely on the Sterimol parameters [23], which describe the size (in Å) of a substituent using three different measurements: (1) L, the maximum length measured along the axis of the bond between the substituent and the parent molecule (2) B1, the minimum width measured perpendicular to the bond axis used to measure L, and (3) B5, the maximum width measured perpendicular to the same bond axis [23]. The Sterimol parameters were very successful in correlating asymmetric induction in various enantioselective organic reactions, especially where the one-value per substituent classic Taft/Charton steric parameters [24-26] have failed to provide adequate correlation [27,28].
The Sterimol parameters for carboxylate (COOMe), either ethyl (Et) and methyl (Me) groups are shown in Table 1. The parameters indicate the decreasing trend of the steric factors, COOMe > Et > COOH > Me. The comparison between gem-diethyl and gem-dimethyl nitroxides, in which the Et is clearly larger than Me, would suggest correlation between the steric factors and the rate of reduction [9,12]. Following this trend, the gem-dicarboxylate nitroxide 4, in which the COOMe is sterically larger group than either Et or Me, should be reduced at the slowest rate.
Table 1.
| Substituent | B1 | B5 | L |
|---|---|---|---|
| Me | 1.70 | 2.19 | 3.07 |
| COOH | 1.70 | 2.76 | 4.14 |
| Et | 1.71 | 3.33 | 4.33 |
| COOMe | 1.80 | 3.52 | 5.00 |
Values of B1, B5, and L are based on 3-substiuted benzene derivatives
The effect of electron withdrawing/donating substituent groups on the rate of reduction of nitroxides has been reported [12,16,18]. For gem-substituted imidazoline and imidazolidine nitroxides, the ethyl groups, compared to the methyl groups, in the 5-membered ring structures leads to a decrease in the rate of reduction with ascorbate. In the other study, it was shown that the 6-membered piperidine nitroxides (mostly spirocyclic), which possess inherently faster rates of reduction than the corresponding 5-membered ring nitroxides, electron withdrawing groups further accelerate the rate of reduction with ascorbate [18].
EPR spectroscopy is the common technique employed in the kinetic studies of nitroxide reductions by ascorbate/GSH, which provide useful information about their suitability for in vivo applications. Meanwhile, electrochemical studies of nitroxides provide an additional insight into the structure-activity relationships that can be correlated with the EPR kinetic studies.
Here we report a study on the reduction kinetics of nitroxides 4 – 8 by ascorbate/GSH by EPR and their electrochemistry. Nitroxides 4 – 7 are examined by cyclic and square wave voltammetries and are found to provide a wide ranges of reduction (0.84 V) and oxidation (0.67 V) potentials. We find good correlations of reduction rates and electrochemical potentials with field/inductive parameters Swain/Lupton F-parameters (and/or Charton σI- parameters)[29]; that is, steric effects play negligible role in the reduction rates of nitroxides 4 – 6, and 8.
Materials and methods
Nitroxides 4 – 7 were prepared according to published methods [10,18,30,31]. Nitroxide 8 was prepared from nitroxide 5 via hydrolysis of the ester groups to carboxylate groups as described in the “EPR spectroscopy” section.
EPR spectroscopy
CW EPR spectra were obtained using X-band Bruker EMX instrument at 295 K. Temperature was controlled with a nitrogen flow system. Parameters for spectra in Figure 2 were as follows: microwave power, 2.046 mW (20 dB); modulation amplitude 0.02 mT; conversion time 40.96 ms; time constant 10.24 ms; resolution in X, 2k points. Spectral simulations were carried out using Symphonia software.
Figure 2.
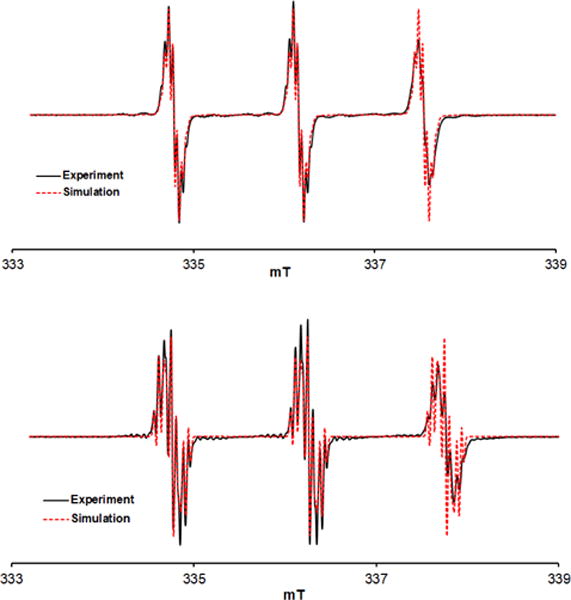
Top plot: EPR (v = 9.4373 GHz) spectrum of 0.1 mM tetraester nitroxide 5 in 25 mM phosphate buffer (pH = 7.4). Simulation: g = 2.0058, A(14N) = 38.70 MHz (n = 1), A(1H) = 1.30 MHz (n = 4), A(1H) = 1.05 MHz (n = 2), A(1H) = 1.087 MHz (n = 2), LW (Lorentzian) = 0.020 mT. Bottom plot: EPR (v = 9.4393 GHz) spectrum of 0.1 mM tetracarboxylic acid nitroxide 8 in 25 mM phosphate buffer (pH = 7.6). Simulation: g = 2.00563, A(14N) = 42.08 MHz (n = 1), A(1H) = 2.26 MHz (n = 2), A(1H) = 1.65 MHz (n = 2), A(1H) = 1.26 MHz (n = 2), LW (Lorentzian) = 0.028 mT.
