

ABSTRACT
Mucosa-associated invariant T (MAIT) cells are a subset of innate T cells that express a semi-invariant Vα chain paired with limited Vβ chains. MAIT cells are activated by riboflavin metabolite derivatives presented by the nonpolymorphic major histocompatibility complex class I (MHC-I)-like molecule MR1. The precise mechanisms required to activate MAIT cells are an area of intense interest. Here we used two closely related intracellular pathogens with distinct inflammasome activation phenotypes to probe the role of innate cytokines in MAIT cell activation. Using an in vitro assay containing transgenic murine MAIT cells, we show that macrophages infected with Francisella novicida, a strong inflammasome activator, released high levels of interleukin-18 (IL-18) and stimulated high levels of MAIT cell gamma interferon (IFN-γ) through a partially MR1-independent pathway. In contrast, macrophages infected with Francisella tularensis live vaccine strain (LVS), a weak inflammasome activator, generated little IL-18 and stimulated low MAIT cell IFN-γ through an MR1-dependent pathway. By manipulating the quantities of IL-18 in these cultures, we show that the IL-18 concentration is sufficient to influence the magnitude of MAIT cell IFN-γ production. Correspondingly, infected IL-18-deficient macrophages failed to induce substantial MAIT cell IFN-γ. In contrast, we found that MAIT cell IFN-γ production in the lungs of IL-18-deficient mice was not significantly different from that in WT mice during F. tularensis LVS pulmonary infection. Overall, we demonstrate that while IL-18 is essential for the MAIT cell IFN-γ response in vitro, it is not essential for MAIT cell IFN-γ production during in vivo LVS pulmonary infection, suggesting that additional signals can drive MAIT cell IFN-γ production in vivo.
KEYWORDS: Francisella, MAIT cells
INTRODUCTION
Mucosa-associated invariant T (MAIT) cells are an innate T cell population that expresses a semi-invariant T cell receptor (TCR) alpha chain (commonly TRAV1-2 combined with TRAJ33, TRAJ20, or TRAJ12 in humans and TRAV1/TRAJ33 in mice) paired with a variety of Vβ chains (1–4). MAIT cells are activated by riboflavin metabolite derivatives presented by the nonpolymorphic major histocompatibility complex class I (MHC-I)-like molecule MR1 (5) and respond by rapidly secreting cytokines such as gamma interferon (IFN-γ) and interleukin-17 (IL-17) (6, 7). The MR1 gene is highly evolutionarily conserved among mammals, suggesting that MAIT cells have an important role in immunity (8, 9). Indeed, MAIT cell-deficient mice are more susceptible to Klebsiella pneumoniae, Mycobacterium bovis BCG, and Francisella tularensis live vaccine strain (LVS) infections, establishing a nonredundant role for MAIT cells in pathogen defense (10–13). In humans, the importance of MAIT cells is implied by their abundance in the blood and mucosa of healthy subjects and their presence at the site of infections (2, 7, 14–16). Since MAIT cells are activated by a wide range of microbes, possess a memory phenotype, and populate mucosal sites, they likely serve as sentinels that detect early signs of infection (7, 17). For these reasons, MAIT cell antigens have the potential to serve as novel vaccine adjuvants, thus promoting an interest in understanding the mechanisms that trigger MAIT cell activation.
Until recently, it was believed that MAIT cells respond only to microbes that possess the riboflavin biosynthetic pathway. Indeed, mutagenesis studies have shown that specific genes of the vitamin B2 pathway are required for MAIT cell activation by Salmonella enterica serovar Typhimurium and Escherichia coli (18–20). However, recent work demonstrated that recombinant cytokines stimulated MAIT cell activation independent of MR1 signaling. In particular, IL-12 activated MAIT cells in the absence of bacterial antigen (13), while the combination of IL-12 and IL-18 was especially potent in stimulating IFN-γ production (21, 22). Release of the mature form of IL-18 requires activation of the inflammasome, a macromolecular complex that plays a key role in innate immune responses (23). Activation of the inflammasome is initiated when host pattern recognition sensors detect cytosolic signs of infection, such as bacterial double-stranded DNA (dsDNA) and lipopolysaccharide (23). Upon detection of infection, host cytosolic sensor molecules oligomerize with an adaptor protein (apoptosis-associated speck-like protein [ASC]) and recruit pro-caspase-1 to form the inflammasome (23). Pro-caspase-1 subsequently undergoes autocatalytic processing to form an active protease, resulting in cleavage and secretion of IL-1β and IL-18 (23, 24). Active caspase-1 also induces a proinflammatory form of cell death called pyroptosis and the release of IL-1α (25).
Many intracellular pathogens have evolved virulence mechanisms to limit proinflammatory responses by dampening signals that lead to inflammasome activation (26, 27). Since the repertoire of virulence factors for each pathogen is unique, a broad range in the magnitude of cytokine responses is observed. The discovery that MAIT cell activation occurs in response to IL-12 and IL-18 raises the question of whether the strength of inflammasome activation impacts the ability of different pathogens to activate MAIT cells. Here we used two closely related Francisella species with distinct inflammasome activation phenotypes to probe the role of cytokines and cognate antigen recognition in MAIT cell activation. Francisella tularensis LVS is an attenuated intracellular pathogen derived from the fully virulent F. tularensis subsp. holarctica, the causative agent of tularemia in humans. Like its virulent counterpart, F. tularensis LVS suppresses inflammasome activity early in infection, while in contrast, Francisella novicida produces a significantly more robust inflammasome response (28). Although F. tularensis and F. novicida share 98% identity at the nucleotide level, F. novicida rarely causes human disease but produces a rapidly lethal pulmonary infection in mice (29, 30). Importantly, both Francisella species possess the riboflavin biosynthetic pathway and share the same intracellular lifestyle. Upon macrophage infection, both pathogens escape the phagosome and replicate in the host cell cytosol, where inflammasome sensors detect the bacteria (31, 32). Due to its robust ability to activate the inflammasome, F. novicida macrophage infection is a well-established model for the study of inflammasome activation via the cytosolic sensor AIM2 (33). In contrast, F. tularensis LVS has been studied to identify virulence factors that suppress inflammasome activation (28, 34). Suppression of the inflammasome by F. tularensis LVS is at least partially attributed to its repression of Toll-like receptor 2 (TLR2)-dependent signaling and activation of NF-κB (28). This leads to distinct differences in the levels of several cytokines produced by macrophages infected with F. tularensis LVS and F. novicida, including IL-18 and IL-1β.
