

Abstract
Disease represents a specific case of malfunctioning within a complex system. Whereas it is often feasible to observe and possibly treat the symptoms of a disease, it is much more challenging to identify and characterize its molecular root causes. Even in infectious diseases that are caused by a known parasite, it is often impossible to pinpoint exactly which molecular profiles of components or processes are directly or indirectly altered. However, a deep understanding of such profiles is a prerequisite for rational, efficacious treatments. Modern omics methodologies are permitting large-scale scans of some molecular profiles, but these scans often yield results that are not intuitive and difficult to interpret. For instance, the comparison of healthy and diseased transcriptome profiles may point to certain sets of involved genes, but a host of post-transcriptional processes and regulatory mechanisms renders predictions regarding metabolic or physiological consequences of the observed changes in gene expression unreliable. Here we present proof of concept that dynamic models of metabolic pathway systems may offer a tool for interpreting transcriptomic profiles measured during disease. We illustrate this strategy with the interpretation of expression data of genes coding for enzymes associated with purine metabolism. These data were obtained during infections of rhesus macaques (Macaca mulatta) with the malaria parasite Plasmodium cynomolgi or P. coatneyi. The model-based interpretation reveals clear patterns of flux redistribution within the purine pathway that are consistent between the two malaria pathogens and are even reflected in data from humans infected with P. falciparum.
Keywords: Biochemical Systems Theory, Dynamic Model, Generalized Mass Action System, Malaria, Metabolic Modeling, Transcriptomics
Introduction
In contrast to many other complex diseases, such as cancer, Crohn’s disease, or metabolic syndrome, infectious diseases have the distinction of a clear root cause: a pathogen has invaded the body and was not stopped by the host’s natural immune defenses. If the pathogen can be eliminated with medical or pharmaceutical means, the disease has a straightforward cure. However, in many cases this is not directly possible, or it requires a relatively long period of time, during which the patient is at risk of deteriorating, with possibly lethal consequence. In these cases, the root cause becomes almost immaterial, and it is the complex system of interactions between the pathogen and the host that needs to move to the center of attention [1]. The intriguing aspect of these interactions is that we often have no real understanding of which specific subsystems in the host or the pathogen are turned on or off, so that any molecular characterization of the disease, or any attempt of a targeted intervention, becomes an enormous challenge. As an illustration, we use in this paper a case study of malaria, which afflicts more than 200 million people world-wide and kills about half a million individuals per year, many of whom are children [2]. While malaria is initially a disease of the blood, it quickly affects other tissues and organ functions and triggers uncounted responses of the host’s defense systems. To measure the molecular state of a person is therefore an unsurmountable problem. The Malaria Host Pathogen Interaction Center (MaHPIC; www.systemsbiology.emory.edu/) actually offers the great advantage of allowing us to collect very large datasets on infections of different monkey species with different malaria parasites. These non-human primates (NHPs) present with symptoms of malaria that are very similar to those in humans and permit experiments that would not be possible in humans, due to ethical and other considerations.
The complexity of host responses to parasites poses a grand challenge. Namely, it is not even clear what exactly should optimally be measured, even if one had the luxury of being able to obtain measurements frequently. Of course, some experimental targets are quite evident. For instance, one can certainly measure the numbers of pathogens on a regular basis during an infection in order to characterize the extent of the disease. Such measurements can then be used to develop models of specific aspects of the disease [3–5]. One can also measure generic physiological markers like body temperature and blood cell counts, which reflect the severity of the disease at a high level. However, if the goal is to identify specific drug targets, a much deeper understanding of the molecular events during an infection is required. This necessity of a better characterization of processes is problematic, because it is generally much more difficult to measure processes than states. As a consequence, disease investigations must usually resort to measuring molecular profiles. The good news is that the –omics revolution has rendered it possible to assess molecular profiles incomparably more comprehensively than just a couple of decades ago. For instance, we can relatively easily and reliably measure the expression of most genes, and in the process distinguish between host and pathogen genes. Modern mass spectrometry has rendered it possible to establish profiles of many thousands of native and foreign metabolites and their break-down products, even though it is not always entirely clear how such high-throughput results are to be interpreted. While not yet as definitive as genome analyses, proteomics, metabolomics, and lipidomics offer a glimpse into the abundances of subclasses of metabolites, proteins, and lipids. Taken together, modern biology allows us to convert small volumes of biological samples into enormous datasets.
The sheer sizes of -omics datasets pose challenges that are relatively new to the field of biology [6]. Namely, it is no longer easy to discern valid information or true signals in the data from uncertainties, variability, and noise. Every diseased individual is different, and many differences in gene expression within a patient cohort may simply be manifestations of their genetic make-up and health histories. As a pertinent example in our case study, which we will discuss later in this article, seemingly similar macaques responded very differently to malaria infections, with some suffering relatively lightly, some very severely, and one not even surviving [7]. Clearly, humans and monkeys are dynamical entities whose features change over time. Also, of course, all experiments are burdened with certain inaccuracies, which may not be fully characterized. As a consequence of these and other complications, the expression of a given gene or protein at a given time point may be suggestive of a biomarker of disease, but it may also be a spurious event.
In this article, we describe, as a proof of principle, a computational strategy for approaching the complex questions raised in the previous paragraphs. We use malaria as an example and discuss how different types of experiments and computational analyses have shed light on unforeseen aspects of the disease. However, we certainly do not claim to have obtained complete or definitive answers to the questions we had asked at the beginning of our analysis. In other words, this article focuses on strategies rather than results.
Material and Methods
A Brief Background on Malaria
In all types of malaria, the sporozoite form of the Plasmodium parasite enters the human or NHP host through a mosquito bite. Moving quickly with the blood stream, the sporozoites soon reach the liver, where they infect hepatocytes. Depending on the species, the pathogens may remain in the liver for a long time in the form of hypnozoites, or they multiply aggressively and over time release thousands of merozoites into the bloodstream. The merozoites invade red blood cells (RBCs), where they multiply and, after a day or two, depending on the species, are released and infect other RBCs. Eventually, some of the parasites transform into sexual forms, called gametocytes, which are taken up by another mosquito. They mate within the mosquito, thereby completing the pathogen’s life cycle.