The results of kinetic measurements are summarized in Table 2. The ascorbate solution was made with ascorbic acid, diethylenetriaminepentaacetic acid (DTPA, 0.1 mM), sodium hydroxide, and sodium phosphates (<30 ppm transition metals) at pH 7.4, measured with pH/ion analyzer. Phosphate buffer was made with sodium phosphates and DTPA (0.1 mM) at pH 7.4 and used to make nitroxide solutions. Standard linear regression fit to -ln[nitroxide] vs t was employed, to obtain pseudo-first order rate constants, k′ × 104 (s−1), in Table 2.
Table 2.
Detailed summary of second-order rate constants, k (M−1s−1) for initial rates of reduction of 0.005 – 1.0 mM nitroxides with ascorbate (Asc) and glutathione (GSH).
| Compd | Run No. | Data used | Nitrox. Conc. (mM) |
Asc Conc. (mM) |
GSH Conc. (mM) |
k′×
104 (s−1) |
R2 |
k (M−1s−1) |
k mean ± stddev (M−1s−1) |
Time (s) | Radical left (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 4 | 1 | IPH | 0.005 | 0.05 | 0.13 | 106.64 | 0.9862 | 213.3 | 245 ± 31 (IPH) | 230 | 6 |
| PH | 112.74 | 0.9947 | 225.5 | 246 ± 23 (PH) | 230 | 5 | |||||
| 2 | IPH | 0.005 | 0.05 | 0.13 | 116.77 | 0.9862 | 233.5 | 230 | 4 | ||
| PH | 115.91 | 0.9945 | 231.8 | 230 | 4 | ||||||
| 3 | IPH | 0.005 | 0.05 | 0.13 | 143.50 | 0.9886 | 287.0 | 184 | 5 | ||
| PH | 137.66 | 0.9897 | 275.3 | 184 | 4 | ||||||
| 4 | IPH | 0.005 | 0.05 | 0.00 | 123.65 | 0.9950 | 247.3 | 184 | 5 | ||
| PH | 126.54 | 0.9945 | 253.1 | 184 | 5 | ||||||
| 5a | 1 | IPH | 0.005 | 0.05 | 0.13 | 59.104 | 0.9980 | 118.2 | 113.5 ± 5.2 (IPH) | 510 | 5 |
| PH | 64.987 | 0.9938 | 130.0 | 123.4 ± 6.8 (PH) | 510 | 4 | |||||
| 2 | IPH | 0.005 | 0.05 | 0.13 | 57.261 | 0.9931 | 114.5 | 510 | 6 | ||
| PH | 61.808 | 0.9970 | 123.6 | 510 | 5 | ||||||
| 3 | IPH | 53.941 | 0.9986 | 107.9 | 525 | 6 | |||||
| PH | 0.005 | 0.05 | 0.13 | 58.238 | 0.9973 | 116.5 | 525 | 5 | |||
| 6 | 1 | IPH | 1.0 | 20 | 25 | 45.680 | 0.9904 | 0.2284 | 0.2348 ± 0.0093 (IPH) | 340 | 24 |
| PH | 44.740 | 0.9897 | 0.2237 | 0.2308 ± 0.0091 (PH) | 340 | 24 | |||||
| 2 | IPH | 1.0 | 20 | 25 | 49.077 | 0.9907 | 0.2452 | 330 | 24 | ||
| PH | 48.216 | 0.9906 | 0.2411 | 330 | 24 | ||||||
| 3 | IPH | 1.0 | 20 | 25 | 46.107 | 0.9902 | 0.2305 | 350 | 26 | ||
| PH | 45.543 | 0.9907 | 0.2277 | 350 | 27 | ||||||
| 7 | 1 | IPH | 0.7 | 14 | 17.5 | 0.1981 | 0.7079 | 0.001409 | 0.00144 ± 0.00017 (IPH) | 180 | >99 |
| PH | 0.1207 | 0.9798 | 0.0008584 | 0.00112 ± 0.00032 (PH) | 180 | >99 | |||||
| 2 | IPH | 0.7 | 14 | 17.5 | 0.1769 | 0.7118 | 0.001258 | 200 | >99 | ||
| PH | 0.2067 | 0.8068 | 0.001470 | 270 | >99 | ||||||
| 3 | IPH | 0.7 | 14 | 17.5 | 0.2040 | 0.7840 | 0.001451 | 230 | >99 | ||
| PH | 0.2016 | 0.8524 | 0.001434 | 230 | >99 | ||||||
| 4 | IPH | 0.7 | 70 | 17.6 | 1.1468 | 0.9492 | 0.001634 | 210 | 99 | ||
| PH | 0.8302 | 0.8499 | 0.001183 | 230 | 99 | ||||||