Using the aforementioned Francisella strains, we show that inflammasome activity is a critical component of the MAIT cell response to intracellular infection in vitro. We found that macrophages infected with the strong inflammasome activator F. novicida released high levels of IL-18 and stimulated high levels of MAIT cell IFN-γ production through a partially MR1-independent pathway. In contrast, macrophages infected with the weak inflammasome activator F. tularensis LVS generated low levels of IL-18, stimulated less MAIT cell IFN-γ production, and operated through an entirely MR1-dependent pathway. We further demonstrate that macrophage caspase-1/11 and IL-18 are essential for MAIT cell IFN-γ production in this system. Overall, our data demonstrate that different pathogens differentially activate MAIT cells by cytokine-driven and cognate-antigen-driven signals and that the levels of IL-18 modulate the magnitude of the MAIT cell IFN-γ response in vitro. In contrast, we found that while MAIT cell IFN-γ responses to LVS pulmonary infection in vivo can be augmented by addition of recombinant IL-18, MAIT cell IFN-γ responses in IL-18−/− mice were not significantly different from those of WT mice, suggesting that additional signals found in vivo can compensate for the loss of IL-18.
RESULTS
IL-12 synergizes with IL-18 or IL-33 to stimulate Vα19iTg T cell IFN-γ production.
IL-12 synergizes with IL-18 to activate MAIT cell IFN-γ production in an MR1-independent manner (21, 22). Since IL-18 is a member of the IL-1 family of cytokines, we examined the ability of several IL-1 family cytokines to synergize with IL-12 to stimulate MAIT cell IFN-γ production. As a source of MAIT cells, we used Thy1.2+ cells isolated from transgenic mice that exclusively express the canonical MAIT cell TRAV1/TRAJ33 TCRα chain (Vα19iTgCα−/− MR1+/+ mice). Purified Vα19iTg Thy1.2+ cells were cultured in the presence of recombinant IL-12 in combination with recombinant IL-18 (rIL-18), rIL-1α, rIL-1β, or rIL-33; IFN-γ production was examined by enzyme-linked immunosorbent assay (ELISA) and intracellular cytokine staining (ICS) after 20 h of culture. Consistent with previously published evidence, IL-12 stimulated low levels of Vα19iTg T cell IFN-γ production that synergized with IL-18 to yield high levels of IFN-γ by ELISA (Fig. 1A) (22). In addition, IL-33 synergized with IL-12 to yield high levels of IFN-γ (Fig. 1A). To determine whether the IFN-γ measured in the supernatants was derived exclusively from MAIT cells, we used the MR1 5-OP-RU tetramer to identify tetramer-reactive cells in the cultures, and IFN-γ production was assessed by intracellular cytokine staining. We found that Thy1.2+ cells isolated from the spleens of Vα19iTgCα−/− MR1+/+ mice contained two substantial TCRβ+ populations that were MR1 5-OP-RU tetramer+ and tetramer−; staining with negative-control nonantigenic acetyl-6-formylpterin-loaded MR1 tetramers (6-FP) failed to show a population of tetramer+ cells (<1% of the total number of TCRβ+ cells; not depicted). Both of the TCRβ+ 5-OP-RU MR1 tetramer+ and tetramer− populations expressed high levels of IL-18 receptor that were further augmented by exposure to IL-12 (Fig. 1B); attempts to measure the IL-33 receptor (ST2) levels showed only low levels of positively stained cells (approximately 0.6 to 1% of total TCRβ+ cells) (not depicted). The combinations of IL-12 and IL-18 as well as IL-12 and IL-33 induced significant increases in IFN-γ production by both the TCRβ+ tetramer+ and the TCRβ+ tetramer− populations compared to the individual cytokine treatments (Fig. 1C and D). Not surprisingly, IL-1α and IL-1β failed to significantly augment IL-12-induced IFN-γ production when measured via intracellular cytokine staining (Fig. 1D), although a small but significant increase was reproducibly detected when measured via ELISA (Fig. 1A). These data demonstrate that both the 5-OP-RU MR1 tetramer+ and tetramer− Vα19iTg T cell populations produce IFN-γ in response to IL-12 coupled with IL-33 or IL-18 in an MR1-independent manner.
FIG 1.

Vα19iTg T cells produce IFN-γ in response to recombinant IL-12, IL-12 + IL-18, and IL-12 + IL-33 in the absence of cognate antigen. Naive Thy1.2+ cells purified from Vα19iTgCα−/− MR1+/+ mice were cultured for 20 h in the presence of recombinant cytokines, as indicated. (A) ELISA analysis of IFN-γ production by Vα19iTg T cells in the culture supernatants. (B) Flow cytometry analysis of Vα19iTg T cell IL-18Rα expression after treatment with recombinant IL-12. Results show reactivity to MR1-5-OP-RU tetramer (MR1 tetramer). The percentages of IL-18Rα+ cells for the 5-OP-RU MR1 tetramer+ (Tet+) and tetramer− (Tet−) populations are depicted graphically. (C and D) IFN-γ intracellular cytokine staining of Vα19iTg T cells cultured for 20 h in the presence of recombinant cytokines, as indicated. The percentages of intracellular IFN-γ+ cells for the 5-OP-RU MR1 tetramer+ and tetramer− populations are depicted graphically. Brefeldin A was added during the final 4 h of culture. All flow cytometry dot plots depict TCRβ+ lymphocytes with B220+ and F4/80+ cells excluded by electronic gating. No treatment, culture medium without recombinant cytokines; isotype Ab, intracellular cytokine staining using a nonspecific control Ab. Data represent values ± standard errors of the means (SEM) from three replicates and are representative of three independent experiments. *, P < 0.05 compared to “No treatment”; **, P < 0.05 compared to IL-12.
The magnitude of Vα19iTg T cell IFN-γ production reflects the level of pathogen inflammasome activation.
Since release of IL-18 requires activation of the inflammasome and different pathogens exhibit various capacities for inflammasome activation, we were interested in determining whether this property affects their ability to activate MAIT cells. To test this hypothesis, we infected wild-type (WT) macrophages with F. tularensis LVS (a poor inflammasome activator), F. novicida (a strong inflammasome activator), and an F. novicida mutant that is unable to escape the phagosome (ΔpdpA; no inflammasome activation) (35). Inflammasome activation in macrophages infected with these bacteria was confirmed by Western blotting for active caspase-1 (p10) and corresponded to the levels of IL-18, IL-1α, and IL-1β produced (Fig. 2A and B). F. novicida-infected macrophages produced high levels of active caspase-1, IL-18, IL-1α, and IL-1β after 24 h, which were undetectable in macrophages infected with F. novicida ΔpdpA. In contrast, LVS-infected macrophages produced low levels of active caspase-1 and correspondingly low levels of IL-18, IL-1α, and IL-1β. Of note, we were unable to detect IL-33 in any of the cultures (data not shown).
FIG 2.

