
Fig. 8.
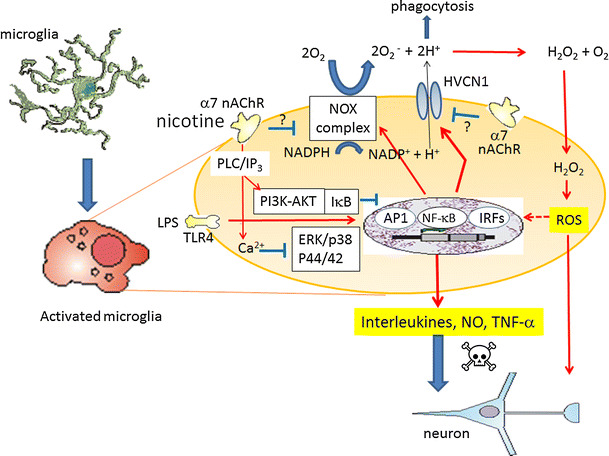
Proposed schema on inhibitory effects of nicotine on LPS-induced microglial activation. LPS, glycolipids found in the outer membrane of some types of Gram-negative bacteria, bind to Toll-like receptor 4 (TLR4) and activate signaling pathways; extracellular signal-regulated kinase (ERK)/p38 mitogen-activated protein kinase (MAPK), AP1, nuclear factor-κB (NF-κB), or IRFs (IRF3/IRF7), and hence production and release of pro-inflammatory cytokines, nitric oxide (NO) via inducible NO synthase and tumor necrosis factor-α (TNF-α). LPS also upregulates NADPH oxidase (NOX) assembly. The voltage-gated H+ channel, HVCN1, enables NOX function by compensating cellular loss of electrons with protons, which are required for phagocytosis. Furthermore, HVCN1 was required for NOX-dependent ROS generation. Nicotine binds to α7 nAChR in microglia, causing transient increase in intracellular Ca2+ in phospholipase C (PLC)/inositol 1,4,5-trisphosphate (IP3)-dependent manner [69], negatively modulates LPS-induced release of TNF-α. Cholinergic protection via α7 nAChR and PI3K-Akt pathway in LPS-induced neuroinflammation is also reported [70]. Nicotine may inhibit LPS-induced NOX. On the other hand, nicotine inhibits H+ current without affecting LPS-increased expression of HVCN1. Presumably, α7 nAChR signaling inhibits function of HVCN1 either directly or by inhibiting NOX, hence attenuating ROS production and further stimulation of pro-inflammatory cytokines, NO and TNF-α