| 5 | IPH | 0.7 | 70 | 17.6 | 0.8912 | 0.9686 | 0.001270 | 230 | 99 | ||
| PH | 0.4603 | 0.9412 | 0.0006557 | 250 | 99 | ||||||
| 6 | IPH | 0.7 | 70 | 17.6 | 1.1480 | 0.9843 | 0.001635 | 290 | 98 | ||
| PH | 0.7980 | 0.9765 | 0.001137 | 290 | 98 | ||||||
| 8 | 1 | IPH | 0.060 | 0.600 | 0.600 | 14.788 | 0.9963 | 2.4647 | 2.04 ± 0.58 (IPH) | 570 | 50 |
| PH | 0.060 | 0.600 | 0.600 | 16.078 | 0.9942 | 2.6796 | 2.05 ± 0.65 (PH) | 570 | 50 | ||
| 2 | IPH | 0.054 | 0.540 | 0.540 | 6.7092 | 0.9992 | 1.2424 | 480 | 79 | ||
| PH | 0.054 | 0.540 | 0.540 | 6.4416 | 0.9974 | 1.1929 | 480 | 80 | |||
| 3 | IPH | 0.054 | 0.540 | 0.540 | 8.9329 | 0.9997 | 1.6542 | 470 | 73 | ||
| PH | 0.054 | 0.540 | 0.540 | 8.7206 | 0.9991 | 1.6149 | 470 | 74 | |||
| 4 | IPH | 0.049 | 0.489 | 0.489 | 12.996 | 0.9998 | 2.6576 | 470 | 63 | ||
| PH | 0.049 | 0.489 | 0.489 | 12.858 | 0.9995 | 2.6294 | 470 | 63 | |||
| 5 | IPH | 0.054 | 0.541 | 0.541 | 11.670 | 0.9998 | 2.1590 | 480 | 66 | ||
| PH | 0.054 | 0.541 | 0.541 | 11.594 | 0.9998 | 2.1450 | 480 | 66 | |||
| 1b | 1 | IPH | 1 | 20 | 5 | 0.2870 | 0.9937 | 14.35d | 0.00126 ± 0.00030 | 660 | 98 |
| PH | 0.2195 | 0.9628 | 10.98d | 0.00108 ± 0.00015 | 660 | 99 | |||||
| 2 | IPH | 1 | 20 | 5 | 0.2372 | 0.9667 | 11.86d | 360 | 99.5 | ||
| PH | 0.2277 | 0.8665 | 11.39d | 330 | 99.5 | ||||||
| 3 | IPH | 1 | 20 | 5 | 0.2329 | 0.9832 | 11.65d | 360 | 99 | ||
| PH | 0.1996 | 0.9389 | 9.980d | 360 | 99 | ||||||
| 1 | IPH | 0.2 | 4.0 | 5 | 2.435 | 0.9997 | 608.8d | 0.0608 ± 0.0004 (IPH) | 480 | 89 | |
| 2c | PH | 2.361 | 0.9990 | 590.3d | 0.0603 ± 0.0025 (PH) | 480 | 89 | ||||
| (Proxyl) | 2 | IPH | 0.2 | 4.0 | 5 | 2.438 | 0.9997 | 609.6d | 480 | 89 | |
| PH | 2.410 | 0.9996 | 602.4d | 540 | 89 | ||||||
| 3 | IPH | 0.2 | 4.0 | 5 | 2.423 | 0.9998 | 605.6d | 600 | 86 | ||
| PH | 2.461 | 0.9996 | 615.2d | 480 | 86 | ||||||
| 3c | 1 | IPH | 1 | 20 | 25 | 0.2750 | 0.9950 | 13.75d | 0.00140 ± 0.00002 (IPH) | 970 | 97 |
| PH | 0.2414 | 0.9922 | 12.07d | 0.00122 ± 0.00003 (PH) | 970 | 97 | |||||
| 2 | IPH | 1 | 20 | 25 | 0.2814 | 0.9883 | 14.07d | 980 | 97 | ||
| PH | 0.2465 | 0.9919 | 12.33d | 800 | 97 | ||||||
| 3 | IPH | 1 | 20 | 25 | 0.2838 | 0.9940 | 14.19d | 980 | 97 | ||
| PH | 0.2410 | 0.9950 | 12.05d | 850 | 97 |
For fast reducing nitroxides 4 and 5, typical experiments were carried out on 0.005 mM nitroxides with 0.05 mM (10-fold molar excess) ascorbate and 0.065 mM GSH in pH 7.4 20 mM phosphate buffer. Prior to a typical experiment, solutions of nitroxide and ascorbate/GSH were combined in equal portions, vortexed for 6 seconds, and then the resultant mixture was drawn into a 2-mm OD EPR-tube. The peak height (PH, Table 2) and the integrated peak height (IPH, Table 2) of the low-field line of the triplet were measured as a function of time. Microwave power was kept under 20 mW and temperature was controlled at 295 K with nitrogen flow system.