The responses of the human and NHP hosts to the Plasmodium parasite are multifold, complicated and in very many aspects ill understood. It is not surprising that the invasion and destruction of RBCs typically leads to anemia. However, the details of how the disease affects the erythropoietic system and thus the dynamics of new RBC formation, as well as their aging and removal, are not well understood. A second clearly affected host subsystem is the immune system. The presence of the pathogen very definitively triggers numerous cellular and humoral immune responses, but the chains of events leading to these responses have remained obscure.
The reasons for these gaps in our understanding are manifold. First and foremost, the immune system is exceedingly complicated. It contains uncounted components in the form of different immune cells and specific proteins, such as immunoglobulins and cytokines, whose roles are not always exactly known. Confounding the situation is the difficulty of obtaining sufficiently large high-quality datasets. Yet another puzzle is the observation that the host often seems to clear an infection, only to suffer a relapse, which has different characteristics than the initial infection. Even this very coarse overview of a few aspects of malaria will render it evident that the disease is systemic and that large numbers of physiological subsystems interact in a life-or-death effort to control the disease.
Generic Data-Based Characterization of a Complex Disease
Extracting information from large omics datasets has been compared to “drinking from a firehose.” Yet, even comprehensive attempts to measure pertinent data are not always sufficient. Within the context of our case study, our Malaria Host Pathogen Interaction Center (MaHPIC; www.systemsbiology.emory.edu/) has been collecting frequent –omics datasets to characterize the process accompanying malarial infections in NHPs. Although well-equipped and well-funded, this effort has been encountering complicated obstacles that are typical for investigations of complex diseases.
Even specifically with respect to the –omics of blood, which is much easier to obtain than measurements from other tissue samples, the following limitations arose. First, issues of ethics and animal welfare restrict blood draws from macaques, for example, to 10 milliliters/kilogram/month, or to 6 milliliters/kilogram/month if the animal is anemic. This regulation results in a spacing of measurement time points that obviously precludes the assessment of immediate metabolic host responses to the emergence of pathogens in the bloodstream. In fact, one is led to assume that metabolism, measured in this manner, is always in a steady state. It is permissible to obtain blood from the monkeys daily through standard procedures involving ear pricks, where no anesthesia is required, but it surprisingly turned out that blood from this source is metabolically quite different from venous blood [8].
Second, multi-omics approaches are often envisioned to include genomics, proteomics, metabolomics, and maybe other measurements from the same source at the same time. In our case, it is of course possible to subject blood samples to these different omics measurements. However, these measurements shed light on different blood components. RBCs, which are affected most directly, have no nuclei or mitochondria and therefore no DNA. Thus, “blood genomics” automatically and necessarily excludes about 99% of all blood cells, as it is restricted to white blood cells and parasites. By contrast, plasma proteomics is overshadowed by typical proteins like albumins, while membrane-proteomic measurements from infected RBCs are dominated by the host RBC membrane proteins, simple due to their sheer number. Metabolomics suffers from the fast time scale of metabolic profiles that change very quickly, even with quenching. Thus, the “different omics from the same source” turn out to yield a heterogeneous gemisch of information. Notwithstanding, this information is very valuable, but it is by far not as straightforward, comprehensive and indicative of the same molecular events as one might naïvely assume.
In spite of this unavoidable, natural heterogeneity, the different –omics measurements can be used to identify correlations and associations with methods of statistical machine learning. As a case in point, we analyzed the transcriptional response in the bone marrow (BM) of macaques of the species Macaca mulatta to infection with the malaria parasite Plasmodium cynomolgi to test the overarching hypothesis that infected animals during relapses exhibit substantial molecular changes similar to those observed during the primary infection [9]. Contrary to this hypothesis, we found very little impact on the transcriptional profiles in the BM during relapses. We then correlated gene- and pathway-level changes in the transcriptional profiles during peak parasitemia with immunological and chemical profiles and found that differentially expressed genes in the BM were primarily related to ongoing inflammatory responses that were measurable in the periphery and dominated by interferon signatures. The analysis ultimately suggested that malarial anemia may be driven by monocyte-associated suppression of the transcription factors GATA1/2 in erythroid progenitors, resulting in disrupted erythropoiesis and insufficient erythropoietic output. Further study led to the creation of an atlas of tissue-specific Multi-Omics Relatedness Networks (MORNs) of malaria, which exhibit differential dynamics of the host-immune response to P. cynomolgi infection in the peripheral blood and BM. When the MORNs were integrated with immunological profiles—including cytokine profiles—and clinical traits to bridge molecular mechanisms with disease outcomes, we found a positive correlation of PD-1 and mTOR signaling with PD-1+ central memory CD8+ T cells and PD-1+ B cells, which suggested a possible involvement of the combination of these signals in immune memory.
Direct Data Generation and Analysis
1. Details of Induced Malaria Infections
Details of data acquisition and raw data analysis for infections of rhesus macaques (M. mulatta) with P. cynomolgi were recently described in [7]. Here, we use, for easy comparisons, the same names for the macaques as in [7], namely, RFa14, RFv13, RIc14, RMe14, and RSb14. Corresponding details for infections with P. coatneyi will be published elsewhere. The names of the macaques in this study were RCs13, RTi13, RUn13, RWr13 and RZe13. For the following, it is sufficient to present the most pertinent highlights.
1.1. Parasites
Infections were introduced using P. coatneyi or P. cynomolgi B/M strain sporozoites and carried out for approximately 100 days. Baseline control measurements were taken before infections were introduced. Each infection was started with an inoculum of about 100 P. coatneyi or 2,000 P. cynomolgi sporozoites that were freshly dissected from the salivary glands of Anopheles mosquitos bred at a laboratory of the Centers of Disease Control and Prevention (CDC). Some auxiliary data analyzed here pertained to human infections with P. falciparum, which corresponds closely to the macaque parasite P. coatneyi.