Differences in the magnitude of inflammasome activation by F. tularensis LVS and F. novicida-infected macrophages reflect the magnitude of Vα19iTg T cell IFN-γ production. (A) WT macrophages infected with either no bacteria (Mock), LVS, F. novicida (FN), or F. novicida ΔpdpA (MOI, 100:1) for 20 h. Western blot analysis of culture supernatants was performed to visualize active caspsase-1 (caspase-1 p10). The blot was reprobed for GAPDH, which was used as a loading control. (B) WT macrophages were infected with either LVS, F. novicida, or F. novicida ΔpdpA (MOI, 100:1). The levels of IL-18, IL-1α, and IL-1β were examined in the supernatants after 20 h of culture. *, P < 0.01 compared to LVS; ND, not detected. (C) WT macrophages were infected with either LVS (MOI, 1:1), F. novicida (MOI, 1:1), or F. novicida ΔpdpA (a MOI of 100:1 was used since ΔpdpA does not grow well in macrophages) and cocultured with Vα19iTg T cells in the presence and absence of blocking anti-MR1 Ab or matched-isotype control Ab (25 μg/ml). IFN-γ was measured in the supernatants after 20 h of culture. *, P < 0.01 compared to LVS; **, P < 0.01 compared to wild-type F. novicida. (D) IL-12 p40 levels in the supernatants of macrophage cultures infected with either LVS (MOI, 1:1), F. novicida (MOI, 1:1), or F. novicida ΔpdpA (MOI, 100:1) in the presence (+) and absence (−) of Vα19iTg T cells for 20 h. Data represent values ± SEM from three replicates and are representative of three independent experiments.
We next cocultured Vα19iTg Thy1.2+ cells with macrophages infected with the three bacterial strains. To assess the MR1 dependence of Vα19iTg T cell activation, IFN-γ production was measured in the supernatants after culture with macrophages in the presence and absence of blocking anti-MR1 antibody (Ab). As shown in Fig. 2C, Vα19iTg T cells cultured with LVS-infected WT macrophages produced relatively low levels of IFN-γ, which were abrogated in the presence of anti-MR1 Ab. In contrast, Vα19iTg T cells cultured with F. novicida-infected WT macrophages generated significantly higher levels of IFN-γ than those produced with LVS; importantly, this IFN-γ production was largely unaffected by the presence of anti-MR1 Ab (identical results were obtained using MR1−/− macrophages [data not shown]). Finally, Vα19iTg T cells cultured with the F. novicida ΔpdpA mutant generated only marginal amounts of IFN-γ, indicating that vacuolar escape, which is critical for inflammasome activation, is required for productive Vα19iTg T cell activation by F. novicida. Of note, IL-12 levels in the supernatants of cocultures containing WT macrophages infected with F. novicida or the F. novicida ΔpdpA mutant were not significantly different (Fig. 2D), demonstrating that the observed differences in IFN-γ production were not likely due to inequalities in IL-12 production.
To determine whether the IFN-γ measured in the supernatants was derived from MAIT cells, we used the MR1 5-OP-RU tetramer to identify tetramer-reactive cells in the cocultures, and IFN-γ production was assessed by intracellular cytokine staining. Staining with the negative-control 6-FP-loaded MR1 tetramers did not yield a population of tetramer+ cells (<0.5% positive cells) (not depicted). As shown in Fig. 3A, both the MR1 5-OP-RU tetramer+ and the tetramer− populations produced IFN-γ during coculture with LVS-infected macrophages. Interestingly, IFN-γ production by the two cell populations was strongly inhibited by coculture with MR1−/− macrophages (82% and 85% reduction for the TCRβ+ tetramer+ and TCRβ+ tetramer− populations, respectively) (Fig. 3B), demonstrating that cells within the tetramer− population recognized antigen via MR1 (identical results were obtained using WT macrophages and a blocking anti-MR1 Ab [data not shown]). In contrast, IFN-γ production by both the TCRβ+ tetramer+ and the TCRβ+ tetramer− populations cultured with F. novicida-infected macrophages was less reliant on MR1; the TCRβ+ tetramer+ and TCRβ+ tetramer− populations exhibited only 41% and 54% reductions, respectively, in IFN-γ production 24 h after culture with MR1−/− macrophages compared to WT macrophages (Fig. 3A and B). Of note, both the TCRβ+ tetramer+ and TCRβ+ tetramer− populations cultured with F. novicida-infected macrophages produced significantly higher levels of IFN-γ than did cells cultured with LVS-infected macrophages, which was consistent with the IFN-γ levels measured by ELISA (Fig. 2C). Overall, these data demonstrate that both MR1 5-OP-RU tetramer+ and tetramer− Vα19iTg T cells exhibit MR1-dependent IFN-γ production and that cells exposed to macrophages infected with F. novicida and LVS possess different requirements for MR1 to induce maximum IFN-γ production.
FIG 3.
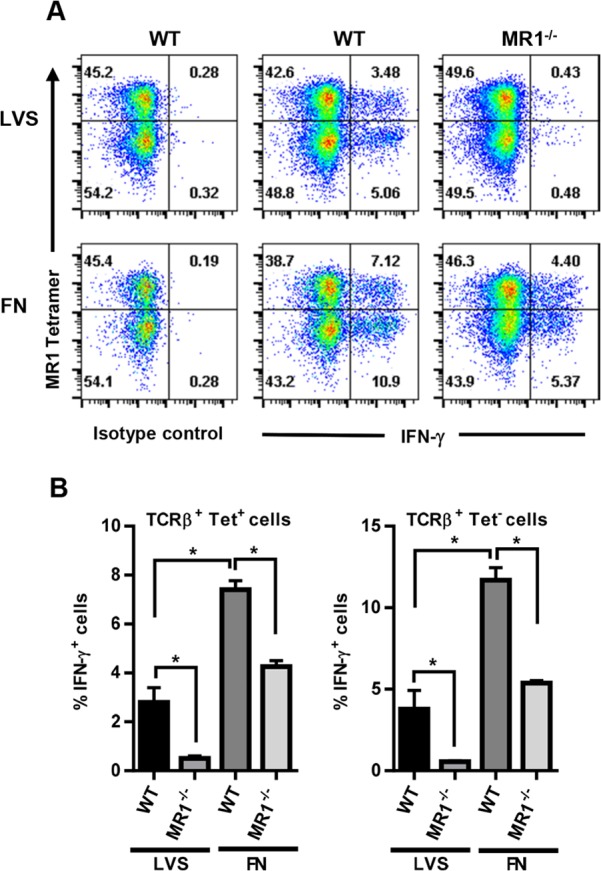
MR1-5-OP-RU tetramer+ and tetramer− Vα19iTg T cells exhibit MR1-dependent IFN-γ production in response to F. tularensis LVS and F. novicida. WT and MR1−/− macrophages were infected with either LVS or F. novicida (FN) at an MOI of 1:1 and cocultured with Vα19iTg T cells. After 20 h of culture, the T cells were removed, labeled with MR1-5-OP-RU tetramer, and stained for IFN-γ by ICS. (A) Representative flow cytometry dot plots showing the percentage of IFN-γ+ cells present in the 5-OP-RU MR1 tetramer+ and tetramer− populations. All flow cytometry dot plots depict TCRβ+ lymphocytes with B220+ and F4/80+ cells excluded by electronic gating. Isotype control consists of samples stained via ICS with a control antibody isotype matched to the α-IFN-γ Ab. (B) The percentages of intracellular IFN-γ+ cells for the 5-OP-RU MR1 tetramer+ and tetramer− populations are depicted graphically. Brefeldin A was added during the final 4 h of culture. *, P < 0.01 compared to the indicated population. Data represent values ± SEM from three replicates and are representative of three independent experiments.