For slow reducing nitroxides 6 and 7, typical experiments were carried out on 0.7 – 1.0 mM nitroxides with 14 – 70 mM (20-100-fold molar excess) ascorbate and 17.5 – 25 mM GSH in pH 7.4 40 mM phosphate buffer, using similar procedures as outlined in the preceding paragraph. Maximum solubility of gem-diethyl nitroxide 7 in 40 mM PBS was only about 1 mM.
Typical procedure for intermediate reducing nitroxide 8 was as follows. Solution of lithium hydroxide (15.5 mg, 0.368 mmol, 25 equiv) in water (0.5 mL) was added to nitroxide 5 (5.07 mg, 0.0147 mmol) suspended in water (0.5 mL). After a brief sonication and stirring for 1 – 2 h at room temperature, a yellow and homogeneous reaction mixture was diluted with a 25 mM phosphate buffer (pH 7.4) to obtain 0.108 mM solution of 8 (pH 7.6) as measured by EPR spin counting (Figure 2). Prior to a kinetic experiment, solution of 8 and solution of ascorbic acid (1.08 mM)/GSH (1.08 mM) in 25 mM phosphate buffer (pH 7.4) were combined in equal portions, vortexed for 6 seconds, and then the resultant mixture (pH ~7.3) was drawn into a quartz capillary (0.9-mm ID, 1.1-mm OD). The data were obtained as described in the preceding paragraphs.
The kinetic studies in oxygen-free environment were carried out on 0.005 mM nitroxide 5 with 0.05 mM (10-fold molar excess) ascorbate and 0.065 mM GSH (13-fold molar excess) in pH 7.4 25 mM degassed phosphate buffer. The solution of nitroxide 5 was filtered and bubbled with nitrogen gas for at least 15 min; spin concentration of the solution was measured by EPR spectroscopy. Similarly, the phosphate buffer and the ascorbate/GSH solution mixture (0.1 mM/0.13 mM) were bubbled with nitrogen gas for at least 15 min. After that, all solutions and glassware were transferred to a nitrogen glove bag, and then the nitroxide solution was diluted with the phosphate buffer to 0.01 mM. The solutions of nitroxide (0.01 mM) and ascorbate/GSH (0.1 mM/0.13 mM) were combined in equal portions, vortexed for 6 seconds, and then the resultant mixture was drawn into a quartz capillary (0.9-mm ID, 1.1-mm OD), which was sealed by CRITOSEAL and placed into a 4-mm EPR tube. The tube was capped and sealed with parafilm before removal from the glove bag for the EPR measurements.
Electrochemical studies
The voltammetry experiments were carried out in a glove bag under argon gas atmosphere [32]. (The supply gas was a commercial ultra-high purity argon, certified to contain <1 ppm of O2 and <1 ppm of H2O.) The custom-made electrochemical cell, all solid reagents, syringes, needles, etc. were extensively evacuated in Schlenk vessels (typical pressure ≤ 1 mTorr, temperature 20–70 °C), before placing into the glove bag. Acetonitrile (MeCN) was obtained doubly distilled from calcium hydride under high purity argon gas, and stored in the absence of light in a Schlenk vessel on a vacuum line; just prior to the use, the solvent was vacuum transferred as needed. A commercial potentiostat/galvanostat was used. Three electrodes were employed: quasi-reference (Ag-wire), counter (Pt-foil), and working (100-μm Pt-disk). The concentration of an electroactive solute was about 1 mM and concentration of the tetrabutylammonium hexafluorophosphate supporting electrolyte ([n-Bu4N]+[PF6]−) was about 0.1 M. The solution volume was about 2.4 mL. A set of background data was obtained from series of cyclic (and/or square wave) voltammograms, with potential increments of 2 – 3 mV. Then, a solution of nitroxide in supporting electrolyte was added and a series of cyclic (and/or square wave) voltammograms) was recorded. After that, a small amount (0.1 – 0.15 mL) of solution of ferrocene (0.7 – 1.0 mg of Cp2Fe, loaded to a Schlenk vessel in a glovebox under an argon atmosphere in the supporting electrolyte (ca. 0.4 mL), was added to the cell, to provide reference potentials (+0.40 V vs. SCE for Cp2Fe/Cp2Fe+ in MeCN) [33]. Cyclic voltammograms with the scanning rates in the 50–500 mV/s or 50–2000 mV/s range and square wave voltammograms (frequency 2 – 20 Hz, pulse height 25 mV) were obtained.
Results and discussion
Tetracarboxylic acid nitroxide 8 was prepared by lithium hydroxide-catalyzed hydrolysis of ester groups in nitroxide 5. As expected for a nitroxide with more electron donating substituents, EPR spectrum for 8 in 25 mM phosphate buffer showed significantly smaller g-value and larger A(14N) (as well as A(1H)) values, compared to those for nitroxide 5 (Figure 2).