1.2. Monkeys
The rhesus macaques (M. mulatta) used for the infections under investigation were malaria-naïve males, which had been born and raised at the Yerkes National Primate Research Center at Emory University. The limitation to one gender eliminated potential blood loss issues associated with the female menstrual cycle, which could have confounded efforts to characterize malarial anemia. All experimental methods followed standard protocols that were approved by Emory’s Institutional Animal Care and Use Committee (IACUC). The monkeys were followed throughout the infections and through relapses in the case of P. cynomolgi, if they occurred within the 100-day experimental period. Details for P. cynomolgi were described in [7], and the corresponding details for P. coatneyi will be published elsewhere.
1.3. Time Point Sampling
In each experiment, the first sample was taken at time point 1 (TP1), which served as a baseline control. TP2 was chosen at peak parasitemia and TP3 immediately after this peak. TP4 – TP7 were spread out over the remaining days during the 100-day experiments; some were coarsely aligned with relapses, others with phases in between.
2. Transcriptome Analysis
2.1. Library preparation for RNASeq
RNAs were extracted from whole blood using Tempus-Spin RNA isolation kits and from BM mononuclear cells using Qiagen RNEasy Mini-Plus kits, according to the manufacturer’s instructions. Sufficient quality of RNA samples was confirmed using an Agilent Bioanalyzer. Approximately 1 μg of total RNA per sample was reverse-transcribed into double-stranded cDNA. Strand-specific libraries were generated using Illumina TruSeq Stranded mRNA Sample Prep kits. For quality control, spike-in RNAs with known GC proportion and concentration (ERCC Spike-In Control, Life Technologies) [10] were added to each library to constitute 1% of total RNA.
2.2. Quantification of gene expression
Libraries were sequenced on the Illumina HiSeq 2000 at the Yerkes Genome Core. RNASeq reads were aligned to a reference genome (assembly of M. mulatta version 4.0, GenBank accession number PRJNA214746 ID: 214746) using Tophat2 with default parameters [11, 12]. Reads mapping to multiple genomic locations were excluded from the analysis to ensure high-confidence mapping. Transcript abundance was inferred at the level of annotated genes using HTSeq v0.5.4 [13]. Data reliability was assessed by quality control: linear correlation of spike-in control abundance with known concentration; confirmation of strand-specificity of controls as 99.9%; and confirmation of the absence of 3’ bias in the controls with the RSeqC software [14]. Gene expression was normalized to library size with the R package DESeq (version1.10.1; [15]), using default parameters.
2.3. RNASeq Data Processing
To minimize noise, features below a minimum FPKM (Fragments Per Kilobase of transcript per Million mapped reads) cutoff value of 32 were excluded from further analysis. Gene expression data were log2-transformed. A large variance in gene expression was observed among animals. Supervised Normalization of Microarrays (SNM) [16] was used to remove the variance associated with the animal effect. The SNM model was used with “longitudinal time points” as the biological variable, representing the course of pre-infection, acute primary infection, post-peak, inter-relapse and relapses, and “animal” as the adjustable variable.
2.4. Differential Expression Analysis (DEA)
DEA analysis was performed using analysis of variance in the JMP Genomics software (SAS Institute Inc., Cary, NC). “Individual animal” was set as random effect and the “longitudinal time point” as a fixed effect. Differences in gene expression across all genes among the fixed effects were tested. Benjamini-Hochberg false discovery rate (FDR; [17]) corrections were used to adjust for multiple hypothesis testing; FDR ≤ 0.05 was used as the significance threshold.
2.5. Human transcriptome analysis
In order to assess whether the NHP results on changes in purine metabolism might be relevant for human malaria, we took advantage of a human malaria study [18] and downloaded the human transcriptome data of this study from the Gene Expression Omnibus (GEO accession number GSE67184). Briefly, 12 individuals in this study volunteered in a challenge trail. Blood samples were taken at two time points, namely prior to a P. falciparum challenge (baseline) and at the day of diagnosis (infection). Data processing and normalization were described in the original paper. Normalized expression values for selected genes were used for modeling in the same way as described for non-human primate models.
3. Gene Set Enrichment Analysis
To identify pathway-level changes in gene expression, we performed Gene Set Enrichment Analysis (GSEA; [19, 20]), which determines the statistical significance of the frequency of changes in expression for specific sets of genes within a larger set of genes. GSEA leads to a ranked list of pathways that are likely to be most affected, as judged by differentially expressed genes.
4. Dynamic Modeling
To interpret the implications of transcriptional changes during the infection, we employed a kinetic model of purine metabolism that we had developed, diagnosed, and validated a number of years ago [21–24]. This model has the format of a Generalized Mass Action system, where all processes are represented as products of power-law functions. It contains 16 metabolites and 37 fluxes, as well as a large number of regulatory signals. A diagram of the model structure is shown in Figure 1 and the equations are presented in the Supplements. It is evident that the pathway system is tightly regulated through numerous inhibiting and activating signals. The mathematical formulation of the model was established for purine metabolism in humans, and we assume here that this implementation is applicable to NHPs as well. We therefore use the model without changes.
Figure 1. Model diagram of purine metabolism (adapted from [23]).

Peach-colored boxes contain metabolites or pools of metabolites. Blue arrows indicate enzyme-catalyzed reactions or transport steps. Subscripted quantities v denote enzymes primarily responsible for associated steps. Solid green and dashed red arrows represent activating or inhibiting signals, respectively. Abbreviations of metabolite pools: PRPP, phosphoribosylpyrophosphate; IMP, inosine monophosphate; S-AMP, adenylosuccinate; Ado, adenosine; AMP, adenosine monophosphate; ADP, adenosine diphosphate; ATP, adenosine triphosphate; SAM, S-adenosyl-L-methionine; Ade, adenine; XMP, xanthosine monophosphate; GMP, guanosine monophosphate; GDP, guanosine diphosphate; GTP, guanosine triphosphate; dAdo, deoxyadenosine; dAMP, deoxyadenosine monophosphate; dADP, deoxyadenosine diphosphate; dATP, deoxyadenosine triphosphate; dGMP, deoxyguanosine monophosphate; dGDP, deoxyguanosine diphosphate; dGTP, deoxyguanosine triphosphate; RNA, ribonucleic acid; DNA, deoxyribonucleic acid; HX, hypoxanthine; Ino, inosine; dIno, deoxyinosine; Xa, xanthine; Gua, guanine; Guo, guanosine; dGuo, deoxyguanosine; UA, uric acid; R5P, ribose-5-phosphate. The Supplements contain a list of the reactions names and their abbreviations.
