

Abstract
We develop two complementary pipelines, “Zhang-Server” and “QUARK”, based on I-TASSER and QUARK pipelines for template-based modeling (TBM) and free modeling (FM), and test them in the CASP12 experiment. The combination of I-TASSER and QUARK successfully folds three medium-size FM targets that have more than 150 residues, even though the interplay between the two pipelines still awaits further optimization. Newly developed sequence-based contact prediction by NeBcon plays a critical role to enhance the quality of models, particularly for FM targets, by the new pipelines. The inclusion of NeBcon predicted contacts as restraints in the QUARK simulations results in an average TM-score of 0.41 for the best in top five predicted models, which is 37% higher than that by the QUARK simulations without contacts. In particular, there are seven targets that are converted from non-foldable to foldable (TM-score >0.5) due to the use of contact restraints in the simulations. Another additional feature in the current pipelines is the local structure quality prediction by ResQ, which provides a robust residue-level modeling error estimation. Despite the success, significant challenges still remain in ab initio modeling of multi-domain proteins and folding of β-proteins with complicated topologies bound by long-range strand-strand interactions. Improvements on domain boundary and long-range contact prediction, as well as optimal use of the predicted contacts and multiple threading alignments, are critical to address these issues seen in the CASP12 experiment.
Keywords: Protein structure prediction, CASP12, contact prediction, ab initio folding, threading, residue quality estimation
INTRODUCTION
In every two years, the community-wide CASP experiment provides an objective platform to critically assess the progress and challenges in the field of protein structure prediction. The methods for protein structure prediction are generally categorized into template-based modeling (TBM) and free-modeling (FM), depending on whether homologous templates could be detected from the PDB library. Considerable progress has been witnessed in recent CASP experiments in TBM for modeling distant-homologous proteins and for refining templates closer to the native structure,1-3 which have been mainly driven by the use of multiple threading templates.4 While the progress in FM seems more difficult and slower, excitements have been recently brought about by the success in the co-evolution based contact predictions and their utilization for guiding the folding of small- to medium-size protein targets.5-8
Although the TBM and FM methods have been primarily developed for modeling different categories of protein targets, based on templates or ab initio folding, a recent trend shows that the integration of their complementarity can be useful in improving the structure modeling accuracy for both categories of protein targets. In CASP10 and CASP11, for instance, the interplay between the template-based I-TASSER9,10 method and the ab initio folding QUARK11,12 method has demonstrated enhancements of accuracy of the final models for FM targets.13,14 In this approach, the structures of QUARK based models are compared with those of the templates identified by LOMETS, and the templates are re-ranked based on their similarity to the QUARK models. These templates are then used in the I-TASSER structure-assembly simulations to predict the final models. Meanwhile, the integration of the QUARK models into the I-TASSER structure assembly also showed the improvement of local structure accuracy for the TBM targets15. However, constructing ab initio folds for larger proteins with complicated topologies, in particular for β-proteins that have long-range β-strand contacts, is still a challenge.5,13,14,16,17
One of the primary reasons for the difficulty of ab initio modeling in predicting large proteins with complicated topologies is the lack of precise long-range contact interaction information in the force field during protein structure assembly simulation. Recently, sequence-based contact prediction has attracted considerable interest to capture long-range contact interactions. In particular, sequence-based contact predictions based on co-evolution18-20 and machine learning21,22 have demonstrated the usefulness of contact-maps in assisting folding of larger-size proteins.7,8,23 The major advantage for contact based folding simulation methods is that long-range contacts provide a constraint to reduce conformational space to be sampled, and help in folding the proteins with a more complicated topology.23 The success is however contingent upon precise contact map prediction that in turn depends on high volume of sequence homologs, particularly in co-evolution based contact prediction methods. In order to enhance the robustness of the contact map prediction, we recently developed NeBcon24, a contact prediction pipeline which combines multiple sources of contact maps from both co-evolution and machine learning through a novel naïve Bayes classifier model. The posterior probabilities of the classifiers are then trained with intrinsic structural features using neural network to generate the final contact map.
While continuous progress is on-going in protein structure prediction, a reliable estimation of the quality of the predicted structure models is critical to guide the biologist users to better use the model predictions in their experimental research.25 In this regard, it is of particular importance to identify the trustable regions of the predicted models by estimating the residue-level quality. ResQ26 is a recently developed algorithm that has been designed to assess the residue-level quality of the predicted structure models by combining the structural variations in the assembly simulation with the local features of secondary structure prediction and sequence conservation search.
In CASP12 experiment, we combined NeBcon predicted contacts with I-TASSER and QUARK to fold proteins that are distantly- or non-homologous to the experimentally solved structures. Additionally, we used ResQ to assess the residue-level quality of the predicted protein models based on I-TASSER and QUARK. The focus of this manuscript is mainly on the analysis of the results generated by the automated servers, “Zhang-Server” and “QUARK”. The models in the “QUARK” group are constructed by QUARK-based ab initio folding programs guided by NeBcon predicted contacts, while those in “Zhang-Server” are generated based on the I-TASSER pipeline, where NeBcon and QUARK are incorporated to enhance the accuracy of the models.
METHODS
The pipelines of I-TASSER9,10 and QUARK11 have been described previously. Here, we briefly outline the two pipelines that are used in CASP12, followed by some detailed discussion about the recently developed components added to the pipelines for protein structure prediction.
Outline of the QUARK pipeline
The “QUARK” server group in CASP12 is based on a modified version of QUARK ab initio protein structure prediction pipeline shown in Figure 1A. At first, if the target protein is detected as a multi-domain protein by ThreaDom27, the full length sequence is split into individual domains. The sequence of the domains (or the target for single domain proteins) is threaded through a non-redundant set of 6,023 high-resolution PDB structures by gapless threading to generate position-specific fragment structures with continuous lengths ranging from 1 to 20 residues. The scoring function for the gapless threading comprises profile-profile, secondary structure, solvent accessibility, and torsion angle matches between the target and the templates.12 A histogram of distances dij for each residue pair (i and j) of the target is derived from the top 200 fragments at ith and jth positions if the fragments are from the same PDB structure. The histogram that has a peak at the position of dij <9 Å is converted to a distance profile for the residue pair. In addition to obtaining distance profiles for the residue pairs, we predict contacts between the residues that are within 8 Å using NeBcon, a sequence-based contact predictor24. The distance profile restraint, sequence-based contact restraint, the inherent knowledge-based and physical potential terms are used to guide the assembly of the fragments into full structural models by replica-exchange Monte Carlo (REMC) simulations; the NeBCon-based contact restraints are applied again at a later stage for decoy filtering.
Figure 1.
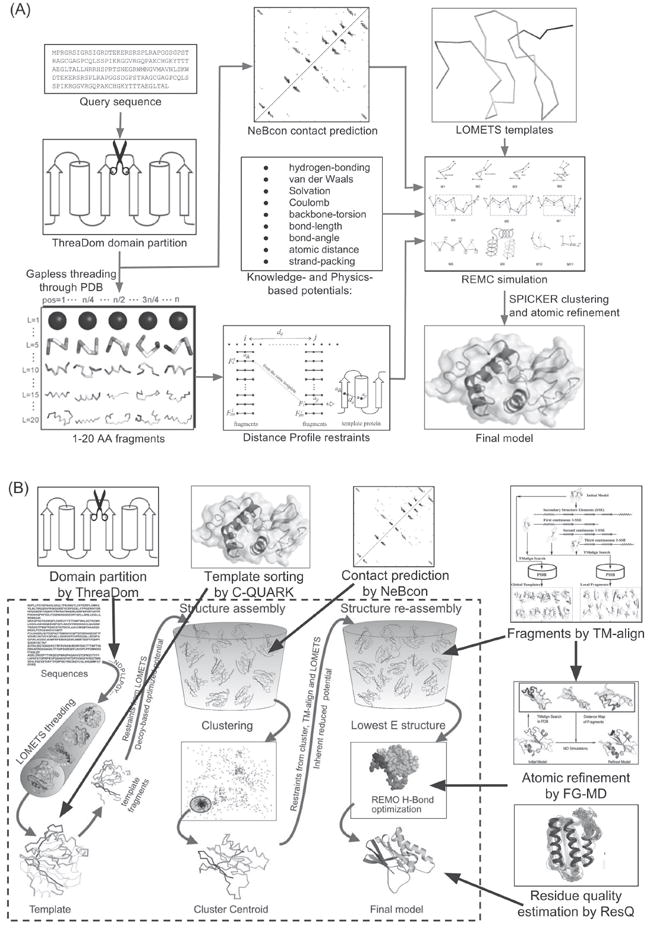
Automated protein structure prediction pipelines in CASP12. (A) Flowchart of “QUARK” server extended from original QUARK program with added contact prediction by NeBcon and threading templates from LOMETS. (B) The “Zhang-Server” pipeline is based on the classical I-TASSER pipeline (dashed box) with newly introduced components, including ThreaDom, NeBcon, FG-MD and ResQ.
It is noted that if the target is identified as “trivial” or “easy”, based on the significance and consensus of LOMETS28 threading alignments (as described in Eq. 1 of our CASP10 report13), the initial structures and distance restraints are obtained from the LOMETS templates, which are also used for guiding the QUARK-based REMC simulations.