In the EPR kinetic studies, rates of reduction for pyrroline nitroxides 4 – 8 are determined under pseudo-first-order conditions using a 10-20 fold excess of ascorbate in pH 7.4 PBS buffer at 295 K. For comparison, the rates of reduction of for nitroxides 1 – 3 are measured under similar conditions [9-11,14]. Second-order rate constants, k, are obtained by monitoring the decay of the low-field EPR peak height of nitroxides at 295 K (Figure 3, top panels and Tables 2 and 3). Also, decays of EPR single integrated peak height are examined and found to produce similar values of k for most nitroxides (Table 2).
Figure 3.
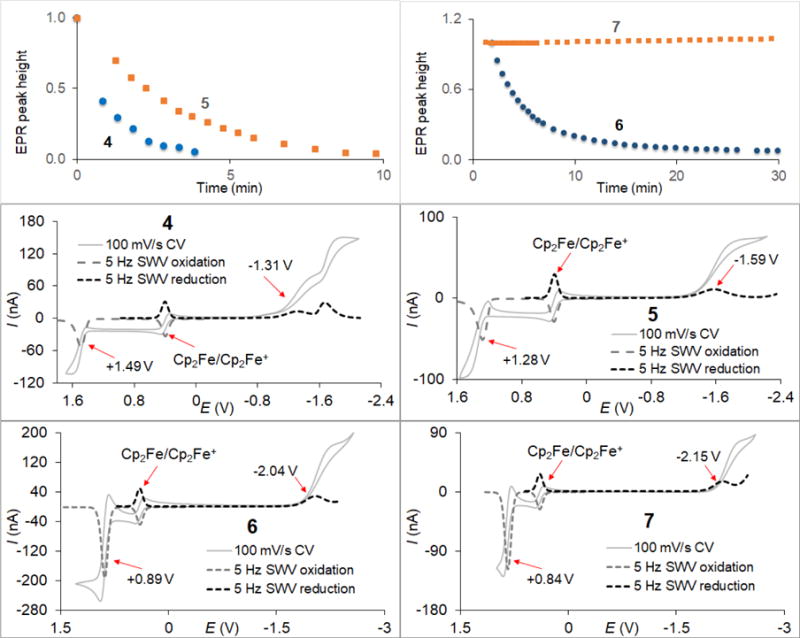
Left upper panel: reduction profiles for 0.005 mM nitroxides 4 and 5 with 0.05 mM ascorbate and 0.13 mM GSH in 25 mM PBS pH 7.4 at 295 K; right upper panel: reduction profiles for 0.7 mM nitroxides 6 and 7 with 14 mM ascorbate and 17.5 mM GSH in 40 mM PBS pH 7.4 at 295 K; middle panels: CV and SWV plots for 4 and 5; bottom panels: CV and SWV plots for 6 and 7. CV had scan rates of 100 mV/s and scan increments of 3 mV; SWV had frequencies of 5 Hz and scan increments of either 2 (oxidation) or 3 (reduction) mV. CV potential scans start at about −0.5 V, then run toward the positive (oxidation) potentials, then go to the negative potentials (reduction), and finally return to the starting point.
Table 3.
Summary of parameters of kinetics nitroxide reduction with ascorbate, electrochemical potentials, and field/inductive descriptors.
| Nitroxide |
ka (M−1s−1) |
Eoreda,b (V) |
Eooxida,b (V) |
F or σI |
|---|---|---|---|---|
| 4 | 246 ± 23 | −1.35 ± 0.07c | 1.51 ± 0.03 | 0.34 × 4 |
| 5 | 123 ± 7 | −1.55 ± 0.06 | 1.31 ± 0.04 | (0.34+0.19) × 2 |
| 6 | 0.231 ± 0.009 | −2.03 ± 0.07 | 0.885 ± 0.005 | 0.01 × 4 |
| 7 | 0.00112 ± 0.00032 | −2.19 ± 0.04 | 0.851 ± 0.005 | 0.00 × 4 |
| 8 | 2.05 ± 0.65 | - | - | (0.19–0.10) × 2 |
mean ± stddev
potentials vs saturated calomel electrode (SCE), obtained from square wave voltammetry (SWV).
Another intense peak at Eored = −1.68 ± 0.04 V was observed (Figure 3).
In the electrochemical studies of 4 – 7, the reduction (Eored) and oxidation (Eooxid) potentials span the wide ranges from −2.19 to −1.35 V and from +0.85 to +1.51, respectively (Table 3). The reduction and oxidation potentials obtained from cyclic voltammetry (CV) and square wave voltammetry (SWV) are similar. In Table 3, potentials from SWV are listed only [21], since they typically possess smaller standard deviations. Reduction waves in CV and broadened peaks in SWV correspond to nonreversible electrode processes for all studied nitroxides [22]. Oxidation waves in CV and sharp peaks in SWV correspond to reversible electrode processes for 6 and 7 but they are nonreversible for 4 and 5 (Figure 3).