To achieve a coarse interpretation of the transcriptomics data in this study, we supposed that fold-changes in the expression of pertinent coding genes correspond to the same fold-changes in enzyme amounts. Though this assumption ignores issues of post-transcriptional regulation, it is expected that it may be qualitatively, and possibly quantitatively, appropriate, at least approximately. Thus, the differential expression of each gene was incorporated as a corresponding change in enzyme amount. For missing values, where a transcript had not been measured, we retained the original parameters. In cases of isozymes, the gene expression values were averaged.
In the original model formulation, the enzyme activities were lumped into apparent rate constants. Therefore, the differential expression of each gene was modeled as a corresponding change in its respective reaction rate constant parameter. All other parameters were retained at the same values as at the original steady state. The model equations were then integrated to obtain a new steady state, and the variable concentrations and fluxes of the system were studied. This analysis was performed for every monkey and every time point during its infection.
As an example, consider the dynamics of xanthosine monophosphate (XMP; X7), an intermediate between inosine monophosphate (IMP; X2) and the pool of guanosine phosphates (GTP, GDP, GMP; “GXP”; X8). The production of XMP depends on the substrate, IMP, and is inhibited by both XMP and GXP. It is catalyzed by the enzyme IMP dehydrogenase (IMPD). The dynamics of XMP is formulated in the Generalized Mass Action (GMA) model as
(see Supplements for other equations). In GMA models, which are based on Biochemical Systems Theory [24–27], all factors contributing to a process are modeled as power-law functions, in which the exponent, called the kinetic order, quantifies the strength of the effect of the variable on the production term. The kinetic order associated with the substrate, IMP, in the term for the production of XMP is positive, whereas the two inhibitory signals, from XMP and GXP, are negative. The product of these power-law functions is multiplied by a rate constant. The amount or activity of the catalyzing enzyme, IMPD, is not explicit in the synthesis term, but subsumed in this rate constant. The degradation term of X7 is constructed similarly.
To account for a change in the expression of the gene coding for IMPD, we replace the value 1.2823 of the rate constant with 1.2823 × (fold-change in IMPDH1) (see Figure 2). This procedure was repeated for all significant changes in transcriptomics at a given time point for a given animal.
Figure 2. Heat map of changes in gene expression at different time points (TP2 – TP7) during P. cynomolgi infection.

Shown here are transcriptomic changes in the bone marrow of monkey RSb14, relative to gene expression at TP1. TP3, which immediately follows the peak of infection (TP2), exhibits the strongest changes. Other expression patterns were similar for different monkeys, infected with the same parasite (P. cynomolgi; see Supplement Figure S1) or even a different parasite (P. coatneyi). Results are shown on a log-10 scale.
Results
Interpretation of Blood Transcriptomics
Transcriptomics is generally viewed to be the most reliable source of omics information. However, its output is not always easy to interpret in terms of phenotypical outcomes, as gene expression is two or three steps removed from metabolic, immunological, or physiological manifestations. Faced with this challenge, we employed computational modeling to interpret transcriptomic information.
Gene Set Enrichment Analysis of transcriptional data during malarial infection showed significant changes in the expression of genes associated with purine pathway, leading us to choose this pathway as an example for interpreting transcriptomic changes with metabolic models. This pathway is furthermore of interest, as it has been implicated in a variety of inflammation studies (see Discussion).
Specifically, we considered gene expression at seven time points (TPs). Measurements at TP1, which preceded the infections, were used as “baseline control” values to which other TP measurements were normalized. TP2 coincided with the peak of infection. Interestingly, the most significant changes in purine metabolism transcriptional profiles occurred at TP3, a few days after peak parasitemia (Figure 2). The expression patterns more or less returned to the baseline at later time points.
Quite similar transcriptomic patterns were obtained for NHPs infected with different Plasmodium species (P. cynomolgi and P. coatneyi), and even with data from a human cohort (see later). In the cases of rhesus macaques, the strength of the pattern correlated with the severity of parasitemia [7]: animals with higher parasitemia at TP3 showed more pronounced patterns than the animals with lower parasitemia.
The altered expression of numerous genes coding for enzymes within purine metabolism is interesting, but difficult to interpret, because altered gene expression levels do not reveal much regarding changes in the metabolic concentration or flux profile, which in turn could lead to new functional insights or hypotheses. For example, simultaneous up-regulation of separate genes coding for the production and for the degradation of a metabolite could result in no net changes in the level of that metabolite.
Thus, our first goal was to assess to what degree transcriptomic changes alone are predictive of changes in metabolic fluxes. Such predictions are complicated due to the fact that changes in enzyme activities lead to changes in fluxes, which cause alterations in metabolite concentrations, which in turn affect the magnitudes of fluxes. Because purine metabolism constitutes a complex, highly regulated pathway system, intuition regarding the ultimate consequences of quantitative alterations in transcripts is quickly overwhelmed.
We therefore employed the kinetic model of purine metabolism described in the Section Materials and Methods and implemented significant fold-changes in the expression of pertinent coding genes as the same fold-changes in enzyme amounts (Figure 2). A priori, one might expect that a plot of the magnitudes of changed fluxes at the steady state would exhibit a clear positive trend. However, this trend, while it does exist, is quite weak (Figure 3), with a correlation coefficient of 0.1228; the difference between this value and 0 itself is significant with a correlation coefficient of 0.00069. These findings suggest that transcriptomic data alone are insufficient to predict flux distributions in this system.
Figure 3. Limited predictability of fluxes from transcriptomics data alone.