Capase-1/11, IL-12, and IL-18 are required for Vα19iTg T cell IFN-γ production during culture with Francisella-infected macrophages.
Since the magnitude of Vα19iTg T cell IFN-γ production in response to Francisella-infected macrophages mirrored caspase-1 activation (Fig. 2 and 3), we assessed the ability of infected macrophages derived from caspase-1-deficient mice (caspase-1/11−/−) to stimulate Vα19iTg T cell IFN-γ production. As observed in Fig. 4A, IFN-γ production was abrogated in cultures containing caspase-1/11−/− macrophages infected with either LVS or F. novicida, establishing that caspase-1/11 is critical for the ability of macrophages to stimulate both tetramer+ and tetramer− Vα19iTg T cell activation. These data also demonstrate that an inflammasome-dependent cytokine is critical for Vα19iTg T cell IFN-γ production in the Francisella cultures.
FIG 4.

IL-18 modulates the level of MR1-5-OP-RU tetramer+ and tetramer− Vα19iTg T cell IFN-γ responses to F. tularensis LVS and F. novicida. (A) WT and caspase-1/11−/− macrophages were infected with either LVS or F. novicida (FN) at an MOI of 1:1 and cocultured with Vα19iTg T cells. IFN-γ was measured in the supernatants after 20 h of culture. *, P < 0.01 compared to WT macrophages infected with the same organism. (B) WT, IL-12p35−/−, IL-12p40−/−, and IL-18−/− macrophages were infected with either LVS or F. novicida (FN) at an MOI of 1:1 and cocultured with Vα19iTg T cells. IFN-γ was measured in the supernatants after 20 h of culture. *, P < 0.01 compared to WT macrophages infected with the same organism. (C) WT macrophages were infected with F. novicida at an MOI of 1:1 and cocultured with Vα19iTg T cells. Neutralizing anti-IL-18 Ab was added at the indicated concentrations at the start of coculture, and IFN-γ was measured in the supernatants after 20 h of culture. *, P < 0.01 compared to cultures containing no anti-IL-18 Ab. (D) Levels of IL-18 measured in the supernatants of cultures depicted in panel C. *, P < 0.01 compared to cultures containing no anti-IL-18 Ab. (E) WT macrophages were infected with F. tularensis LVS at an MOI of 1:1 and cocultured with Vα19iTg T cells. Recombinant IL-18 was added at the indicated concentrations at the start of coculture, and IFN-γ was measured in the supernatants after 20 h of culture. *, P < 0.01 compared to cultures containing no recombinant IL-18. (F) IFN-γ intracellular cytokine staining of Vα19iTg T cells cultured with F. tularensis LVS-infected WT macrophages for 20 h in the presence of 1 ng/ml recombinant IL-18, or no additional IL-18 (“no treatment”), as indicated. The percentages of intracellular IFN-γ+ cells for the 5-OP-RU MR1 tetramer+ and tetramer− populations are depicted graphically. *, P < 0.01 compared to cultures containing no additional recombinant IL-18. Brefeldin A was added during the final 4 h of culture. All flow cytometry dot plots depict TCRβ+ lymphocytes with B220+ and F4/80+ cells excluded by electronic gating. No treatment, culture medium without recombinant cytokines; isotype control, intracellular cytokine staining using a nonspecific control Ab matched to the isotype of the IFN-γ Ab. Data represent values ± SEM from three replicates and are representative of three independent experiments.
We next sought to determine whether IL-18 is critical for IFN-γ production during coculture with Francisella-infected macrophages. To this end, Vα19iTg T cells were cultured with LVS and F. novicida-infected macrophages derived from WT, IL-18−/−, IL-12p40−/−, and IL-12p35−/− mice. Vα19iTg T cell IFN-γ production was assessed in the supernatants after 24 h of coculture. As shown in Fig. 4B, Vα19iTg T cell IFN-γ production in response to both LVS and F. novicida was largely abrogated in cultures containing IL-12p40−/−, IL-12p35−/−, and IL-18−/− macrophages; only a small amount of IFN-γ remained detectable in the F. novicida IL-18−/− macrophage cultures. These data show that both IL-12 and IL-18 are necessary to induce high levels of Vα19iTg T cell IFN-γ production by both the TCRβ+ tetramer+ and the TCRβ+ tetramer− populations; further, this phenomenon was observed for both pathogens, indicating that IL-12 and IL-18 are critical regardless of the requirement for MR1.
Our data showed that Vα19iTg T cells produced significantly different amounts of IFN-γ in response to F. novicida and LVS-infected macrophages and that these two pathogens differentially induced high and low levels of macrophage IL-18 production, respectively. To determine whether these differences in IL-18 are sufficient to influence the magnitude of Vα19iTg T cell IFN-γ production, we used neutralizing anti-IL-18 antibodies to reduce, but not eliminate, the high quantities of IL-18 present in cultures containing F. novicida-infected WT macrophages. As shown in Fig. 4C, addition of anti-IL-18 antibodies significantly reduced Vα19iTg T cell IFN-γ production in a dose-dependent manner that correlated with the amount of IL-18 detected in the cultures (Fig. 4D). Similarly, addition of recombinant IL-18 to the LVS cultures significantly increased Vα19iTg T cell IFN-γ production in a dose-dependent manner (Fig. 4E). Of note, addition of 125 ng/ml of rIL-18 to the LVS cultures resulted in a 7-fold increase in Vα19iTg T cell IFN-γ production. Given that F. novicida-infected macrophages induced approximately 150 ng/ml more IL-18 than did LVS-infected macrophages, this result is consistent with the 10-fold increase in IFN-γ produced by Vα19iTg T cells cocultured with F. novicida compared to LVS (Fig. 2B and C). Flow cytometry analyses demonstrated that the increased IFN-γ observed in the LVS cultures was derived from both the tetramer+ and tetramer− Vα19iTg T cell populations (Fig. 4F). These data confirm that the magnitude of Vα19iTg T cell IFN-γ production is modulated by the levels of IL-18 present in the cultures, which is a direct result of the ability of the pathogen to activate the inflammasome.
IL-18 is not required for MAIT cell IFN-γ production during in vivo pulmonary Francisella infection.