It was as unexpected that 4 and 5 undergo very fast reduction with ascorbate/GSH, with second order rate constants that are five orders of magnitude greater than those for gem-diethyl pyrroline nitroxides (Tables 2 and 3). This result suggests that, beside the steric factors, there are other significant contributions in the reduction kinetics of nitroxides.
To have a better understanding of these results, we consider the field/inductive parameters, such as Swain/Lupton F-parameters and/or Charton σI-parameters [29]. We note that Kirilyuk, Bagryanskaya, and their colleagues used multiple regression to correlate rates of reduction and reduction potentials of methyl/ethyl substituted imidazoline and imidazolidine nitroxides with both σI-parameters and modified Taft steric constant Es [12]. It was noted that the reduction potentials correlated much more strongly with σI than Es [12]. As shown in Table 3, Figure 4, and Equations 1 – 3, the field/inductive parameters provide good correlations for the rates of reduction of – 6 and 8, and electrochemical potentials of 4 – 7; that is, for these series of nitroxides in this studies, there is no need to invoke correlation with steric parameters – simple regressions do suffice. Notably, inclusion of the slowest reducing nitroxide 7 in the correlation for the rate of reduction lowers adjusted R2 from 0.94 to 0.69 and increases significance F from 0.022 to 0.050. There are many precedencies in the literature for correlations between electrochemical potentials and Hammett σp (and σm) or field/inductive parameters σI (Taft-ortho parameters) [36-39].
Figure 4.
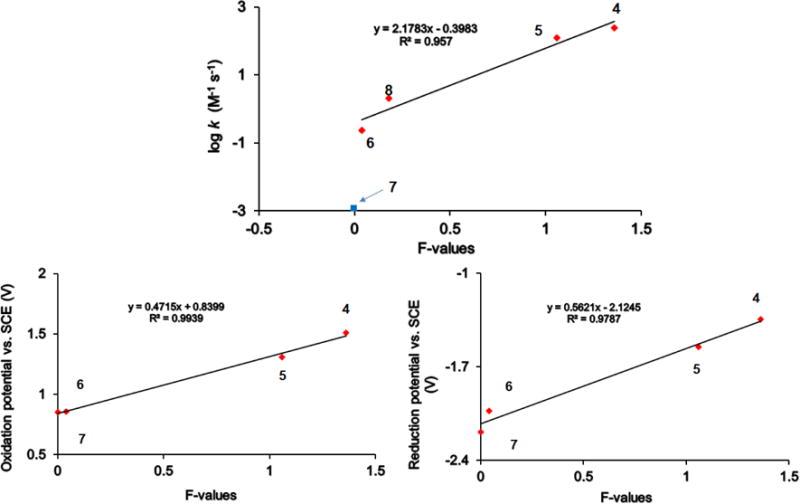
Correlations of the rates of reduction (upper panel and eq. 1) for nitroxides 4 – 6 and 8 and electrochemical potentials (lower panels, oxidation, eq. 2; reduction, eg. 3) for nitroxides 4 – 7 with the field/inductive parameters (F and/or σI).
| (1) |
| (2) |
| (3) |
In conclusion, we anticipate that polar substituents with field/inductive parameters very close to zero or less than zero will provide hydrophilic nitroxides that are reduced at relatively slow rates. Because a CH2CH2OCH3 group has the Charton σI = 0.00 [29], groups such as CH2CH2O-PEG, where PEG = polyethylene glycol, should provide suitable gem-substituents for hydrophilic nitroxides that are resistant to reduction. In addition, these substituents will be sterically larger than Et groups.
Acknowledgments
Funding information
We thank the National Institutes of Health for support of this work through Grants NIBIB R01EB019950-02, NIGMS R01GM124310-01, and NIGMS U54GM087519-06.
Footnotes
Disclosure statement
We declare no conflict of interest between the authors or with any institution in relation to the contents of this article.