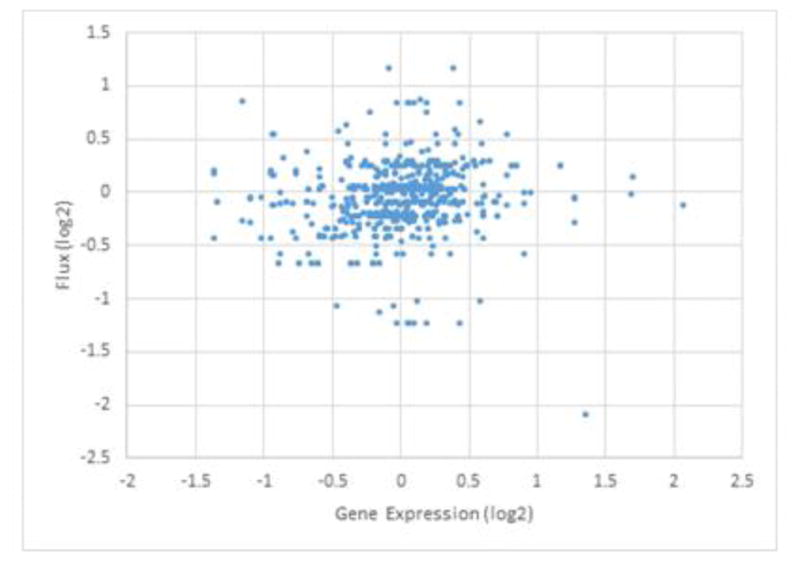
Fold-changes in steady-state fluxes, computed with the dynamic model, are plotted against fold-changes in gene expression in monkey RIc14 for time points TP2, …, TP7 during infection with P. cynomolgi. Both axes are presented on a log-2 scale, so that 1 represents a two-fold change. One notes horizontal lines, which correspond to several genes associated with the same pathway.
Changes in Purine Metabolism Following Peak Infection
Faced with the observation of unreliable predictability of metabolic fluxes from transcriptional changes, we explored what the kinetic model, implemented with altered transcriptional profiles, might suggest about metabolic responses during malarial infection. While we analyzed changes in purine metabolism for all measured time points, but the most substantial changes were noticed immediately following the peak of infection (TP3).
Although quite coarse, the model-based interpretation of the transcriptomics data revealed striking results. In the experiments with P. cynomolgi [7], two particular animals (RFa14 and RMe14) displayed severe symptoms and needed and received sub-curative treatment during the acute primary infection in order to aid in the control of the parasites and disease manifestations; one monkey (RFv13) actually had to be euthanized. By contrast, two other animals of the same cohort, RSb14 and RIc14, recovered without treatment, while experiencing much higher parasitemia. It is to be expected that these two untreated animals probably suffered from more severe inflammation at TP3. Particularly strong changes were observed in the fluxes from PRPP to IMP, from GMP/GDP/GTP to IMP, from IMP to HX/Ino/dIno, from HX/Ino/dIno to Xa, and the excretion of HX and Xa, which were elevated more than two-fold at TP3 compared to baseline. The concentration of the HX/Ino/dIno pool also increased more than two-fold at TP3 compared to baseline. By contrast, the two animals with lower parasitemia showed no changes in these fluxes or metabolite concentrations (data not shown).
In a similar experiment with P. coatneyi infections (manuscript in preparation), the macaques RWr13 and RTi13 exhibited higher parasitemia at TP3 and also showed much more pronounced changes in fluxes in the purine model than two other animals with lower parasitemias (Figure S2 and S3). Intriguingly, the same purine fluxes were increased as in the P. cynomolgi infection: from PRPP to IMP, from GMP/GDP/GTP to IMP, from IMP to HX/Ino/dIno, from HX/Ino/dIno to Xa, and the excretion of Xa and HX. These fluxes at TP3 were elevated more than two-fold compared to the baseline value before the infection (TP1). Also as in the P. cynomolgi infection, the concentration of HX/Ino/dIno increased more than two-fold at TP3 compared to baseline.
Taken together, the profile analysis of purine fluxes and metabolites computed with the model, based on transcriptomics data, reveals a distinct signature in the affected purine pathway. In the P. coatneyi infection experiment at TP3, clustering of the profiles of the 37 simulated fluxes in the model grouped the animals with high and low parasitemias separately (Figure 5A). Similar results were seen in clustering profiles of simulated concentrations of the 16 metabolites (Figure 5B). The P. cynomolgi infection experiments yielded the same results: both TP3 profiles of model fluxes (Figure 5C) and metabolite concentrations (Figure 5D) clustered the high-parasitemia and low-parasitemia animals separately.
Figure 5. Heat maps of simulated fluxes and metabolites immediately following the peak of infection.

- Heat map of model-inferred fluxes in P. coatneyi infections.
- Heat map of model-inferred metabolites in P. coatneyi infections.
- Heat map of model-inferred fluxes in P. cynomolgi infections.
- Heat map of model-inferred metabolites in P. cynomolgi infections.
Changes in Human Purine Metabolism during Malaria
To assess whether these results on macaques had any relevance for human malaria, we analyzed transcriptomics data from a human study [18], in which volunteers were enrolled for a challenge trial with P. falciparum, which is closely related to the macaque parasite P. coatneyi. RNASeq analysis was performed on 12 individuals. Blood samples were taken before the challenge (baseline) and at the day of diagnosis, which here is considered as a time point during infection. Normalized expression values of pertinent genes were used for simulation. Quite strikingly, we observed similar pattern of purine metabolism in some of individuals as in the severe animals (Figure 6). In particular, the same fluxes we observed in macaques exhibited significant alterations in humans, namely, the fluxes from GMP/GDP/GTP to IMP, from IMP to HX/Ino/dIno, from HX/Ino/dIno to Xa, as well as the excretion of HX and Xa. These results suggest that molecular mechanisms perturbing purine metabolism during malaria are conserved from non-human primates to humans.
Figure 6. Changes in fluxes and concentrations within purine metabolism before and during infection in a human volunteer challenge trial with P. falciparum.