Next, “Decoy” conformations from the QUARK simulation trajectories are clustered by SPICKER29 to identify cluster centroids, which correspond to low free-energy states. Before clustering, the decoys that do not satisfy a large portion of the NeBcon predicted contacts are filtered out for the “hard” and “very hard” targets. The cluster centroids from the five largest clusters are refined by ModRefiner30 or fragment-guided molecular dynamics (FG-MD)31 to obtain five final models. Here, the sequence-based contact restraints are not used, where the only external restraints in the ModRefiner and FG-MD simulations are those derived from the initial input models to the programs. The models from the corresponding clusters are ranked in descending order of the size of the SPICKER clusters. For multi-domain proteins, the final structures of the individual domains are assembled together with appropriate orientations by a rigid-body Metropolis Monte Carlo simulation (see Eq. 6 below). Finally, the residue-level quality is predicted by ResQ.26
Outline of the I-TASSER pipeline
The “Zhang-Server” in CASP12 is based on the classical I-TASSER structure prediction pipeline as shown in the dashed box of Figure 1B. Similar to the QUARK pipeline, multi-domain proteins are first partitioned into individual domains by ThreaDom27. The sequence of the individual domain or the target is threaded through a representative template library of the PDB using LOMETS28. If the target is classified as “hard” or “very-hard” based on the significance and consensus of the templates identified by LOMETS, the templates are re-ordered by their structural similarity to the QUARK models of the target. Fragments are extracted from the continuously aligned regions of the template structures and assembled into full-length structural models by a modified REMC simulation procedure32. A composite force field,10,33,34 which combines the distance restraints calculated from the templates and the NeBcon derived contact maps with the inherent knowledge-based energy terms, is used to guide the structural assembly simulations. The decoys from the trajectories of the simulations are clustered by SPICKER.29
Next, the cluster centroids are aligned against the structures in the PDB library using TM-align.35 The spatial restraints extracted from the TM-align templates are used for the second round of REMC simulations. The re-assembled structure models are reconstructed into full atomic models by REMO36 and further refined by FG-MD31 to generate the final structure models. For each of these models, we obtain different rankings from five Model Quality Assurance Programs (MQAPs)14: C-score37, structural consensus (the average TM-score of the target model to all other models), and three statistical energy functions (RWplus38, GOAP39, and DOPE40). The final ranking of the models is determined by ascending order of overall MQAP score, calculated as with rm,p being the ranking of the mth model by the pth program. The models of the multi-domain proteins are assembled together to form the full-length structure of the proteins. The residue-level quality of these models is finally estimated by ResQ.26
The “Zhang” human group in CASP12 adopts the same pipeline as “Zhang-Server” group, except that structure models from other CASP12 servers are used as an additional set of templates together with the LOMETS detected templates in the simulations.
New components in recent developments of QUARK and I-TASSER pipelines
Template re-ordering based on QUARK models
Structural assembly simulations in the classical I-TASSER pipeline oftentimes cannot accurately fold distantly- or non-homologous proteins due to the lack of accurate long-range interaction information. Therefore, if a target is categorized as “hard” or “very-hard”, we use ab initio models built by QUARK to re-rank the LOMETS templates in two steps, with the purpose of identifying the low-scoring templates that have correct folds. First, for each of the identified LOMETS templates, we compute its structural similarity to the top-five QUARK models by:
where TMscorem is the TM-score41 between the LOMETS template and mth QUARK model. TMscoreinterplay for each of the templates that indicates the structural similarity between the QUARK models and the templates is used to sort all the identified LOMETS templates in descending order. Second, the QUARK models are inserted at the [(m-1)M+1]-th position of the sorted template list, where M is the total number of threading programs in the LOMETS meta-threading program. Since higher ranked templates have stronger weights in template-structure-derived distance restraint collection, such ordering helps to balance the impact of threading the templates and QUARK models to the I-TASSER structure assembly simulations.
Integration of sequence-based contact prediction by NeBcon in structure assembly
In an effort to capture the long-range interaction information based on contacts between residues, sequence-based residue contact prediction is performed by NeBcon24. An initial set of contact maps are predicted by eight state-of-the-art contact prediction programs: PSICOV19, BETACON21, SVMcon42, SVMSEQ43, CCMpred44, mfDCA45, STRUCTCH46 and MetaPSICOV47. The confidence scores of the predicted contacts from these predictors are then combined by naïve Bayes classifiers (NBC) to obtain posterior probabilities of the contacts. The contact map derived from the NBC model is further refined by neural network training, where the NBC posterior probabilities are coupled with a variety of sequence-based features, including amino acid composition, Shannon entropy, residue separation, predicted solvent accessibility and secondary structure.24
In order to reduce the number of the falsely predicted contacts that may lead to inaccurate folding of the protein, we discard the contacts between residue pairs (i and j) that have a raw confidence score of NeBcon, Cscoreij, lower than a confidence score cut-off, which is set as 0.5, 0.4 and 0.3 for short (|i − j| ≤ 11), medium (12 ≤ |i − j| ≤ 24) and long (|i − j| > 24) range contacts, respectively. We further remove the contacts with low confidence scores until the number of contacts in each range is equal to an estimated number of contacts, as predicted for each range by a separate neural network predictor that is trained based on the length and secondary structure composition of the query sequence. The remaining contacts are used in the following sequence-based contact restraints, together with other energy terms in QUARK and I-TASSER based REMC simulations for structural assembly:
| (1) |
Here, dij is the distance between the Cα atoms of ith and jth residues during the simulations. The upper and lower bounds of the contact potentials and , respectively, are defined as
| (2) |
where Kb is the Boltzmann constant and T is the temperature of the replicas in the REMC simulations. ACCij is the posterior probability of residue i and j being in contact given the raw NeBcon confidence score Cscoreij, i.e., ACCij = P(ij in contact∣Cscoreij), with P(x) calculated based on a training set of 517 proteins. This training set was also used to optimize the above-mentioned confidence score cut-offs and the contact potential in the recent benchmark studies (Mortuza et al, in preparation).
Decoy filtering based on contact prediction by NeBcon
In QUARK, the NeBcon contact maps are further used after the structural assembly simulations to filter out decoy conformations that strongly violate the NeBcon derived contact predictions. For each of the decoy structures obtained from the simulations, a Gaussian-like score is calculated by
| (3) |
which is obtained by summing over contact of every residue pair (i and j) that has a raw confidence score, Cscoreij, greater than a confidence score cut-off, :
| (4) |
Here, wij is the weight for the contact pair (i, j) in the decoy structure that varies with the sequence separation as
| (5) |
Additionally, dij is the Cα distance between residue i and j in the decoy structure, and μ(Cscoreij) and σ(Cscoreij) are the mean and standard deviation, respectively, of the residue-residue Cα distance given the confidence score Cscoreij; both of which are trained based on the training set mentioned before (Mortuza et al, in preparation).
Scontact in Eq. (3) is calculated for each decoy structure generated by QUARK simulation, which is used to sort the decoy set. Only the top 20% of the decoy structures are retained for the subsequent SPICKER clustering.
Domain assembly for multi-domain proteins
For both “Zhang-Server” and “QUARK” groups, a full length multi-domain protein sequence is split into single domain sequences using ThreaDom.27 The structure of individual domains is then predicted by I-TASSER or QUARK pipeline. In order to assemble these domains to form the structure of the full protein, at first, a rough whole-chain structure is modeled by I-TASSER that provides a reference template to identify the orientation of the domains. The domain structures are then docked together with appropriate orientations by a quick Metropolis Monte Carlo simulation run, which is guided by a simple energy function:
| (6) |
Here, RMSD is the root-mean-square deviation between an individual domain and the rough whole-chain structure, and dij is the Cα distance between residue i of the first domain and residue j of the second domain. In the simulation, we consider those distances dij that are smaller than dcut = 3.7 Å. Finally, FG-MD simulation31 is applied to remove steric clashes (mainly between side-chains) between the domains in the assembled full-length structure.
Residue-level structural error estimation by ResQ
In order to assess residue-specific quality of the structure models, we use a recently developed algorithm, ResQ.26 Briefly, the algorithm first extracts the following residue-level features for a target protein: i) coverage and structural variations of the LOMETS templates, ii) consistency between the solvent accessibility of the model residues and that predicted from the sequence by the SOLVE program from the I-TASSER suite,10 iii) difference between the predicted secondary structure by PSSpred48 from the sequence and the secondary structure of the model, iv) structural variations among the decoys obtained from the REMC simulations, and v) the deviations of the final model structures from the templates resulted from TM-align structural alignment search of the model through the PDB database. These features are trained by Support Vector regression to predict the deviation of each residue position in the models from the native residue position.
RESULTS AND DISCUSSION
96 domains from 71 protein chains are assessed in CASP12. Based on the modeling difficulty, the CASP12 assessors classified the 96 domains into 39 FM targets, 38 TBM targets, and 19 FM/TBM (or TBM-hard) targets. Since the “Zhang” human group uses essentially the same pipeline as our server groups, the following discussion mainly focuses on the results obtained by the “Zhang-Server” and “QUARK” server pipelines, with the comparison of the server and human predictions briefly summarized at the end of the section.