References
- 1.Tebben L, Studer A. Nitroxides: applications in synthesis and in polymer chemistry. Angew Chem Int Ed. 2011;50:5034–5068. doi: 10.1002/anie.201002547. [DOI] [PubMed] [Google Scholar]
- 2.Krishna MC, Russo A, Mitchell JB, Goldstein S, Dafni H, Samuni A. Do nitroxide antioxidants act as scavengers of O2-. or as SOD mimics? J Biol Chem. 1996;271:26026–26031. doi: 10.1074/jbc.271.42.26026. [DOI] [PubMed] [Google Scholar]
- 3.Hubbell WL, Lopez CJ, Altenbach C, Yang Z. Technological advances in site-directed spin labeling of proteins. Curr Opin Struct Biol. 2013;23:725–733. doi: 10.1016/j.sbi.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weaver J, Burks SR, Liu KJ, Kao JPY, Rosen GM. In vivo EPR oximetry using an isotopically-substituted nitroxide: Potential for quantitative measurement of tissue oxygen. J Magn Reson. 2016;271:68–74. doi: 10.1016/j.jmr.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rajca A, Wang Y, Boska M, Paletta JT, Olankitwanit A, Swanson MA, Mitchell DG, Eaton SS, Eaton GR, Rajca S. Organic Radical Contrast Agents for Magnetic Resonance Imaging. J Am Chem Soc. 2012;134:15724–15727. doi: 10.1021/ja3079829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sowers MA, McCombs JR, Wang Y, Paletta JT, Morton SW, Dreaden EC, Boska MD, Ottaviani MF, Hammond PT, Rajca A, Johnson JA. Redox-responsive branched-bottlebrush polymers for in vivo MRI and fluorescence imaging. Nature Commun. 2014;5:5460. doi: 10.1038/ncomms6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kirilyuk IA, Bobko AA, Grigor’ev IA, Khramtsov VV. Synthesis of the tetraethyl substituted pH-sensitive nitroxides of imidazole series with enhanced stability towards reduction. Org Biomol Chem. 2004;2:1025–1030. doi: 10.1039/b400252k. [DOI] [PubMed] [Google Scholar]
- 8.Morozov DA, Kirilyuk IA, Komarov DA, Goti A, Bagryanskaya IY, Kuratieva NV, Grigor’ev IA. Synthesis of a Chiral C2‑Symmetric Sterically Hindered Pyrrolidine Nitroxide Radical via Combined Iterative Nucleophilic Additions and Intramolecular 1,3-Dipolar Cycloadditions to Cyclic Nitrones. J Org Chem. 2012;77:10688–10698. doi: 10.1021/jo3019158. [DOI] [PubMed] [Google Scholar]
- 9.Paletta JT, Pink M, Foley B, Rajca S, Rajca A. Synthesis and Reduction Kinetics of Sterically Shielded Pyrrolidine Nitroxides. Org Lett. 2012;14:5322–5325. doi: 10.1021/ol302506f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Y, Paletta JT, Berg K, Reinhart E, Rajca S, Rajca A. Synthesis of Unnatural Amino Acids Functionalized with Sterically Shielded Pyrroline Nitroxides. Org Lett. 2014;16:5298–5300. doi: 10.1021/ol502449r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jagtap AP, Krstic I, Kunjir NC, Hänsel R, Prisner TF, Sigurdson ST. Sterically shielded spin labels for in-cell EPR spectroscopy: Analysis of stability in reducing environment. Free Radical Res. 2015;49:78–85. doi: 10.3109/10715762.2014.979409. [DOI] [PubMed] [Google Scholar]
- 12.Kirilyuk IA, Bobko AA, Semenov SV, Komarov DA, Irtegova IG, Grigor’ev IA, Bagryanskaya EG. The effect of sterical shielding on the redox properties of imidazoline and imidazolidine nitroxides. J Org Chem. 2015;80:9118–9125. doi: 10.1021/acs.joc.5b01494. [DOI] [PubMed] [Google Scholar]
- 13.Roser P, Schmidt MJ, Drescher M, Summerer D. Site-directed spin labeling of proteins for distance measurements in vitro and in cells. Org Biomol Chem. 2016;14:5468–5476. doi: 10.1039/c6ob00473c. [DOI] [PubMed] [Google Scholar]
- 14.Vianello F, Momo F, Scarpa M, Rigo A. Kinetics of nitroxide spin label removal in biological systems: An in vitro and in vivo ESR study. Magn Reson Imaging. 1995;13:219–226. doi: 10.1016/0730-725x(94)00121-i. [DOI] [PubMed] [Google Scholar]
- 15.Davis RM, Sowers AL, DeGraff W, Bernardo M, Thetford A, Krishna MC, Mitchell JB. Magnetic resonance imaging of organic contrast agents in mice: capturing the whole-body redox landscape. Free Radical Biol Med. 2011;51:780–790. doi: 10.1016/j.freeradbiomed.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morris S, Sosnovsky G, Hui B, Huber CO, Rao NU, Swartz HM. Chemical and electrochemical reduction rates of cyclic nitroxides (nitroxyls) J Pharm Sci. 1991;80:149–152. doi: 10.1002/jps.2600800212. [DOI] [PubMed] [Google Scholar]
- 17.Marx L, Chiarelli R, Guiberteau T, Rassat A. A comparative study of the reduction by ascorbate of 1,1,3,3-tetraethylisoindolin-2-yloxyl and of 1,1,3,3-tetramethylisoindolin-2-yloxyl. J Chem Soc Perkin Trans 1. 2000;8:1181–1182. [Google Scholar]