All metabolites or metabolite pools are color-coded to show fold-increases or decreases in concentrations between infection and baseline: red colors represent increased concentrations at post-peak, while green colors represent decreased concentrations at post-peak, according to the log-2 color bar. White boxes indicate no significant changes. Red and green colors of fluxes represent up- or down-regulation at post-peak compared to baseline. Only fluxes with fold-changes greater than 2 are colored; others are shown in blue. The degree of change is indicated by the line thickness. For simplicity, regulatory signals are not shown (cf. Figure 1).
Changes in Purine Metabolism during Chronic Infection
In contrast to P. cynomolgi, P. coatneyi infections can become chronic, thereby causing persistent inflammation. Model simulations across all time points during primary and chronic infection with P. coatneyi showed purine-related responses in monkeys with higher parasitemia (RTi13 and RWr13) that were different from those with low parasitemia. Specifically, we found prolonged increases in fluxes from IMP to HX/Ino/dIno and the excretion of HX/Ino/dIno and Xa during the entire time from acute primary infection to recrudescence patency. These two animals also showed prolonged increases in the concentration of HX/Ino/dIno across all seven time points (Figures S4A and S4B). These changes were not observed for acute infections with P. cynomolgi. These findings are interesting, because HX and Ino have been shown to be associated with inflammation [28, 29].
Discussion and Conclusions
Disease reflects the malfunctioning of a very complex system. To combat disease effectively, we need to develop a deep understanding of the molecular signatures and events that the malfunctioning system exhibits. Especially within the setting of personalized disease and medicine, these signatures and their trends must be characterized in individualized detail, so that population-averaged information may be substituted with patient-specific parameters [30, 31]. In the past, experimental and clinical limitations allowed the quantification of only very restricted subsets of relevant biomarker signatures. The omics revolution has fundamentally changed this situation, because single experiments can easily yield thousands of data points, each of which could potentially be a biomarker. The new challenge accompanying these experiments is that it is often difficult to interpret the phenotypical ramifications of changes in patterns of molecular biomarkers. Here, we take as a pertinent example high-throughput transcriptional data, widely viewed to be comprehensive and useful. However, clinical manifestations in terms of metabolic or physiological aberrations, which may have their root causes in transcriptional changes, are several steps removed from the expression of genes so that the specific consequences of altered gene expression are difficult to intuit. Here, we address this issue by proposing an interpretation of genomics information through a dynamic model that allows us to translate changes at the transcript level into alterations in metabolic signatures.
We used as an example for our demonstration the responses of hosts to infections with malaria parasites. More precisely, we focused on purine metabolism, which was identified by gene set enrichment analysis as significantly affected during infection. While the expression of many genes associated with this pathway system was indeed changed to some degree, the functional impacts of this expression profile could not be interpreted easily. By contrast, using the transcriptome changes as corresponding changes in enzyme amounts in a dynamic model of purine metabolism and integrating this model to the steady state revealed a pattern of flux rearrangement within the pathway system. This pattern was consistent among different monkeys and even among infections with different Plasmodium parasites and in part correlated with parasitemia levels. The main results of this flux rearrangement were identified as an increased production and excretion of inosine (IN), hypoxanthine (HX) and xanthine (XA), as well as less dramatic changes in a few other compounds.
The fact that the same pattern was seen in many of the infected monkeys, and was somewhat corroborated by human malaria data, directly leads to new hypotheses regarding the synthesis and utilization of specific purine compounds. This type of hypothesis generation is valuable, but it is clear that the mathematical model alone is not in a position to provide a possible rationale for such changes. Nonetheless, the literature documents a number of observations regarding the roles of purines in malaria, and these may or may not turn out be relevant and/or explanatory. Two classes of observations with particular pertinence are the following.
1. Purine compounds are often associated with inflammation
It has been known for a while that the end product of the pathway, uric acid, is known to stimulate immune activation, even in the absence of bacteria or other stimuli [28, 29]. At the same time, IN inhibits inflammatory cytokine production [32] and, upon metabolic stress, cells release IN into the extracellular space, where elevated inosine levels are present in various inflammatory states. Along the same lines, HX and XA are increased in synovial fluid during inflammatory arthritis [33]. It has also been shown that adenosine and IN have anti-inflammatory effects [34–36]. More generically, a recent review described the role of purinergic signaling in the immune system [37].
Specifically with respect to malaria, hypoxanthine accumulates in infected RBCs [38]. When these release schizonts, the parasites precipitate uric acid, which is released into the blood stream [39]. HX is also released into circulation, where it may be converted into uric acid. Uric acid precipitates are considered key inflammation signals in malaria [38]. They are highly inflammatory molecules and serve as danger signals for the innate immune system [28]. In particular, uric acid precipitates move into micro-vessels, where they stimulate immune cells to produce IL-6, IL-8, IL-10, TNFα, sTNFRII, MCP-1, and IP-10 [40]. These inflammatory cytokines are considered important components of the host’s inflammatory reaction to Plasmodium infection and a major cause of malaria pathogenesis.
2. The Plasmodium parasite needs large amounts of purines for proliferation
Plasmodium does not have the metabolic machinery to synthesize purines de novo [41]. However, it propagates very quickly within RBCs and thus requires rapid large-scale synthesis of DNA, RNA and of ubiquitous factors like ATP and GTP. In particular, HX is essential for Plasmodium growth and commonly used as a required reagent in parasite cultures; its concentration in infected RBCs is much higher than in uninfected ones [38, 39]. An article by Downie et al. [41] summarized alternative mechanisms in Plasmodium for providing purines. Most importantly, the parasite relies on salvaging purine compounds, which are mainly funneled through HX to IMP. HX is transported from plasma into the cytoplasm and can also be produced from adenosine or IN. To augment the typical salvage observed in mammals, which is facilitated by the enzymes HGXPRT and APRT, P. falciparum has an additional enzyme, PfHGXPRT, that utilizes XA instead of HX or adenine, respectively. As an aside, enzymes involved in purine metabolism have been proposed as potential drug targets [42].