We next sought to determine whether IL-18 influences MAIT cell IFN-γ production during in vivo infection. Low-dose pulmonary F. novicida infection induces a cytokine storm that is rapidly lethal for WT mice, resulting in death 4 to 5 days after inoculation (36). Since we were unable to detect IFN-γ-producing MAIT cells in the lungs of F. novicida-infected mice prior to death (data not shown), we instead employed the F. tularensis LVS intranasal infection model, which allows mice to survive the infection to later time points. To determine whether augmentation of IL-18 influences MAIT cell IFN-γ production in vivo, WT mice were administered supplemental rIL-18 during LVS intranasal infection. IFN-γ production by MR1 5-OP-RU tetramer+ MAIT cells in the lungs was assessed on day 11 after LVS inoculation, at a time when MAIT cells have accumulated in large numbers. To determine whether loss of IL-18 influences MAIT cell IFN-γ production in vivo, we assessed IFN-γ production by MR1 5-OP-RU tetramer+ MAIT cells in the lungs of IL-18−/− mice after infection. Since we found that IL-18−/− mice succumbed to LVS intranasal infection on days 9 to 10 after inoculation (data not shown), MAIT cell IFN-γ production was assessed on day 8. MAIT cell numbers in the lungs were not significantly affected by either augmentation or loss of IL-18 (Fig. 5A and B and data not shown). Administration of supplemental rIL-18 significantly increased MAIT cell IFN-γ production (Fig. 5A). Surprisingly, MAIT cell IFN-γ production was not significantly reduced in IL-18−/− mice compared to WT mice (Fig. 5B), in contrast to our in vitro studies. These data demonstrate that IL-18 is not essential for MAIT cell IFN-γ production during in vivo F. tularensis LVS pulmonary infection, suggesting that additional costimulatory signals that are not present in our in vitro cultures are available during in vivo infection.
FIG 5.

MR1-5-OP-RU tetramer+ T cell IFN-γ responses during in vivo F. tularensis LVS pulmonary infection. (A) Wild-type C57BL/6 mice were infected with a sublethal intranasal dose of 2 × 102 CFU F. tularensis LVS and administered supplemental recombinant IL-18 (rIL18) delivered intranasally on days 7 and 10. IFN-γ intracellular cytokine staining of 5-OP-RU MR1 tetramer+ TCRβ+ cells in the lungs of mice given supplemental rIL-18 or PBS (no treatment) was assessed on day 11 after infection. (B) Wild-type C57BL/6 mice and IL-18−/− mice were infected with a sublethal intranasal dose of 2 × 102 CFU F. tularensis LVS. IFN-γ intracellular cytokine staining of 5-OP-RU MR1 tetramer+ TCRβ+ cells was assessed in the lungs on day 8 after infection. Control tetramer (6-FP) staining is shown for comparison. The total numbers of intracellular IFN-γ+ 5-OP-RU MR1 tetramer+ cells are depicted graphically. *, P < 0.01 compared to control mice. Data represent values ± SEM from 3 to 5 individual mice and are representative of 3 or 4 independent experiments.
DISCUSSION
Adult human MAIT cells (CD161hi CD8α+ cells) require costimulation through either CD28 or innate cytokine receptors in order to fully respond to TCR signaling (37). This stringent regulation of TCR signaling is proposed to prevent undesirable responses to microbiota-derived riboflavin metabolites. Thus, secondary signals, such as inflammatory cytokines, are a critical checkpoint that ensures that MAIT cell activity is directed toward pathogens. Since many pathogens evade intracellular detection and inhibit proinflammatory responses, including inflammasome activation and IL-18 production, we were interested in determining whether this virulence strategy impacts MAIT cell activation. Here we used two closely related Francisella species that have distinct inflammasome activation phenotypes to probe the roles of cytokines and cognate antigen recognition in MAIT cell activation in the Vα19iTgCα−/− MR1+/+ mouse model.
We found that both IL-12 and IL-18 were critical to support Vα19iTg T cell IFN-γ responses toward macrophages infected with the intracellular pathogens F. tularensis LVS and F. novicida. Importantly, while F. novicida-infected macrophages produced high levels of IL-18 and correspondingly high levels of Vα19iTg T cell IFN-γ, an F. novicida mutant that cannot activate the inflammasome failed to induce macrophage IL-18 and substantial IFN-γ production. In contrast, LVS cultures contained low levels of IL-18 and correspondingly low levels of IFN-γ. By manipulating the quantities of IL-18 in these cultures, we showed that changing the concentration of IL-18 was sufficient to influence the magnitude of Vα19iTg T cell IFN-γ production. The ability of IL-18 to synergize with IL-12 to induce TH1 and MAIT cell IFN-γ is well established (22, 38), and recent studies demonstrated that increasing the levels of recombinant IL-18 in the presence of a constant concentration of recombinant IL-12 increased MAIT cell IFN-γ production in a dose-dependent manner in vitro (22). The data presented here support these observations in a more complex biological in vitro system. However, we found that MAIT cell IFN-γ responses were not reduced in IL-18−/− mice compared to WT mice during in vivo pulmonary LVS infection. This suggests that additional signals can promote MAIT cell IFN-γ responses in vivo; indeed, our in vitro studies found that Vα19iTg T cells potently produced IFN-γ in response to recombinant IL-12 and IL-33, confirming that other mediators can be substituted for IL-18 to induce MAIT cell IFN-γ. Further, recent studies demonstrated that several unexpected cytokine combinations can mediate IFN-γ production by memory CD8+ T cells, such as IL-12 in combination with IL-2, tumor necrosis factor (TNF), and TNF-like cytokine 1A (TL1A) (39). Whether these cytokines drive MAIT cell IFN-γ production in the absence of IL-18 during in vivo LVS pulmonary infection remains to be investigated.
Our source of MAIT cells for these studies was Vα19iTgCα−/− MR1+/+ mice, which exclusively express the canonical TRAV1-TRAJ33 TCRα gene and are highly enriched for MAIT cells. Surprisingly, we found that both the 5-OP-RU MR1 tetramer+ and tetramer− T cell populations in these mice exhibited MR1-dependent IFN-γ production. Since the proportion of splenic TCRβ+ 5-OP-RU MR1 tetramer+ cells detected in our experiments (approximately 50%) is corroborated by other studies using the same mouse strain (17, 22), we do not believe that our data are the result of suboptimal tetramer staining. In addition, the observed results were not altered by increasing the tetramer concentration. One possible reason for the failure of some MAIT cells to bind the tetramer is downregulation of the TCR following activation; however, since we gated on TCRβ+ cells prior to our assessment of tetramer binding, it seems unlikely that TCR downregulation can account for these results.