- 18.Yamasaki T, Mito F, Ito Y, Pandian S, Kinoshita Y, Nakano K, Murugesan R, Sakai K, Utsumi H, Yamada K. Structure−Reactivity Relationship of Piperidine Nitroxide: Electrochemical, ESR and Computational Studies. J Org Chem. 2011;76:435–440. doi: 10.1021/jo101961m. [DOI] [PubMed] [Google Scholar]
- 19.Huang S, Paletta JT, Elajaili H, Huber K, Pink M, Rajca S, Eaton GR, Eaton SS, Rajca A. Synthesis and electron spin relaxation of tetra-carboxylate pyrroline nitroxides. J Org Chem. 2017;82:1538–1544. doi: 10.1021/acs.joc.6b02737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meyer V, Swanson MA, Clouston LJ, Boratynski PJ, Stein RA, McHaourab HS, Rajca A, Eaton SS, Eaton GR. Room-temperature distance measurements of immobilized spin-labeled protein by DEER/PELDOR. Biophys J. 2015;108:1213–1219. doi: 10.1016/j.bpj.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krumkacheva O, Bagryanskaya E. EPR-based distance measurements at ambient temperature. J Magn Reson. 2017;280:117–126. doi: 10.1016/j.jmr.2017.02.015. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen HAT, Chen Q, Paletta JT, Harvey P, Jiang Y, Zhang H, Boska MD, Ottaviani MF, Jasanoff AP, Rajca A, Johnson JA. Nitroxide-based macromolecular contrast agents with unprecedented transverse relaxivity and stability for magnetic resonance imaging of tumors. ACS Central Science. 2017;3:800–811. doi: 10.1021/acscentsci.7b00253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verloop A. Drug Design. III Academic Press; New York: 1976. [Google Scholar]
- 24.Taft RW., Jr Polar and steric substituent constants for aliphatic and o-benzoate groups from rates of esterification and hydrolysis of esters. J Am Chem Soc. 1952;74:3120–3128. [Google Scholar]
- 25.Taft RW., Jr Linear steric energy relationships. J Am Chem Soc. 1953;75:4538–4539. [Google Scholar]
- 26.Charton M. Steric effects. 7. Additional V constants. J Org Chem. 1976;41:2217–2220. [Google Scholar]
- 27.Harper KC, Bess EN, Sigman MS. Multidimensional steric parameters in the analysis of asymmetric catalytic reactions. Nature Chem. 2012;4:366–374. doi: 10.1038/nchem.1297. [DOI] [PubMed] [Google Scholar]
- 28.Santiago CB, Milo A, Sigman MS. Developing a Modern Approach To Account for Steric Effects in Hammett-Type Correlations. J Am Chem Soc. 2016;138:13424–13430. doi: 10.1021/jacs.6b08799. [DOI] [PubMed] [Google Scholar]
- 29.Hansch C, Leo A, Taft RW. A Survey of Hammett Substituent Constants and Resonance and Field Parameters. Chem Rev. 1991;91:165–195. [Google Scholar]
- 30.Rozantzev EG, Krinitzkaya LA. Free iminoxyl radicals in the hydrogenated pyrrole series. Tetrahedron. 1965;21:491–499. [Google Scholar]
- 31.Haugland MM, El-Sagheer AH, Porter RJ, Peña J, Brown T, Anderson EA, Lovett JE. 2′-Alkynylnucleotides: A Sequence- and Spin Label-Flexible Strategy for EPR Spectroscopy in DNA. J Am Chem Soc. 2016;138:9069–9072. doi: 10.1021/jacs.6b05421. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Zhang H, Pink M, Olankitwanit A, Rajca S, Rajca A. Radical Cation and Neutral Radical of Aza-thia[7]helicene with SOMO−HOMO Energy Level Inversion. J Am Chem Soc. 2016;138:7298–7304. doi: 10.1021/jacs.6b01498. [DOI] [PubMed] [Google Scholar]
- 33.Connelly NG, Geiger WE. Chemical Redox Agents for Organometallic Chemistry. Chem Rev. 1996;96:877–910. doi: 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- 34.Osteryoung J. Voltammetry for the future. Acc Chem Res. 1993;26:77–83. [Google Scholar]
- 35.Batchelor-McAuley C, Kätelhön E, Barnes EO, Compton RG, Laborda E, Molina A. Recent Advances in Voltammetry. ChemistryOpen. 2015;4:224–260. doi: 10.1002/open.201500042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kunana T, Bublitz D, Hoh G. Chronopotentiometric Studies on the Oxidation of Ferrocene, Ruthenocene, Osmocene and Some of their Derivatives. J Am Chem Soc. 1960;82:5811–5817. [Google Scholar]
- 37.Little WF, Reilley CN, Johnson JD, Lynn KN, Sanders AP. Chronopotentiometric Studies of Ferrocene Derivatives. I. Determination of Substituent Constants with Substituted Phenylferrocenes. J Am Chem Soc. 1964;86:1376–1381. [Google Scholar]
- 38.Hall DW, Russell CD. Substituent Effects in the Chronopotentiometric Oxidation of Ferrocene Derivatives. Internal Solvation of Certain Substituted Ferricenium Ions. J Am Chem Soc. 1967;89:2316–2322. [Google Scholar]
- 39.Britton WE, Kashyap R, El-Hashash M, El-Kady M, Herberhold M. The Anomalous Electrochemistry of the Ferrocenylamines. Organometallics. 1986;5:1029–1031. [Google Scholar]