Prediction of FM targets remains challenging
We present a summary of the results based on the “Zhang-Server” models for the 39 FM targets in Figure 2A, where it is shown that 11 targets are successfully modeled with a TM-score >0.5 by the “Zhang-Server” pipeline. Additionally, there are seven targets, which are reasonably folded with a TM-score in [0.40, 0.5]. While the majority of the successfully modeled targets are small-size proteins (<150 residues), there are three correctly predicted medium-size FM targets, T0915-D1, T0905-D1 and T0899-D1, which have more than 150 residues (marked by the arrows in Figure 2A).
Figure 2.
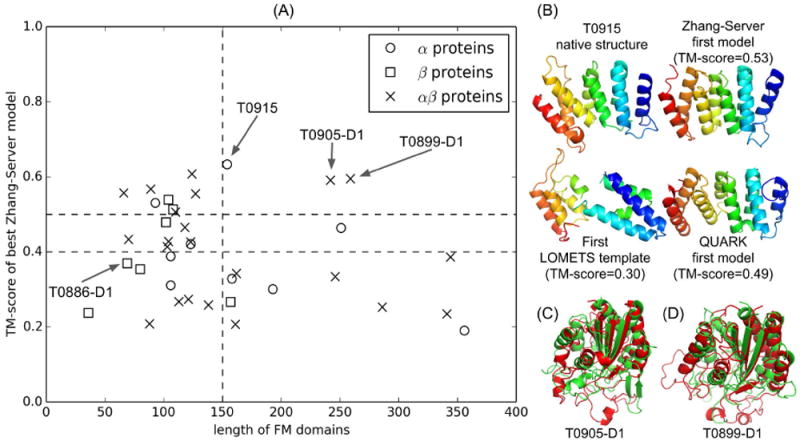
(A) TM-score of the best “Zhang-Server” model for the FM domains versus the length of each domain. The vertical dashed line represents the length cutoff of 150 residues. The horizontal dashed lines represent the TM-score cutoff of 0.4 and 0.5, respectively. Global structure folds of three medium size FM targets with length > 150, T0915, T0905-D1 and T0899-D1, are correctly predicted (TM-score > 0.5) by “Zhang-Server” as highlighted by the arrows. (B) Native structure and server models for T0915. All structures are colored in spectrum, with blue to red indicating N- to C- terminal. (C, D) “Zhang-Server” models (red) superposed onto the corresponding native structures (green) of T0905-D1 and T0899-D1, respectively.
T0915-D1, which is an α-protein of 161 residues with an eight-helix bundle topology (Figure 2B), is of special interest to discuss. Before the incorporation of QUARK based models in “Zhang-Server”, the first LOMETS template (4l8tA domain 2) for this target has a low TM-score of 0.30 (GDT_TS=24) to the native due to significant structural differences of the last four helices between the template and the native structure, as shown in Figure 2B. As a result, the target is considered as a “hard” target, and QUARK models, which have the correct topology of eight-helix bundle with adjacent helices anti-parallel to each other as in native, are used to re-order the LOMETS identified templates in the “Zhang-Server” pipeline. Re-ordering the templates based on the QUARK models significantly improves the quality of the top LOMETS template (3woyA, TM-score=0.43 and GDT_TS=36), which was ranked as 21st in the template list before the re-ordering. As discussed in Methods, the first QUARK model (TM-score=0.49 and GDT_TS=42) is placed above the first template in the re-ordered list for the I-TASSER. Due to the unique advantage of combining and refining multiple templates, the final “Zhang-Server” model has a TM-score =0.53 (GDT_TS=45) that is higher than the best of the QUARK models and LOMETS templates. This particular target highlights the efficacy of incorporation of QUARK based ab initio modeling in “Zhang-Server” to fold FM targets.
T0905-D1 (Figure 2C) and T0899-D1 (Figure 2D) are two other medium-size α/β FM targets with 242 and 259 residues, respectively, which are correctly folded by the “Zhang-Server” pipeline into typical Rossman folds with a TM-score=0.59 in both cases (GDT_TS= 39 and GDT_TS= 36, respectively). The successful models of these targets are attributed to the templates 4wk0B (TM-score=0.55 and GDT_TS=37) and 3t3pB (TM-score=0.51 and GDT_TS=31), which are identified by LOMETS for T0905-D1 and T0899-D1, respectively. It is noted that due to the use of LOMETS templates in QUARK based simulations, “QUARK” group was also able to correctly fold these targets.
While approximately fifty percent of the FM targets are modeled either correctly or reasonably by “Zhang-Server”, the pipelines still face difficulties in modeling of several small-size FM targets. For instance, the TM-score of the target T0886-D1, a β-protein with a length of 69 residues, is 0.37 (GDT_TS=48). This is due to its complicated topology with multiple pairs of long-range β-strand pairings, which is difficult to fold using our current pipelines.
Another significant unsolved issue to us (as well as to the protein structure prediction community49) is the ranking and selection of the best predicted models. For the FM targets, for instance, the average TM-score of the first models is 0.34 (average GDT_TS=31), while it is 0.40 (average GDT_TS=36) for the best models by Zhang-Server. The failure can be essentially attributed to the inaccuracy of the QUARK and I-TASSER force fields which fail to rank the best models as the lowest free-energy clusters in the structural assembly simulations, where the models for the FM domains are mainly ranked by the size of the SPICKER clusters.
The failure in model selection was also observed for the TBM targets, which occurs most frequently for the cases when the best templates are detected only by a minority of the LOMETS programs. Since the model selection for the TBM domain is dominated by the consensus score of the models, the automated model selection process tends to select the consensus but less accurate models for these targets, an issue which has been extensively discussed in the previous CASP studies13,15,50. Here these data highlight again the remarkable gap that the current model ranking process remains to fill up.
LOMETS template sorting by QUARK models is not always beneficial
Given the templates with correct topologies (TM-score>0.5) for a target, I-TASSER is usually able to utilize multiple template information to construct the structure model that is often closer to the target. As shown in Figure 3A, out of 53 the target domains, for which at least one correct or roughly correct (TM-score>0.4) LOMETS templates are available in top 20 hits, the first “Zhang-Server” models for 41 targets have a greater TM-score than that of the best templates. The TM-score difference between the first “Zhang-Server” models and the best in top 20 LOMETS templates is no more than 0.03 for the rest of the 11 targets, except in two cases, T0890 and T0868. For these two targets, the qualities of the predicted first models are significantly worse than that of the best LOMETS templates, which have a reasonably correct topology.
Figure 3.
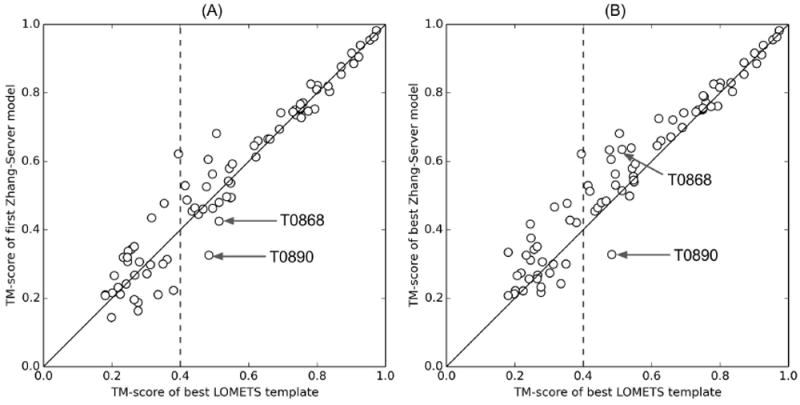
TM-score of the “Zhang-Server” models versus that of the best in top 20 LOMETS templates. (A) the first “Zhang-Server” model, (B) the best “Zhang-Server” model submitted. The vertical dashed line represents the TM-score cutoff of 0.4 for the best in top 20 LOMETS templates. For T0868 and T0890, the quality of the predicted first models is significantly worse than that of the best threading templates as highlighted by the arrows.
Here, we note that we lack the prior knowledge of CASP assessors’ domain boundary definition during CASP12. Therefore, the template identification and the construction of QUARK models for re-ordering the templates in the “Zhang-Server” pipeline have been made based on the domains predicted by ThreaDom. To make a fair assessment the effect of the internal QUARK sorting process, Figure 3 has used the same domain definition utilized by our servers in the following discussion. Nevertheless, we also provide the corresponding data based on the domain defined by the CASP assessors in Table S1 in the Supplementary Material, to facilitate comparisons with the models by other groups when needed.
T0890 is a two-domain protein, where the domains are partitioned as T0890-D1 and T0890-D2 by the CASP assessors. However, ThreaDom incorrectly predicted it as a single-domain protein as shown in Figure 4A. As a result, the first LOMETS template from the second domain of 3td7A, which covers only the second domain of the target structure with a TM-score of 0.74 (GDT_TS=70), has TM-score =0.46 (GDT_TS=41) for the full-length sequence as shown in the figure. Due to the incorrect domain prediction, the first QUARK model also has a low TM-score of 0.32 (GDT_TS=27) with respect to the full-length native structure, where only the first domain of the target was correctly modeled (TM-score=0.56, GDT_TS=62). Therefore, re-ordering the LOMETS templates based on the QUARK models leads to the selection of those templates to be used in the I-TASSER simulations that have as low as TM-score of 0.32 (GDT_TS=27), which in turn drives the construction of the first “Zhang-Server” model with incorrect topology (TM-score=0.32, GDT_TS=28). This indicates that the incorporation of QUARK based ab initio models in “Zhang-Server” pipeline may have less usefulness in modeling multi-domain proteins, if the domains are not correctly predicted.