Another reason for differences in MAIT cell reactivity to the 5-OP-RU tetramer may be variations in the TCR. Although MAIT cells in Vα19iTgCα−/− MR1+/+ mice all possess the same Vα chain, some variability in the Vβ chain repertoire has been reported, which may contribute to the phenomenon that we observed (17, 22, 40). However, studies conducted to date suggest that most MAIT cells recognize any stable MR1 tetramer bound to an activating ligand (2, 17). Reantragoon et al. demonstrated that cell lines expressing different TRAV1-2 TCRs all effectively recognized an rRL-6-CH2OH-loaded MR1 tetramer, including those with alternate T cell receptor alpha joining (TRAJ) genes, such as TRAJ20 and TRAJ12 (2). Thus far, differences in Vβ-chain usage by TRAV1-2+ MAIT cells have been associated not with significant loss in recognition of MR1 tetramers bound to activating ligands (2, 17, 41) but instead with a gain in reactivity to other antigens (e.g., 6-FP) or empty tetramers. Nevertheless, human TRAV1-2+ MAIT cells exhibit different staining intensities for MR1 tetramers, and the affinity of MR1-Ag binding is not equivalent among all MAIT TCRs (2, 41). Further, unique CDR3β loop sequences have been associated with reactivity to different pathogens, revealing that the TCRβ chain likely participates in ligand discrimination (4). This is supported by studies examining the interaction of the MAIT TCR with MR1, whereby the CDR3β loop was positioned above the ligand binding groove, mediated contacts with the ligand, and modulated the affinity of binding to MR1 (42–46). Thus, it is possible that differences in the Vβ chain repertoire of Vα19iTg MAIT cells may affect recognition of a specific MR1-Ag tetramer. Whether the observations that we describe here are relevant in wild-type mice and humans or limited to the transgenic mouse model remains to be determined.
Here we found that IFN-γ production by tetramer+ MAIT cells cultured with F. novicida-infected macrophages was at least partially MR1 independent but required IL-12 and IL-18. Activation of human MAIT cells by Enterococcus faecalis was entirely TCR independent and required both IL-12 and IL-18; however, since this organism lacks the riboflavin biosynthetic pathway, MAIT cell activation necessarily operated through cytokine-dependent signals (21). Similarly, human MAIT cells responded to influenza virus-infected cells in an MR1-independent and IL-18-dependent manner (47). In contrast to these examples, F. novicida possesses the genes for riboflavin biosynthesis. Our data are comparable to those from studies showing that M. bovis BCG, which also contains the riboflavin biosynthetic pathway, induced Vα19iTg T cell IFN-γ production in an IL-12-dependent and MR1-independent manner (12). Additionally, cells treated with fixed E. coli stimulated human liver MAIT cells in a partially MR1-dependent and IL-12/IL-18-dependent manner (48). The importance of riboflavin metabolites as MAIT cell antigens has been confirmed by deletion of riboflavin biosynthesis genes, which abrogated MAIT cell activation by S. Typhimurium, Lactococcus lactis, and E. coli (18, 19). Although derivatives of riboflavin metabolites are potent MAIT cell antigens, our data suggest that the requirement for these metabolites in MAIT cell activation may need to be assessed on a strain-by-strain basis. The differences observed between F. novicida and F. tularensis LVS may depend on the availability of antigen or the composition of the inflammatory milieu. Based on the data provided here, it is tempting to speculate that low levels of IL-18 and/or IL-12 act to amplify TCR-mediated signals, as seen during LVS macrophage infection, while high levels may overcome the need for TCR signaling, as observed during F. novicida macrophage infection. Although further studies will be needed to assess this hypothesis, experiments using recombinant IL-12 and IL-18 confirm that inflammatory cytokines are sufficient to overcome the requirement for TCR-mediated signals.
Of note, Ussher et al. found differences in MR1-dependent human MAIT cell activation that depended on the time of culture. Using fixed bacteria, they demonstrated that short-term culture (5 h) with MAIT cells was MR1 dependent, while long-term culture (20 h) was MR1 independent (21). Although we did not investigate the effects of culture times shorter than 20 h in our model system, we incubated blocking MR1 antibody with the infected macrophages prior to the addition of MAIT cells to ensure that MR1 was not available to drive early MAIT cell activation, and we further confirmed our findings with MR1−/− macrophages. However, we noted that some IFN-γ responses measured by ELISA and ICS yielded similar trends but somewhat different results. For example, IFN-γ responses to F. novicida were fully MR1 independent when measured via ELISA but only partially MR1 independent when measured via ICS. Since ELISA measures the cumulative levels of IFN-γ produced over the full 20-h incubation period whereas ICS measures IFN-γ production only during the last 4 h of culture, this may indicate that MR1 independence predominated early during culture, in contrast to the results with human cells noted above. Differences between the data presented here and those in previous reports likely reflect the difference between fixed bacteria and live intracellular pathogens or may reflect differences between human and murine antigen-presenting cells (APCs) or MAIT cells.
We found that tetramer+ and tetramer− Vα19iTg T cell IFN-γ production could be induced by the combination of IL-12 and IL-33. IL-33 is a potent proinflammatory cytokine that targets multiple cell types, although its function has been attributed primarily to its ability to stimulate group 2 innate lymphoid cells (ICL2s) to secrete large amounts of IL-5 and IL-13 (49). Since optimal T cell activation requires stimulation by an NF-κB inducer (e.g., an IL-1 cytokine family member, such as IL-18) and a signal transducer and activator of transcription (STAT) activator (e.g., IL-12 or IL-15), multiple cytokines have the potential to synergize to induce MAIT cell IFN-γ. Indeed, recent studies revealed that the combination of IL-15 and IL-18 stimulated human MAIT cell IFN-γ (50). Similar to IL-18, IL-33 induces NF-κB activation and has the potential to synergize with IL-12 to promote IFN-γ production. Although IL-33 is typically considered a type 2-promoting cytokine, IL-12 can upregulate IL-33R expression by iNKT cells and NK cells, and these cells produce IFN-γ in response to IL-12 and IL-33 (51, 52). One interesting aspect of IL-33 is that it is released upon cellular stress or damage and is proposed to be a central initiator of sterile inflammation (53). Since MAIT cells have been implicated in the pathogenesis of various autoimmune disorders, their activation by IL-33 in the absence of cognate antigen may be a critical component in their contribution to these conditions (54, 55).
Since products that induce MAIT cell activation have the potential to become novel vaccine adjuvants, it is important to understand the mechanisms that promote maximum MAIT cell activation. Importantly, a recent study showed that purified 5-OP-RU antigen was not sufficient to drive MAIT cell accumulation in the lungs of mice but instead required coadministration with TLR agonists to elicit MAIT cell responses (20). Here we show that the magnitude of inflammasome activation determines the level of MAIT cell IFN-γ production and that this effect is modulated by IL-18 in vitro. In contrast, we found that IL-18 was dispensable for MAIT cell IFN-γ production during in vivo LVS pulmonary infection, suggesting that additional signals that can promote MAIT cell IFN-γ production exist in vivo. Finally, we show that in addition to IL-18, the alarmin IL-33 can synergize with IL-12 to induce MAIT cell IFN-γ. These data underscore the importance of innate signals in promoting MAIT cell effector functions and expand the repertoire of cytokines that can activate MAIT cells.