The predictions of the model have yet to be validated against independent data which, however, are presently not available. We actually took metabolomic measurements, but limitations in annotation prevented us from a more detailed validation of our predictions. Because of these challenges, we analyzed the human data, which showed consistent trends, and which we consider a type of independent validation. More generally, mechanistic-level validation data would be difficult to obtain. For instance, the increased appearance of purine compounds in the blood stream, or the lack of significant changes in metabolic profile within serum, could both be explained in numerous ways. First, the main driver of blood metabolites is the liver, which dwarfs any action by white blood cells whose transcriptional profiles are being analyzed here, thus limiting interpretability of experimental measurements of serum purine levels. Second, suppose the parasite indeed somehow triggered the production of purines in white blood cells. It would most likely do so in order to utilize these compounds, which it cannot synthesize de novo. Thus these compounds would easily enter the red blood cells, where they would quickly be used up by the parasites, causing them to not be measured and providing a false negative result for validation.
Our results and the corresponding observations documented in the literature are intriguing and puzzling at the same time. The consistency among species and the correlation with the degree of parasitemia suggest that the changes are not merely spurious occurrences. Also, at first glance, the results seem to suggest that more hypoxanthine and inosine are produced because the parasites require them. However, such a casual interpretation of the results requires caution and careful consideration. After all, the parasites invade RBCs, which do not have nuclei and therefore cannot respond with transcriptional changes as they are observed in the white blood cells (WBCs). This difference in involvement in the host response raises a complicated question regarding causality of events: Do the parasites deplete purine compounds like ATP in RBCs, which then signal the need for purines, to which WBCs respond with transcriptomic and metabolic alterations? Of course, the model cannot help with a biological interpretation of such speculations, the computational inferences, or the data themselves. Nonetheless, it is remarkable that a metabolic model analysis of the type shown here can lead to new hypotheses regarding the targeted rerouting of fluxes, and regarding a rationale for changes in transcriptomics. It is now up to wet lab experimentalists to test these hypotheses.
Supplementary Material
Figure 4. Identification of changes in fluxes and concentrations within purine metabolism immediately after peak infection (TP3) in monkey RSb14, which was infected with P. cynomolgi.

All metabolites or metabolite pools are color-coded to show fold-increases or decreases in concentrations between post-peak and baseline: red colors represent increased concentrations at post-peak, while green colors represent decreased concentrations at post-peak, according to the log-2 color bar. White boxes indicate no significant changes. Red and green colors of fluxes represent up- or down-regulation at post-peak compared to baseline. Only fluxes with fold-changes greater than 2 are colored; others are shown in blue. The degree of change is indicated by the line thickness. For simplicity, regulatory signals are not shown (cf. Figure 1).
Highlights.
It is often difficult to identify the molecular root causes of a disease
A molecular understanding of a disease is mandatory for targeted interventions
Changes in gene expression during disease are often difficult to interpret
We show how metabolic modeling aids the interpretation of transcriptomic data
We demonstrate our approach with malaria infection data
Acknowledgments
The authors acknowledge members of the MaHPIC Consortium (http://systemsbiology.emory.edu) for insightful discussion.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Conflict of interest.
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gutierrez JB, Galinski MR, Cantrell S, Voit EO. From within host dynamics to the epidemiology of infectious disease: Scientific overview and challenges. Math Biosci. 2015;270:143–155. doi: 10.1016/j.mbs.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO. World Malaria Report. 2016 http://www.who.int/malaria/publications/world_malaria_report/en/
- 3.Fonseca LL, Alezi HS, Moreno A, Barnwell JW, Galinski MR, Voit EO. Quantifying the removal of red blood cells in Macaca mulatta during a Plasmodium coatneyi infection. Malar J. 2016;15:410. doi: 10.1186/s12936-016-1465-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fonseca LL, Voit EO. Comparison of mathematical frameworks for modeling erythropoiesis in the context of malaria infection. Math Biosci. 2015;270:224–236. doi: 10.1016/j.mbs.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fonseca LL, Joyner C, Galinski MR, Voit EO MaHPIC Consortium. A model of Plasmodium vivax concealment based on Plasmodium cynomolgi infections in Macaca mulatta. Malar J. 2017;16:375. doi: 10.1186/s12936-017-2008-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Voit EO. Models-of-data and models-of-processes in the post-genomic era. Math Biosci. 2002;180:263–274. doi: 10.1016/s0025-5564(02)00115-3. [DOI] [PubMed] [Google Scholar]
- 7.Joyner C, Moreno A, Meyer EVS, Cabrera-Mora M, Kissinger JC, Barnwell JW, Galinski MR The MaHPIC Consortium. Plasmodium cynomolgi infections in rhesus macaques display clinical and parasitological features pertinent to modelling vivax malaria pathology and relapse infections. Malaria J. 2016;15:451. doi: 10.1186/s12936-016-1480-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stein DF, O’Connor D, Blohmke CJ, Sadarangani M, Pollard AJ. Gene expression profiles are different in venous and capillary blood: Implications for vaccine studies. Vaccine. 2016;34:5306–5313. doi: 10.1016/j.vaccine.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 9.Tang Y, Joyner C, Cabrera-Mora M, Saney CL, Lapp SA, Nural MV, Bakala SB, DeBarry JD, Soderberg S, Consortium TM, Kissinger JC, Lamb TJ, Galinski MR, Styczynski MP. Integrative analysis associates monocytes with insufficient erythropoiesis during acute Plasmodium cynomolgi malaria in rhesus macaques. Malar J. 2017;16:384. doi: 10.1186/s12936-017-2029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Devonshire AS, Elaswarapu R, Foy CA. Evaluation of external RNA controls for the standardisation of gene expression biomarker measurements. BMC Genomics. 2010;11:662. doi: 10.1186/1471-2164-11-662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28:2184–2185. doi: 10.1093/bioinformatics/bts356. [DOI] [PubMed] [Google Scholar]