Figure 4.
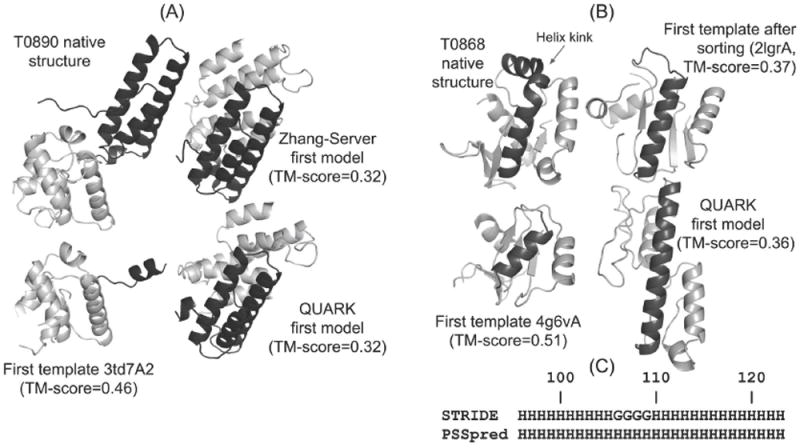
(A) The native structure, best LOMETS template, and the QUARK and Zhang-Server models for T0890, which is a two-domain protein. The domains are distinguished as per the domain assignment by CASP12 assessors, where the first domain is colored in black while the second domain is shown with grey. (B) The native structure, the template structure (before and after sorting) and the first QUARK model for T0868. Residues 96 to 123 are colored in black. (C) Secondary structure of residues 96 to 123 of T0868 assigned by STRIDE using native structure and that predicted by PSSpred. “H” stands for α-helix in STRIDE and any helix type in PSSpred. “G” stands for 3/10 helix in STRIDE.
T0868, shown in Figure 4B, is an example of target mis-categorization based on the significance and consensus of LOMETS templates, where LOMETS identified the first template, 4g6vA, with a correct topology (TM-score=0.51, GDT_TS=47) while the target was categorized as “hard”. As a result, the target is initially modeled by QUARK, and these models are further used in re-ordering the LOMETS templates. Unfortunately, the QUARK based ab initio modeling constructs models with incorrect topologies (TM-score=0.36 and GDT_TS=37 for the first model, and TM-score=0.47 and GDT_TS=49 for the best model in top five), partly due to the prediction of limited types of secondary structure by PSSpred and inappropriate usage of contacts that will be discussed in detail later. The poor quality of the QUARK models, particularly of the first model, leads to the incorrect LOMETS templates being ranked at the top of the list, where the top 15 templates are far away from the native structure (TM-score<0.40). On the other hand, the rank of the correct template, 4g6vA, is dropped from first to 56th place in the template list. As a result, the I-TASSER based simulations in the “Zhang-Server” pipeline fail to correctly construct the first model (TM-score=0.48, GDT_TS=50), since its simulation is mostly driven by the top ranked templates. However, it should be noted that although re-ranking the template list has adverse effects on the quality of the first model by “Zhang-Server”, the TM-score of the best model among the top five models for this target is 0.63 (GDT_TS=59), as indicated by an arrow in Figure 3B. One reason for successful prediction of the best model is the constant utilization of the composite force field to draw the structure closer to the native state during REMC simulations instead of simply satisfying all geometry restraints imposed by the templates. Therefore, while template quality has strong influence on the I-TASSER simulations, the prediction result is not completely biased by the low quality of the templates.
Comparison of templates before and after QUARK-based sorting
Incorrect prediction of the first models for T0890 and T0868 prompts us to further examine the regular LOMETS templates before and after re-ordering them based on QUARK models, as presented in Figure 5. Based on the data shown in Figure 5A, the average TM-score of the first LOMETS templates is 0.28 (average GDT_TS=29) in the original templates list, while it is 0.33 (average GDT_TS=33) after re-ordering the templates, where QUARK models are not included in the list.
Figure 5.
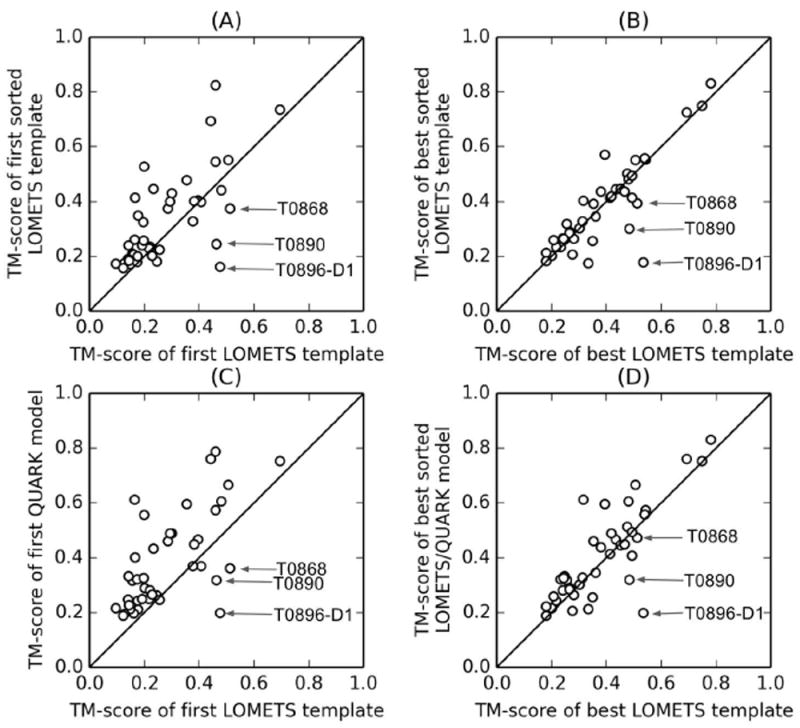
Effect of QUARK-based sorting on the LOMETS templates for the “hard” and “very hard” targets defined by LOMETS. (A) TM-score of the first template after sorting versus that without sorting. (B) TM-score of best out of the top 20 templates after sorting versus that without sorting. (C) TM-score of the first QUARK model versus that of the first LOMETS templates without sorting. (D) TM-score of the best out of the top 20 templates, which include sorted templates and QUARK models and are used by I-TASSER in CASP12, versus that without sorting and are used in the classic I-TASSER pipeline. Three targets (T0868, T0918-D2 and T0896-D1), for which the sorting process significantly reduces the quality of the templates, are marked with arrows.
However, the TM-scores of the best LOMETS templates among the top 20 hits for several targets do not increase significantly after the re-ranking of the templates (Figure 5B); in fact, the re-ordering slightly decreases the average TM-score of the best LOMETS templates from 0.38 to 0.37 (average GDT_TS decreases from 38 to 37). This is understandable because the template structures in the original LOMETS ordering are more diverse than those after QUARK-based re-ordering that are normally converged into the five QUARK models. Therefore, there is a higher possibility for the best in the top 20 templates having a higher TM-score in the original LOMETS ordering. Nevertheless, since the top templates have a higher weight in the restraint collections and therefore are usually more important for the I-TASSER simulations, the QUARK based sorting still turns out to be beneficial to the final I-TASSER modeling results.
The decrease of the TM-scores of the best templates is prominent for several targets, including T0868, T0890 and T0896-D1, which have incorrect QUARK models (TM-score < 0.4). The low TM-scores of the best templates for these targets indicate that the template re-ordering process may occasionally pose a negative effect on the construction of final models due to the lowering of the rank of the good templates when the QUARK models have incorrect topologies, despite its overall benefit to the I-TASSER modeling.
We also compared the TM-score of the first QUARK models and that of the first LOMETS templates before the re-ranking, as shown in Figure 5C. The average TM-score of the first QUARK models is 0.38 (average GDT_TS=37), which is significantly higher than that of the first LOMETS templates (average TM-score=0.28, average GDT_TS=29). This indicates the usefulness of integration of QUARK models into the initial template pool that improve the quality of the top templates and thus guide the I-TASSER simulations to correctly construct the models for the “hard” and “very-hard” targets in “Zhang-Server”. The inclusion of QUARK models in the re-ordered list also leads to the improvement of the TM-score of the best in top 20 templates from 0.37 to 0.40 (GDT_TS improvement from 38 to 39) as shown in Figure 5D. Overall, the data presented in Figure 5 suggests that the insertion of QUARK models into the template list and re-ranking the templates based on the models are often beneficial to further improve the quality of the templates and hence the final models of the I-TASSER simulations, especially for the “hard” and “very-hard” protein targets.
Why does not high-accuracy contact prediction result in correct ab initio structure?
One of the distinct features in our CASP12 pipelines compared to that in the previous CASP experiments is the incorporation of the sequence-based contact prediction by NeBcon24. Here, we examine the contribution of NeBcon predicted contacts to model the structure of “hard” and “very-hard” target domains. For this purpose, in Figure 6A, we show the comparison of final models constructed by QUARK pipeline with NeBcon predicted contacts, i.e. by “QUARK” server, and that by the original QUARK pipeline without NeBcon, which is performed as a post-CASP experiment on the 47 “hard” and “very-hard” targets. Here, since the contact map and the final QUARK models are both created using the internal ThreaDom domains, for the sake of consistency and to better calibrate the difference of the two modeling pipelines we presented the data using again the same domain definition as the contacts and models were predicted. Nevertheless, we also present the corresponding data based on CASP assessor’s definition in the Table S2 in Supplementary Material for the purpose of providing more information.