MATERIALS AND METHODS
Bacteria.
F. tularensis LVS (ATCC 29684; American Type Culture Collection, Rockville, MD), F. novicida U112, and F. novicida ΔpdpA (both originally obtained from Francis Nano, University of Victoria, Victoria, BC, Canada) were grown and frozen as previously described (56). Viable bacteria were quantified by plating serial dilutions on supplemented Mueller-Hinton agar (MHA) plates.
Animals.
Male and female specific-pathogen-free C57BL/6J mice IL-18−/− mice, IL-12p35−/− mice, IL-12p40−/− mice, and caspase-1/11−/− mice were purchased from Jackson Laboratory. Note that the caspase-1/11−/− mice are a knockout for caspase-1 but have an incidental caspase-11 deficiency. MR1-deficient mice (57) and Vα19iTgMR1+/+ and Vα19iTgMR1−/− transgenic mice (40) were obtained from Ted Hansen (WUSTL, St. Louis, MO) and bred at CBER/FDA. Animals were housed in a barrier environment at CBER/FDA, and all procedures were performed according to approved protocols under Animal Care and Use Committee guidelines.
Infection of bone marrow-derived macrophages (BMMØ) with bacteria and coculture with Vα19iTg T cells.
BMMØ were cultured as previously described (58, 59). Bone marrow was flushed from femurs of healthy wild-type mice with Dulbecco minimal essential medium (DMEM; Life Technologies) supplemented with 10% heat-inactivated fetal calf serum (FCS; HyClone), 10% L-929 conditioned medium, 0.2 mM l-glutamine (Life Technologies), 1 mM HEPES buffer (Life Technologies), and 0.1 mM nonessential amino acids (Life Technologies) (complete DMEM [cDMEM]). Cells were washed, a single cell suspension was prepared, and cells were plated at 1 × 106 in 48-well plates or 2 × 106 in 24-well plates in cDMEM supplemented with 50 μg/ml gentamicin (Life Technologies) and incubated at 37°C in 5% CO2. After 1 day of incubation, the medium was replaced with antibiotic-free cDMEM, and the cells were incubated for an additional 6 days at 37°C in 5% CO2. The medium was replaced with fresh, gentamicin-free cDMEM every 2 days during the 7-day incubation.
Following the 7-day culture period, the BMMØ formed a confluent monolayer, and the concentration of BMMØ was estimated to be 5 × 106 cells/well in 48-well plates or 1 × 107 cells/well in 24-well plates. BMMØ were then infected with F. tularensis LVS, F. novicida U112, or F. novicida ΔpdpA according to the following protocol: bacteria were diluted from frozen stocks in cDMEM and added at a multiplicity of infection (MOI) as indicated in the figure legends. Bacteria were coincubated with BMMØ at 37°C in 5% CO2 for 2 h and then washed three times with phosphate-buffered saline (PBS; Life Technologies). The monolayers were incubated for 45 min to 1 h in cDMEM supplemented with 50 μg/ml gentamicin to eliminate extracellular bacteria. The BMMØ were then washed a further three times with PBS. Following the last wash, PBS was replaced with cDMEM and purified Vα19iTg T cells were added at a ratio of 1 purified T cell per 2 macrophages where indicated. The cells were cultured at 37°C in 5% CO2 for 20 h prior to the collection of supernatants for measurement of cytokine levels.
Vα19iTgCα−/− MR1+/+ mice were used as the source of naive splenic Thy1.2+ T cells for coculture with BMMØ. Total Thy1.2+ T cells were purified from Vα19iTgCα−/− MR1+/+ mouse spleens using a Thy1.2+ Dynabead cell enrichment column (Life Technologies), according to the manufacturer's recommendations. Where indicated, anti-MR1 Ab clone 26.5 (BioLegend) was added to BMMØ cultures at a concentration of 25 μg/ml 1 h prior to coculture and remained present throughout the coculture period. Neutralizing anti-IL-18 Ab (R&D Systems) was added at the indicated concentrations at the same time as addition of Vα19iTg T cells to the infected BMMØ cultures and remained present throughout the coculture period. Recombinant IL-18 (R&D Systems) was added at the indicated concentrations at the same time as addition of Vα19iTg T cells to the infected BMMØ cultures and remained present throughout the coculture period.
In vivo infection of mice and preparation of single-cell suspensions from lungs.
Intranasal (i.n.) infections were performed by delivering 2 × 102 CFU LVS or 5 × 101 CFU F. novicida in a volume of 25 μl per naris to anesthetized mice. To supplement IL-18, recombinant IL-18 (BioLegend) was delivered intranasally to WT mice on day 7 (2 μg) and day 10 (2 μg) after infection. To prepare lung cells for flow cytometry, the lungs from each mouse were excised following perfusion through the right heart ventricle with a solution of DMEM containing 2% FBS, collagenase (0.72 mg/ml), and DNase (10 U/ml). Harvested lungs were subjected to pressure disruption, followed by incubation for 20 min at 37°C in 5% CO2. Released cells were then filtered through a Filtra-bag (Lab Plas), subjected to ammonium-chloride-potassium (ACK) lysis to remove red blood cells, and passed through a 40-μm filter. Cells were then cultured with brefeldin A (10 μg/ml; Sigma-Aldrich) for 4 h and subjected to the intracellular cytokine staining and MR1 tetramer staining protocols described below.
Vα19iTg T cell IFN-γ response to recombinant cytokines.
Vα19iTgCα−/− MR1+/+ mice were used as the source of naive splenic total Thy1.2+ T cells for culture with recombinant cytokines. Total Thy1.2+ T cells were purified from Vα19iTgCα−/− MR1+/+ mouse spleens, using a Thy1.2+ Dynabead cell enrichment column (Life Technologies) according to the manufacturer's recommendations, and cultured in 96-well plates at a concentration of 4 × 106 cells/well. Naive T cells were stimulated with 200 ng/ml of recombinant IL-18 (R&D Systems), IL-12p70 (BioLegend), IL-33 (BioLegend), IL-1α (BioLegend), or IL-1β (BioLegend), as indicated, for 20 to 24 h at 37°C in 5% CO2. IFN-γ levels in the supernatants were determined by ELISA (BD Pharmingen). For intracellular IFN-γ staining, brefeldin A (10 μg/ml; Sigma-Aldrich) was added for the final 4 h of culture.
Quantitation of cytokines in BMMØ culture supernatants.
Culture supernatants were assayed for cytokines by ELISA and quantified by comparison to recombinant standards. Cytokines measured included IFN-γ (BD Pharmingen), IL-18 (eBioscience), IL-12p70 (BD Pharmingen), IL-1α and IL-1β (BioLegend), and IL-33 (eBioscience), and ELISAs were performed according to the manufacturer's instructions.
MR1 tetramer staining and intracellular cytokine staining.