- 15.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mecham BH, Nelson PS, Storey JD. Supervised normalization of microarrays. Bioinformatics. 2010;26:1308–1315. doi: 10.1093/bioinformatics/btq118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benjamini Y, Hochberg Y. Controlling false discovery rate - a practical and powerful approach to multiple testing. J Roy Soc Series B-Methodological. 1995;57:289–300. [Google Scholar]
- 18.Rojas-Pena ML, Vallejo A, Herrera S, Gibson G, Arevalo-Herrera M. Transcription profiling of malaria-naive and semi-immune Colombian volunteers in a Plasmodium vivax sporozoite challenge. PLoS Negl Trop Dis. 2015;9:e0003978. doi: 10.1371/journal.pntd.0003978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hung JH, Yang TH, Hu Z, Weng Z, DeLisi C. Gene set enrichment analysis: performance evaluation and usage guidelines. Brief Bioinform. 2012;13:281–291. doi: 10.1093/bib/bbr049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Curto R, Voit EO, Cascante M. Analysis of abnormalities in purine metabolism leading to gout and to neurological dysfunctions in man. Biochem J. 1998;329(Pt 3):477–487. doi: 10.1042/bj3290477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Curto R, Voit EO, Sorribas A, Cascante M. Validation and steady-state analysis of a power-law model of purine metabolism in man. Biochem J. 1997;324(Pt 3):761–775. doi: 10.1042/bj3240761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Curto R, Voit EO, Sorribas A, Cascante M. Mathematical models of purine metabolism in man. Math Biosci. 1998;151:1–49. doi: 10.1016/s0025-5564(98)10001-9. [DOI] [PubMed] [Google Scholar]
- 24.Voit EO. Computational Analysis of Biochemical Systems: A Practical Guide for Biochemists and Molecular Biologists. Cambridge University Press, Place Published; 2000. [Google Scholar]
- 25.Savageau MA. Biochemical Systems Analysis: A Study of Function and Design in Molecular Biology. Addison-Wesley Pub. Co; 1976. Advanced Book Program (reprinted 2009), Place Published. [Google Scholar]
- 26.Torres NV, Voit EO. Pathway Analysis and Optimization in Metabolic Engineering. Cambridge University Press; 2002. Place Published. [Google Scholar]
- 27.Voit EO. Biochemical Systems Theory: A review. Int Scholarly Res Network (ISRN – Biomathematics) 2013:1–53. Article 897658. [Google Scholar]
- 28.Ghaemi-Oskouie F, Shi Y. The role of uric acid as an endogenous danger signal in immunity and inflammation. Curr Rheumatol Rep. 2011;13:160–166. doi: 10.1007/s11926-011-0162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leyva F, Anker SD, Godsland IF, Teixeira M, Hellewell PG, Kox WJ, Poole-Wilson PA, Coats AJ. Uric acid in chronic heart failure: a marker of chronic inflammation. Eur Heart J. 1998;19:1814–1822. doi: 10.1053/euhj.1998.1188. [DOI] [PubMed] [Google Scholar]
- 30.Voit EO, Brigham KL. The role of systems biology in predictive health and personalized medicine. The Open Path J. 2008;2:68–70. [Google Scholar]
- 31.Voit EO. A systems-theoretical framework for health and disease: inflammation and preconditioning from an abstract modeling point of view. Math Biosci. 2009;217:11–18. doi: 10.1016/j.mbs.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haskó G, Kuhel DG, Németh ZH, Mabley JG, Stachlewitz RF, Virág L, Lohinai Z, Southan GJ, Salzman AL, Szabó C. Inosine inhibits inflammatory cytokine production by a posttranscriptional mechanism and protects against endotoxin-induced shock. J Immunol. 2000;164:1013–1019. doi: 10.4049/jimmunol.164.2.1013. [DOI] [PubMed] [Google Scholar]
- 33.Gudbjornsson B, Zak A, Niklasson F, Hallgren R. Hypoxanthine, xanthine, and urate in synovial fluid from patients with inflammatory arthritides. Ann Rheum Dis. 1991;50:669–672. doi: 10.1136/ard.50.10.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.da Rocha Lapa F, da Silva MD, de Almeida Cabrini D, Santos AR. Anti-inflammatory effects of purine nucleosides, adenosine and inosine, in a mouse model of pleurisy: evidence for the role of adenosine A2 receptors. Purinergic Signal. 2012;8:693–704. doi: 10.1007/s11302-012-9299-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marton A, Pacher P, Murthy KG, Nemeth ZH, Hasko G, Szabo C. Anti-inflammatory effects of inosine in human monocytes, neutrophils and epithelial cells in vitro. Int J Mol Med. 2001;8:617–621. [PubMed] [Google Scholar]
- 36.Takahashi T, Otsuguro K, Ohta T, Ito S. Adenosine and inosine release during hypoxia in the isolated spinal cord of neonatal rats. Br J Pharmacol. 2010;161:1806–1816. doi: 10.1111/j.1476-5381.2010.01002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferrari D, McNamee EN, Idzko M, Gambari R, Eltzschig HK. Purinergic signaling during immune cell trafficking. Trends Immunol. 2016;37:399–411. doi: 10.1016/j.it.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 38.Gallego-Delgado J, Ty M, Orengo JM, van de Hoef D, Rodriguez A. A surprising role for uric acid: the inflammatory malaria response. Curr Rheumatol Rep. 2014;16:401. doi: 10.1007/s11926-013-0401-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lutgen P. [Accessed 2017]; https://malariaworld.org/blog/uric-acid-emerging-key-factor-malaria.
- 40.Lopera-Mesa TM, Mita-Mendoza NK, van de Hoef DL, Doumbia S, Konate D, Doumbouya M, Gu W, Traore K, Diakite SA, Remaley AT, Anderson JM, Rodriguez A, Fay MP, Long CA, Diakite M, Fairhurst RM. Plasma uric acid levels correlate with inflammation and disease severity in Malian children with Plasmodium falciparum malaria. PLoS One. 2012;7:e46424. doi: 10.1371/journal.pone.0046424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Downie MJ, Kirk K, Mamoun CB. Purine salvage pathways in the intraerythrocytic malaria parasite Plasmodium falciparum. Eukaryot Cell. 2008;7:1231–1237. doi: 10.1128/EC.00159-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cassera MB, Zhang Y, Hazleton KZ, Schramm VL. Purine and pyrimidine pathways as targets in Plasmodium falciparum. Curr Top Med Chem. 2011;11:2103–2115. doi: 10.2174/156802611796575948. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.