Figure 6.
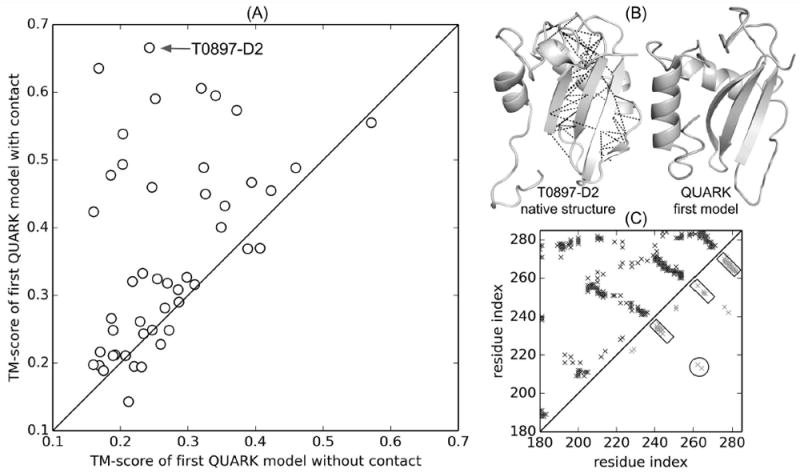
(A) Comparison between TM-score of the first model for “hard” and “very hard” targets generated by QUARK with NeBcon predicted contacts (“QUARK” server group in CASP12) and that by original QUARK without contacts (post-CASP experiment). (B) Native structure of T0897-D2 with the NeBcon predicted contacts shown by black dashed lines and the first QUARK model constructed during CASP12. (C) Native contact map for T0897-D2 (upper-left triangle) and NeBcon predicted contact map (lower-right triangle), which was used in structure assembly simulations. Each cross point represents a native or predicted contact. The NeBcon predicted contacts for parallel β-strand pairings and for interaction between β-strand and α-helix are highlighted by rectangles and circles, respectively.
As shown in Figure 6A, the TM-scores for almost all the targets have been increased with the addition of the NeBcon contacts as restraints in the QUARK simulations. For example, the TM-score for the first model of T0897-D2 by the original QUARK without contact is 0.24 (GDT_TS=25), while it was increased to 0.67 (GDT_TS=52) in the CASP12 “QUARK” server. The significant improvement of the model quality by the “QUARK” server is due to the correct prediction of contacts (Figure 6B dashed lines) that capture the information of β-sheet formation and interaction between β-strand and α-helix (Figure 6C rectangles and circle). Overall, the average TM-score of the first models is 0.27 (average GDT_TS=26) by original QUARK for all the Hard and Very-hard targets, while the addition of contact restraints in the QUARK pipeline increases the average TM-score to 0.36 (average GDT_TS=34). Similarly, the average TM-score of the best model among the five submitted QUARK models is increased from 0.30 to 0.41 (average GDT_TS increases from 29 to 37) due to the inclusion of NeBcon contacts in the QUARK pipeline. The significant increase of TM-scores demonstrates the effectiveness of contacts in improving the quality of models based on QUARK ab initio folding. However, correct folds (TM-score < 0.5) are not obtained for most of the “hard” and “very-hard” targets, which are probably due to the lack of prediction or inappropriate usage of the predicted contacts.
Figure 7A shows the precision of the predicted contacts versus the TM-score of the best QUARK models for the same set of targets. It is shown that the precision of the top L/5 all-range (|i − j| ≥ 6) contacts, where L is the length of the target, is weakly correlated to the TM-score of the best QUARK models with a Pearson correlation coefficient =0.59. While the data is not shown here, the correlation between the precision of top L/5 long-range (|i − j| ≥ 24) predicted contacts and the TM-score of the best QUARK models is also not remarkable (Pearson correlation coefficient =0.49), indicating that the high precision of predicted contacts does not guarantee to generate correct models. In order to further investigate the reason for obtaining less accurate models based on the QUARK simulations with highly accurate top L/5 contacts, we consider T0918-D4 as an example as it has a high contact precision (0.76) but low TM-score (0.34) and low GDT_TS (21), highlighted with the arrow in Figure 7A.
Figure 7.
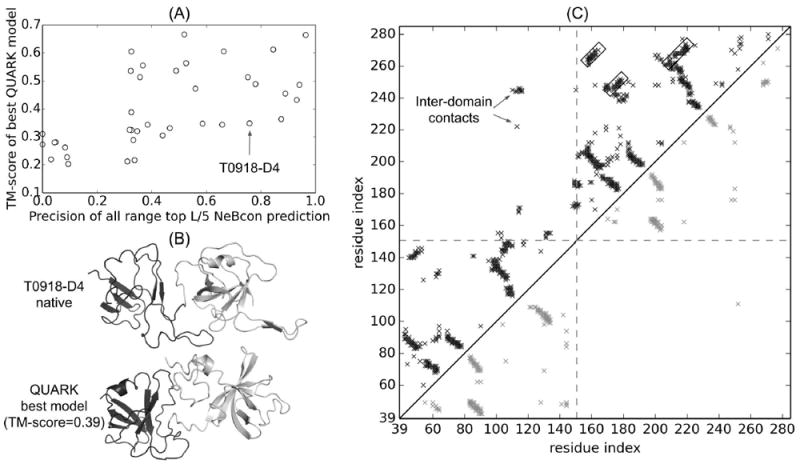
(A) TM-score of the best in top five QUARK models versus the precision of top L/5 contacts predicted by NeBcon for “hard” and “very-hard” targets. The target, T0918-D4, which has a high contact accuracy (0.76) but with a low TM-score (0.34) and GDT_TS (21) is highlighted with an arrow. (B) The native structure and the best QUARK model of T0918-D4. The first domain is colored in black while the second domain is colored in grey as assigned by CASP12 assessors. (C) Native contact map (upper-left triangle), and NeBcon predicted contact map (lower-right triangle) used in structure assembly simulations, for T0918-D4. Each cross point represents a native or predicted contact. The dashed line marks the domain boundary assigned by CASP12 assessors. The parallel β-strand pairings, which are not detected by NeBcon, are highlighted by rectangles. Inter-domain contacts in the native structure are highlighted by arrows.
T0918-D4 is a two-domain target as shown in Figure 7B, while ThreaDom incorrectly predicted it as a single domain target. Each domain in the target is a β-fold with both parallel and anti-parallel β-strand pairings. The precisions of top L all- and long-range predicted contacts are 0.63 and 0.57, respectively, and those of top L/5 all- and long-range contacts are both as high as 0.76. However, NeBcon fails to predict the inter-domain contacts as highlighted with the arrows in Figure 7C, where the black dots in the upper triangle represent the contacts in the native structure of the target and grey dots in the lower triangle represent the NeBcon predicted contacts. As a result, orientation between the domains was not correctly modeled during QUARK simulations. Additionally, NeBcon cannot predict contacts for three long-range parallel β-strand pairings in the second domain as marked with rectangles in Figure 7C. The lack of prediction for these β-strand pairings leads to incorrect modeling of β-pairing in the second domain, as evidenced by low TM-score (0.4) and low GDT_TS (34) of that domain. Therefore, the overall TM-score of the final model is 0.34 (GDT_TS=21), although the first domain is correctly predicted (TM-score = 0.51, GDT_TS=45). The incorrect modeling of this target due to the lack of prediction of long-range contacts emphasizes the importance of accurate prediction of long-range contacts to correctly fold hard targets, especially those with multiple domains. In other words, the long-range contact predictions, although with a high accuracy, are not sufficiently divergent to cover the entire range of the sequence (in particular for the regions critical to determining the overall topology and domain orientations).
We have also investigated how the model quality of T0868, shown in Figure 4B, is affected by the NeBcon predicted contacts that are used as restraints in the QUARK simulations. It is observed that the precisions for top L/5 and top L long-range contacts are 0.65 and 0.35, respectively, while the TM-scores of all the models are low for this target, as mentioned before. A closer check finds out that the QUARK simulations of this target severely lack long-range contact restraints; only three predicted long-range contacts are used in the simulations, since the majority of the predicted long-range contacts are ignored due to low confidence scores. This illustrates the significance of use of optimized number of long-range contacts in ab initio modeling to correctly fold proteins.
Overall, there are multiple reasons that have resulted in the weak correlation between the contact prediction accuracy and the quality of the final models, in particular the observation that high-accuracy contact predictions failed to lead to high-quality model predictions. The major one is probably the lack of correct long-range contacts despite of the accurate short- and medium-range contacts, which are less determinative for the global topology. In some cases, such as T0918-D4, which have even high-accuracy long-range contacts, these contact predictions are not sufficient divergent to cover the entire sequence, especially for the regions that are critical to the global topology and domain orientations. The second important reason is that the integration of the contact restraints with the inherent force fields in I-TASSER and QUARK is not yet optimized. The combination of contact restraints is particularly subtle for an automated pipeline when the target type and the accuracy of contact predictions are unknown, where the weight of contact-map retraints could not be too strong (which could dominate and destroy the correct restraints from the threading templates for the Easy targets) or too weak (which could not be sufficient to guide the folding simulations for the Hard targets that do not have homologous templates).
Current contact predictions cannot improve model quality of Easy targets
While contact plays important roles in ab initio modeling to predict structure of Hard targets, its impact on template-based modeling of Easy targets may not be as strong. To examine this issue, we perform a post-CASP experiment on 38 “Trivial” and “Easy” targets using a modified I-TASSER protocol that is identical to “Zhang-Server” in CASP12, except it does not use NeBcon-derived contacts in the simulations. As shown in Figure 8, TM-scores of the first models from the “Zhang-Server” and the modified I-TASSER without contact do not significantly differ (average TM-scores of 0.70 in both pipelines). This is mainly due to the fact that the quality of the template structures detected by LOMETS for the TBM targets is on average better than that from the ab initio contact predictions. Therefore, the inclusion of the contact predictions in the structural assembly simulations does not result in significant improvement for the easy targets, which is consistent with the observation made previously43.
Figure 8.
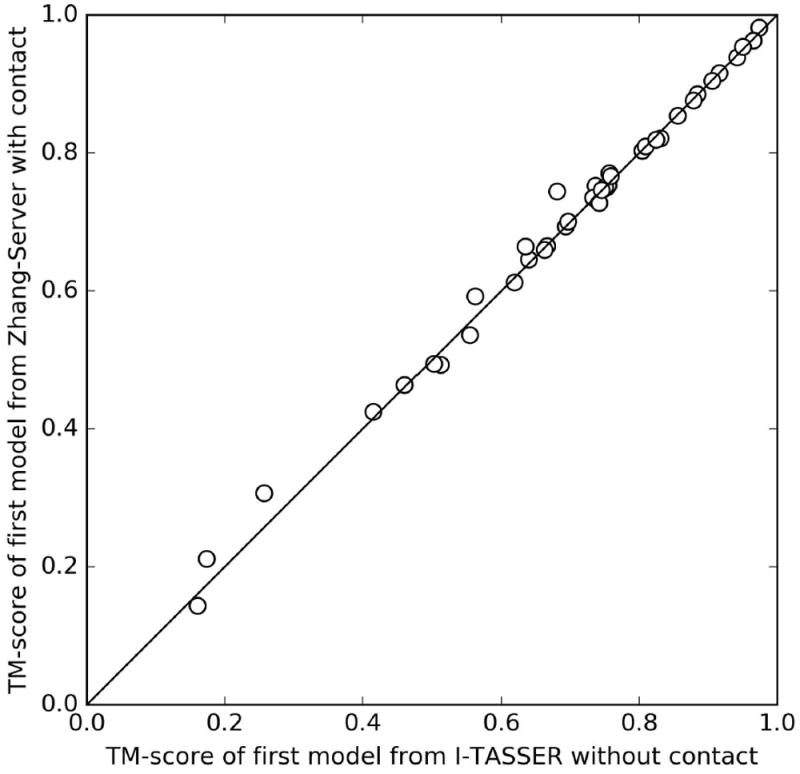
TM-scores of the first models from Zhang-Server with contacts versus those from I-TASSER without contacts for the 38 targets that have significant templates identified. Both of these pipelines use the same pool of templates identified during CASP12.
Prediction of specific type of secondary structure is important
The importance of secondary structure prediction has been discussed in our CASP11 reports14,15. Here, we further examine the importance of specific type of secondary structure prediction to the correct 3D structure prediction. Since the TM-scores of QUARK models for T0868 are low, we have checked the secondary structure of residues 96-123 (highlighted with black color in Figure 4B) in the native structure, the first QUARK model, and the templates. While the secondary structure for this range of residues is predicted to be a helix by PSSpred, shown in Figure 4C, we find that the residues 96-105 and 110-123 are α-helices, and the residues 106-109 correspond to a short 3/10 helix in the native structure as assigned by STRIDE. This subtle difference in helix type induces a helix kink in the native structure as highlighted with the arrow in Figure 4B. It is not possible to capture such a specific type of secondary structure by PSSpred that only predicts three states (helix, strand and coil) secondary structure. As a result, QUARK simulations incorrectly construct an α-helical conformation for the whole residue 96-123 segment in the first QUARK model. Such a limitation of PSSpred necessitates the use of programs that can predict more specific and detailed types of secondary structures,51,52 which are essential to the modeling of the global fold of FM targets such as T0868.
Comparison between server and human predictions
The Zhang-Sever and Zhang human groups used the same pipeline in the CASP12 experiment, where the only difference is that the Zhang-Sever started from the in-house LOMETS templates while the Zhang human group included the models from other CASP servers in the pool of the input templates and models. To examine the impact of the additional templates to the final model prediction, we present in Figure 9A a comparison of the TM-score of the first models by the two groups for all 96 domains as defined by the CASP assessors. Although the overall model quality of the two groups is largely comparable, there are two domains (T0901-D2 and T0905-D2) for which the TM-score of the Zhang models (0.54 and 0.45, respectively) is significantly higher than that of the Zhang-Server models (0.19 and 0.54, respectively). The GDT-TS scores are 24 and 23 versus 59 and 53 for the two targets by Zhang-Sever and Zhang respectively.
Figure 9.
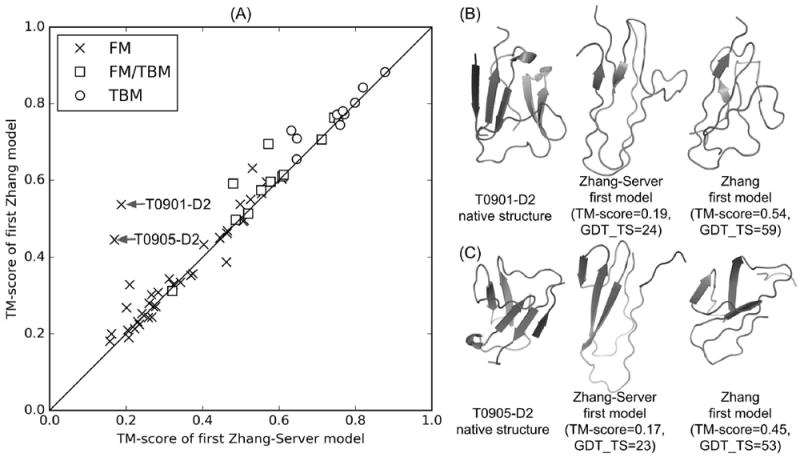
Comparison of the first model obtained by “Zhang-Server” and “Zhang” human group. (A) All-to-all TM-score comparison, with two FM targets (T0901-D2 and T0905-D2) whose qualities of the models by “Zhang-Server” are significantly lower than that by “Zhang”, marked by arrows. (B,C) The native structures, “Zhang-Server” and “Zhang” models for T0901-D2 and T0905-D2.
Interestingly, T0901 and T0905 are a pair of homologous proteins with a pair-wise sequence identity 36.9% and have a similar structure (TM-score=0.70 returned by TM-align35). Both targets are two-domain proteins with the second domain having a discontinuous domain structure, i.e., T0901-D2: 34-41,265-326 and T0905:42-47,290-349. However, ThreaDom failed to detect the discontinuous domain structure and the Zhang-Server thus tried to fold the protein as a single domain which resulted in the completely incorrect structure for these domains because of the lack of correct templates from LOMETS. In the Zhang human group, however, the initial template set includes a correct template from 5dll_A detected by one of the CASP server groups which had probably the correct domain split, where the TM-score for the template of 5dll_A is 0.428 and 0.430 for T0901-D2 and T0905-D2, respectively. After the I-TASSER refinement, the TM-score of the submitted model1 by the Zhang human group was increased to 0.54 and 0.45, respectively (see Figure 9B and 9C), which is apparently attributed to the inclusion of the 5dll_A template. Here, we like to mention that for T0905-D2, the Zhang-Server created a correct model (model3) with a TM-score=0.56 probably due to the integration of the QUARK models in the initial modeling pool, where the TM-score of the first QUARK model is 0.49. But this best model was not ranked as the model1 by the MQAP selection. Overall, these two examples further highlighted the issues of the automatic pipeline in domain split and assembly (especially for the targets of complex continuous domain structures), as well as in the MQAP-based model selection process. The data also indicate that the inclusion of additional complementary threading programs in LOMETS will increase the coverage of the initial template pool, which can further improve the quality of the final model of the I-TASSER pipeline.
ResQ robustly estimates residue-level quality of model structures
In CASP12, we use ResQ to evaluate the quality of the structure of the models at residue-level by estimating distance of the residues of the models from the corresponding residues of the native structures. The estimated residue-level quality is recorded in the “temperature-factor” field of the PDB files that are generated for the output models by the pipelines. Following the protocol of CASP12 assessors (http://www.predictioncenter.org/casp12/doc/help.html#ASE), we calculate the accuracy of ResQ prediction based on Accuracy Self Estimate (ASE) score:
| (7) |
where L is the number of residues in a target protein, tfi is the predicted distance error by ResQ for residue i, and d0 = 5 Å is a scaling constant. Additionally, di is the distance between residue i in the model structure and that in the native structure after the model is superposed onto the native. The superimposition of the model and the corresponding native is performed by the TM-score program41. The value of ASE ranges between 0 and 100, where the value of 100 indicates the perfect prediction by ResQ.
Figure 10A shows a scattering plot of the ASE score of the “Zhang-Server” first models for the CASP12 targets versus the TM-score of the models. As it is seen from the figure, the ASE scores for 90 out of 96 targets are greater than 60 (marked with a dashed line in Figure 10A), indicating the robustness of the ResQ prediction. In particular, ResQ showed remarkable performance in terms of accuracy of the prediction for the TBM and FM/TBM targets, where the average ASE scores are 86.67 and 74.83, respectively, for the targets modeled by “Zhang-Server”. We should emphasize that while the CASP12 assessors evaluate the ResQ prediction for the TBM and FM/TBM targets only, ResQ is also notably accurate for the FM targets. The average ASE score for the FM targets modeled by “Zhang-Server” is 69.24, where 33 out of the 39 FM targets have an ASE >60. Due to the robustness of the ResQ prediction, it can be potentially useful in atomic-level refinement of the models. To further illustrate this point, we discuss about a particular target, T0866-D1 (highlighted with an arrow in Figure 10A), for which the ResQ prediction is reliable as evidenced with the ASE score of 81.5.
Figure 10.
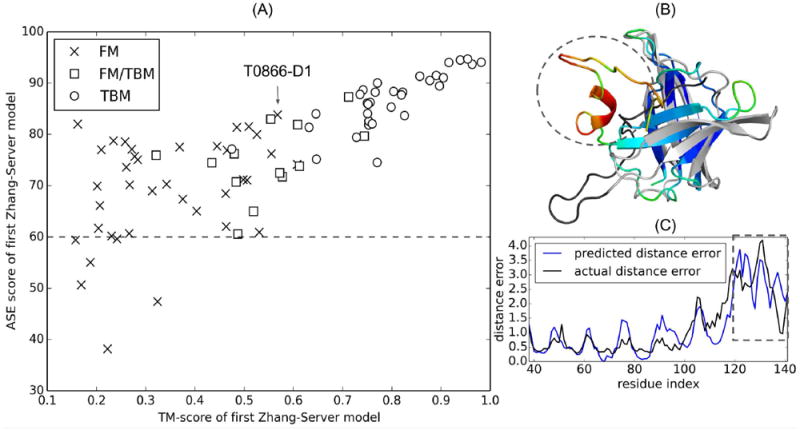
(A) The accuracy of ResQ predicted residue-level quality (ASE score) versus TM-score for the first “Zhang-Server” model. The horizontal dashed line corresponds to ASE score of 60. The FM target T0866-D1 is indicated by an arrow. (B) Superposition of the native structure and the first “Zhang-Server” model of T0866-D1 (TM-score=0.51, GDT_TS=48, ASE=81.51). The native structure is colored in grey, except for the C-terminal tail (residues 119-141) that is colored in black. The same C-terminal tail in the “Zhang-Server” model is highlighted by a dashed circle. The “Zhang-Server” model is colored in spectrum color scheme according to predicted residue quality by ResQ, where blue to red color indicates increasing distance error. (C) Overlay of the distance error predicted by ResQ and the actual distance error (y-axis, higher values indicate worse residue qualities) for T0866-D1. The region of 119-141 with a high distance error is highlighted with a dashed rectangle.
T0866-D1 is an FM target with a β-barrel topology as shown in Figure 10B, where the C-terminal tail (residues 119-141) of the native structure is represented with black color and the rest of the residues are shown with grey color. The superimposed “Zhang-Server” predicted model, which is represented with the spectrum color scheme according to the ResQ prediction with blue to red indicating increased distance error, onto the native structure shows that the C-terminal tail (highlighted with dashed circle in Figure 10B) in the model is far from the native. The ResQ prediction also shows that the residue distance error is high for this region, as highlighted with dashed rectangle in Figure 10C, indicating the region with the worse residue quality. This example shows the usefulness of ResQ in identifying low-quality regions, which may require extra attention during refinement process to enhance the structural quality of the predicted models.
CONCLUSION
We have tested two updated 3D structure prediction pipelines, I-TASSER and QUARK, as “Zhang-Server” and “QUARK” in the CASP12 experiment. One of the most noticeable additions to the pipelines, which have found significant impact to the modeling results, is the incorporation of the sequence-based contact prediction from NeBcon.24 The predicted contact maps were used as soft restraints in the QUARK and I-TASSER simulations that help improve the structural quality of the predicted models. There are seven “hard” targets that were essentially converted from non-foldable to foldable due to the contact restraints used in QUARK simulations. Target T0897-D2 is one of such examples in which contact restraints play important roles to fold a challenging target (discussed in Fig 6).
Nevertheless, the limitation of our contact predictor to correctly predict long-range and divergent contacts and the non-optimum usage of the predicted contacts may reduce the potential usefulness of the contact information to structure modeling. For instance, due to low confidence scores of the majority of predicted contacts for the hard targets, several long-range contacts are ignored that could have been useful in the simulations to correctly fold the targets, as discussed for T0868 (Fig. 4). Therefore, continuous efforts should be given to improve the accuracy of contact map prediction and the optimum integration of the predicted contacts to the QUARK and I-TASSER simulations. One possible solution to incorporate the ignored correct contacts with low confidence scores in the simulations can be the use of ranking, instead of confidence score, as a parameter in the contact potential. Most recently, efforts to integrate contacts into LOMETS threading process have showed promise for non-homologous template selection (Zheng et al, 2017, submitted).
The second noticeable component feature, newly introduced to the CASP12 structure modeling pipelines, is the application of the residue-level quality estimation method, ResQ,26 which shows efficiency and robustness in predicting the quality of local structure of the predicted models. The successful prediction of residue-level quality can be used to identify regions with poor quality that can potentially be improved in the structure refinement stage, as discussed for the case of T0866-D1 (Fig. 10).
While our CASP10 report13 describes the effectiveness of interlay of QUARK and I-TASSER for protein structure prediction, here we further examined its importance, particularly in ab initio structure prediction. For instance, while the quality of the first LOMETS template is considerably bad (TM-score=0.30 and GDT_TS=24) for the FM target, T0915, re-ordering the templates list based on its QUARK models and addition of the models in the list significantly improve the quality of the top-ranked templates. In particular, the TM-scores of the first LOMETS template after re-ranking and the first QUARK model are 0.43 (GDT_TS=35) and 0.49 (GDT_TS=42), respectively, which play vital roles to guide the simulations for correctly folding the target (TM-score=0.53 and GDT_TS=45). However, the use of re-ordered LOMETS templates and QUARK models are occasionally detrimental to the construction of the first model for the cases like T0868, T0890 and T0896-D1 that have completely incorrect QUARK models. One possible way to address this issue may be to determine the template orders based on a combination of original ranking of the templates, estimated quality of the QUARK models, and their structural similarities to the templates.
Predicting structure of multi-domain proteins based on our pipelines remains a significant challenge, specifically for the hard targets, as illustrated for several cases, such as T0890 and T0918-D4. Several limitations are currently prevailing in the pipelines that restrict the correct prediction of structure of multi-domain proteins. First, threading based domain boundary prediction is not always reliable for “hard” targets due to the lack of structural templates with significant alignments. The development of sequence-based domain boundary prediction program can be a possible solution to overcome this issue. Second, although NeBcon predicts contacts with reasonable precision within a domain, it often fails to detect inter-domain contacts, which are helpful to correctly model multi-domain proteins. Therefore, an on-going effort is to expand the capability of NeBcon to correctly predict inter-domain contacts. Third, our current domain assembly protocol, which depends on whole-chain reference structures constructed by I-TASSER simulation, often cannot embed correct domain orientation information. Hence, the development of a specific force field based on domain-domain interactions is needed for ab intio domain assembly.
Finally, folding hard β-proteins with complicated β-strand pairing patterns continues to be a hard, unsolved problem, especially when contact prediction fails to detect long-range β-pairings. In the absence of appropriate contact restraints, the current ab initio QUARK structure assembly process has difficulty in sampling such complicated topologies within the given simulation time. In the future, this deficiency may be addressed by enumerating all possible β-folds as initial conformations for ab initio folding, and by implementing swapping movements between two β-strands in the structure assembly simulations. Studies along this line are in progress.
Supplementary Material
Acknowledgments
The authors want to thank Xiaoqiong Wei and Wei Zheng for insightful discussions. This work was supported in part by the National Institute of General Medical Sciences [GM083107, GM116960] and the National Science Foundation [DBI1564756]. The I-TASSER server uses the Extreme Science and Engineering Discovery Environment (XSEDE)53, which is supported by National Science Foundation grant number ACI-1548562.
References
- 1.Mariani V, Kiefer F, Schmidt T, Haas J, Schwede T. Assessment of template based protein structure predictions in CASP9. Proteins. 2011;79:37–58. doi: 10.1002/prot.23177. [DOI] [PubMed] [Google Scholar]
- 2.Huang YJP, Mao BC, Aramini JM, Montelione GT. Assessment of template-based protein structure predictions in CASP10. Proteins. 2014;82:43–56. doi: 10.1002/prot.24488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Modi V, Xu QF, Adhikari S, Dunbrack RL. Assessment of template-based modeling of protein structure in CASP11. Proteins. 2016;84:200–220. doi: 10.1002/prot.25049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y. Progress and challenges in protein structure prediction. Current Opinion in Structural Biology. 2008;18(3):342–348. doi: 10.1016/j.sbi.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinch LN, Li WL, Monastyrskyy B, Kryshtafovych A, Grishin NV. Evaluation of free modeling targets in CASP11 and ROLL. Proteins-Structure Function and Bioinformatics. 2016;84:51–66. doi: 10.1002/prot.24973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monastyrskyy B, D’Andrea D, Fidelis K, Tramontano A, Kryshtafovych A. New encouraging developments in contact prediction: Assessment of the CASP11 results. Proteins. 2016;84:131–144. doi: 10.1002/prot.24943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu ST, Szilagyi A, Zhang Y. Improving Protein Structure Prediction Using Multiple Sequence-Based Contact Predictions. Structure. 2011;19(8):1182–1191. doi: 10.1016/j.str.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ovchinnikov S, Kim DE, Wang RYR, Liu Y, DiMaio F, Baker D. Improved de novo structure prediction in CASP11 by incorporating coevolution information into Rosetta. Proteins. 2016;84:67–75. doi: 10.1002/prot.24974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nature Protocols. 2010;5(4):725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nature Methods. 2015;12(1):7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu D, Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins-Structure Function and Bioinformatics. 2012;80(7):1715–1735. doi: 10.1002/prot.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu D, Zhang Y. Toward optimal fragment generations for ab initio protein structure assembly. Proteins. 2013;81(2):229–239. doi: 10.1002/prot.24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y. Interplay of I-TASSER and QUARK for template-based and ab initio protein structure prediction in CASP10. Proteins-Structure Function and Bioinformatics. 2014;82:175–187. doi: 10.1002/prot.24341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang WX, Yang JY, He BJ, Walker SE, Zhang HJ, Govindarajoo B, Virtanen J, Xue ZD, Shen HB, Zhang Y. Integration of QUARK and I-TASSER for Ab Initio Protein Structure Prediction in CASP11. Proteins. 2016;84:76–86. doi: 10.1002/prot.24930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang JY, Zhang WX, He BJ, Walker SE, Zhang HJ, Govindarajoo B, Virtanen J, Xue ZD, Shen HB, Zhang Y. Template-based protein structure prediction in CASP11 and retrospect of I-TASSER in the last decade. Proteins. 2016;84:233–246. doi: 10.1002/prot.24918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kinch L, Shi SY, Cong Q, Cheng H, Liao YX, Grishin NV. CASP9 assessment of free modeling target predictions. Proteins-Structure Function and Bioinformatics. 2011;79:59–73. doi: 10.1002/prot.23181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tai CH, Bai HJ, Taylor TJ, Lee B. Assessment of template-free modeling in CASP10 and ROLL. Proteins-Structure Function and Bioinformatics. 2014;82:57–83. doi: 10.1002/prot.24470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weigt M, White RA, Szurmant H, Hoch JA, Hwa T. Identification of direct residue contacts in protein-protein interaction by message passing. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(1):67–72. doi: 10.1073/pnas.0805923106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones DT, Buchan DWA, Cozzetto D, Pontil M. PSICOV: precise structural contact prediction using sparse inverse covariance estimation on large multiple sequence alignments. Bioinformatics. 2012;28(2):184–190. doi: 10.1093/bioinformatics/btr638. [DOI] [PubMed] [Google Scholar]
- 20.Kamisetty H, Ovchinnikov S, Baker D. Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(39):15674–15679. doi: 10.1073/pnas.1314045110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng JL, Baldi P. Three-stage prediction of protein beta-sheets by neural networks, alignments and graph algorithms. Bioinformatics. 2005;21:I75–I84. doi: 10.1093/bioinformatics/bti1004. [DOI] [PubMed] [Google Scholar]
- 22.Wu S, Zhang Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 2008;24(7):924–931. doi: 10.1093/bioinformatics/btn069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kosciolek T, Jones DT. De Novo Structure Prediction of Globular Proteins Aided by Sequence Variation-Derived Contacts. Plos One. 2014;9(3) doi: 10.1371/journal.pone.0092197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He B, Mortuza SM, Wang Y, Shen HB, Zhang Y. NeBcon: protein contact map prediction using neural network training coupled with naive Bayes classifiers. Bioinformatics. 2017;33(15):2296–2306. doi: 10.1093/bioinformatics/btx164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y. Protein structure prediction: when is it useful? Curr Opin Struct Biol. 2009;19(2):145–155. doi: 10.1016/j.sbi.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang J, Wang Y, Zhang Y. ResQ: An Approach to Unified Estimation of B-Factor and Residue-Specific Error in Protein Structure Prediction. Journal of molecular biology. 2016;428(4):693–701. doi: 10.1016/j.jmb.2015.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xue Z, Xu D, Wang Y, Zhang Y. ThreaDom: extracting protein domain boundary information from multiple threading alignments. Bioinformatics. 2013;29(13):i247–i256. doi: 10.1093/bioinformatics/btt209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu ST, Zhang Y. LOMETS: A local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007;35(10):3375–3382. doi: 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Skolnick J. SPICKER: A clustering approach to identify near-native protein folds. Journal of Computational Chemistry. 2004;25(6):865–871. doi: 10.1002/jcc.20011. [DOI] [PubMed] [Google Scholar]
- 30.Xu D, Zhang Y. Improving the Physical Realism and Structural Accuracy of Protein Models by a Two-Step Atomic-Level Energy Minimization. Biophys J. 2011;101(10):2525–2534. doi: 10.1016/j.bpj.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J, Liang Y, Zhang Y. Atomic-level protein structure refinement using fragment-guided molecular dynamics conformation sampling. Structure. 2011;19(12):1784–1795. doi: 10.1016/j.str.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Kihara D, Skolnick J. Local energy landscape flattening: Parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 2002;48:192–201. doi: 10.1002/prot.10141. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Y, Kolinski A, Skolnick J. TOUCHSTONE II: A new approach to ab initio protein structure prediction. Biophys J. 2003;85(2):1145–1164. doi: 10.1016/S0006-3495(03)74551-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu ST, Skolnick J, Zhang Y. Ab initio modeling of small proteins by iterative TASSER simulations. Bmc Biology. 2007;5 doi: 10.1186/1741-7007-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33(7):2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li YQ, Zhang Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 2009;76(3):665–674. doi: 10.1002/prot.22380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y. I-TASSER server for protein 3D structure prediction. Bmc Bioinformatics. 2008;9(1):1. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J, Zhang Y. A Novel Side-Chain Orientation Dependent Potential Derived from Random-Walk Reference State for Protein Fold Selection and Structure Prediction. Plos One. 2010;5(10) doi: 10.1371/journal.pone.0015386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou HY, Skolnick J. GOAP: A Generalized Orientation-Dependent, All-Atom Statistical Potential for Protein Structure Prediction. Biophys J. 2011;101(8):2043–2052. doi: 10.1016/j.bpj.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15(11):2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2004;57(4):702–710. doi: 10.1002/prot.20264. [DOI] [PubMed] [Google Scholar]
- 42.Cheng JL, Baldi P. Improved residue contact prediction using support vector machines and a large feature set. Bmc Bioinformatics. 2007;8 doi: 10.1186/1471-2105-8-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu S, Zhang Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 2008;24(7):924–931. doi: 10.1093/bioinformatics/btn069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Seemayer S, Gruber M, Söding J. CCMpred—fast and precise prediction of protein residue–residue contacts from correlated mutations. Bioinformatics. 2014;30(21):3128–3130. doi: 10.1093/bioinformatics/btu500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaján L, Hopf TA, Kalaš M, Marks DS, Rost B. FreeContact: fast and free software for protein contact prediction from residue co-evolution. Bmc Bioinformatics. 2014;15(1):1. doi: 10.1186/1471-2105-15-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang J, Shen H-B. An ensemble predictor by fusing multiple base predictors composed by both coevolution-based and machine learning-based approaches. Abstract of CASP11 experiment. 2014:209–210. http://www.predictioncenter.org/casp11/doc/CASP11_Abstracts.pdf;
- 47.Jones DT, Singh T, Kosciolek T, Tetchner S. MetaPSICOV: combining coevolution methods for accurate prediction of contacts and long range hydrogen bonding in proteins. Bioinformatics. 2015;31(7):999–1006. doi: 10.1093/bioinformatics/btu791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yan RX, Xu D, Yang JY, Walker S, Zhang Y. A comparative assessment and analysis of 20 representative sequence alignment methods for protein structure prediction. Scientific Reports. 2013;3 doi: 10.1038/srep02619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kryshtafovych A, Barbato A, Monastyrskyy B, Fidelis K, Schwede T, Tramontano A. Methods of model accuracy estimation can help selecting the best models from decoy sets: Assessment of model accuracy estimations in CASP11. Proteins. 2016;84(Suppl 1):349–369. doi: 10.1002/prot.24919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 2009;77(S9):100–113. doi: 10.1002/prot.22588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang S, Peng J, Ma J, Xu J. Protein secondary structure prediction using deep convolutional neural fields. Scientific reports. 2016;6 doi: 10.1038/srep18962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Magnan CN, Baldi P. SSpro/ACCpro 5: almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning and structural similarity. Bioinformatics. 2014;30(18):2592–2597. doi: 10.1093/bioinformatics/btu352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Towns J, Cockerill T, Dahan M, Foster I, Gaither K, Grimshaw A, Hazlewood V, Lathrop S, Lifka D, Peterson GD, Roskies R, Scott JR, Wilkins-Diehr N. XSEDE: Accelerating Scientific Discovery. Computing in Science & Engineering. 2014;16(5):62–74. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.