For intracellular IFN-γ staining, brefeldin A (10 μg/ml; Sigma-Aldrich) was added to the cells for the final 4 h of culture. Approximately 1 × 106 to 2 × 106 cells were removed from the cultures and washed with cold fluorescence-activated cell sorter (FACS) buffer (PBS + 2% FCS). Intracellular IFN-γ staining was performed using the BD Biosciences buffer system according to the manufacturer's instructions using anti-IFN-γ Ab (clone XMG1.2; BioLegend) or an isotype-matched control (clone RTK2071; BioLegend). Cell surface staining was performed as follows: cells were stained with phycoerythrin (PE)-conjugated control 6-FP MR1 tetramer or PE-conjugated 5-OP-RU MR1 tetramer at a dilution of 1:500 for 30 min at room temperature in the dark. 6-FP- and 5-OP-RU-loaded MR1 tetramers were obtained from the NIH Tetramer Core Facility (Atlanta, GA) (18). Immediately thereafter, cells were incubated with Fc block (anti-CD16/32) for 10 min on ice and costained with H57-597 (anti-TCRβ chain), RA3-6B2 (anti-B220), and BM8 (anti-F4/80), obtained from BioLegend. B220 and F4/80 were used as a “dump” channel to gate out all cells that nonspecifically bind tetramers, as well as macrophages (i.e., F4/80+ cells) that may be obtained from the macrophage cocultures. In some experiments, where indicated, BGIL18A (anti-IL-18Rα; BioLegend) and DIH9 (anti-ST2; BioLegend) were included in the surface-staining panel. The Live/Dead Near IR stain was obtained from Life Technologies and included in all staining protocols. Optimal antibody and tetramer concentrations were determined in separate experiments, and appropriate fluorochrome-labeled isotype control antibodies were used throughout. In all cases, cells were first gated on singlets (FSC-W versus FSC-A or -H) and live cells (Live/Dead Near IR negative) prior to further analyses. Cells were analyzed using a Becton-Dickinson Fortessa X-20 flow cytometer and FlowJo software.
Western blotting.
Bone marrow-derived macrophages (3 × 107) grown in 6-well plates were mock infected or infected with LVS, F. novicida, or F. novicida ΔpdpA at an MOI of 100:1. Higher MOIs were used for the Western blot experiments compared to the other experiments in order to allow for optimal detection of caspase p-10. Infected macrophages were left in serum-free DMEM without phenol red (Sigma) for 20 to 24 h. Supernatants were harvested and concentrated using 6 ml Spin-X UF concentrator tubes with a 5-kDa molecular mass cutoff (Corning). Concentrates were treated with 25% (final volume) of StrataClean protein binding resin beads (Agilent) and vortexed for 1 min. Beads were pelleted at 1,000 rpm for 10 min, resuspended in 1× sample loading buffer with reducing agent (Life Technologies), and heated at 98°C on a preheated heat block for 10 min. Beads were separated from the sample by centrifugation prior to loading. Twenty microliters of each sample was loaded onto a Novex NuPAGE 4 to 12% gel (Life Technologies) and run in 2-(N-morpholino) ethanesulfonic acid (MES) SDS buffer (Life Technologies) at 125 V until the dye front reached the bottom. Gel was transferred to a 0.2-μm-pore-size low-fluorescence polyvinylidene difluoride (PVDF) membrane (Thermo Scientific) using the Trans-Blot SD semidry electrophoretic transfer cell (Bio-Rad) according to the manufacturer's instructions. Transfer was carried out at 15 V for 30 min in Bjerrum and Schafer-Nielsen transfer buffer with 1.3 mM SDS. The blot was blocked in PBS–0.1% Tween 20 and 0.375% bovine serum albumin (BSA) for 1 h. Caspase-1 was detected by incubating with 1:1,000 mouse anti-mouse caspase-1 (p10) antibody (Adipogen) in blocking buffer at 4°C overnight, followed by 1:50,000 rabbit anti-mouse IgG antibody (Abcam) at room temperature for 1 h. Visualization was achieved using SignalWest Dura luminol (Thermo Scientific) under a Fluorochem E gel and blot imager (ProteinSimple). The blot was reprobed for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) by incubating with 1:2,000 rabbit anti-mouse GAPDH horseradish peroxidase (HRP)-conjugated antibody (Cell Signaling Technology) for 1 h. The blot was visualized as described above.
Statistical analyses.
All experiments were performed using triplicate samples and repeated at least three times to assess reproducibility. Data were analyzed via one-way analysis of variance (ANOVA) followed by the Student-Newman-Keuls multiple stepwise comparison (for experiments with >2 experimental groups). For experiments with only two comparisons, data were analyzed using a two-tailed Student's t test; P values of 0.05 or less were considered significant.
REFERENCES
- 1.Treiner E, Duban L, Moura IC, Hansen T, Gilfillan S, Lantz O. 2005. Mucosal-associated invariant T (MAIT) cells: an evolutionarily conserved T cell subset. Microbes Infect 7:552–559. doi: 10.1016/j.micinf.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 2.Reantragoon R, Corbett AJ, Sakala IG, Gherardin NA, Furness JB, Chen Z, Eckle SB, Uldrich AP, Birkinshaw RW, Patel O, Kostenko L, Meehan B, Kedzierska K, Liu L, Fairlie DP, Hansen TH, Godfrey DI, Rossjohn J, McCluskey J, Kjer-Nielsen L. 2013. Antigen-loaded MR1 tetramers define T cell receptor heterogeneity in mucosal-associated invariant T cells. J Exp Med 210:2305–2320. doi: 10.1084/jem.20130958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tilloy F, Treiner E, Park SH, Garcia C, Lemonnier F, de la Salle H, Bendelac A, Bonneville M, Lantz O. 1999. An invariant T cell receptor alpha chain defines a novel TAP-independent major histocompatibility complex class Ib-restricted alpha/beta T cell subpopulation in mammals. J Exp Med 189:1907–1921. doi: 10.1084/jem.189.12.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gold MC, McLaren JE, Reistetter JA, Smyk-Pearson S, Ladell K, Swarbrick GM, Yu YY, Hansen TH, Lund O, Nielsen M, Gerritsen B, Kesmir C, Miles JJ, Lewinsohn DA, Price DA, Lewinsohn DM. 2014. MR1-restricted MAIT cells display ligand discrimination and pathogen selectivity through distinct T cell receptor usage. J Exp Med 211:1601–1610. doi: 10.1084/jem.20140507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L, Bhati M, Chen Z, Kostenko L, Reantragoon R, Williamson NA, Purcell AW, Dudek NL, McConville MJ, O'Hair RA, Khairallah GN, Godfrey DI, Fairlie DP, Rossjohn J, McCluskey J. 2012. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 491:717–723. doi: 10.1038/nature11605. [DOI] [PubMed] [Google Scholar]
- 6.Le Bourhis L, Guerri L, Dusseaux M, Martin E, Soudais C, Lantz O. 2011. Mucosal-associated invariant T cells: unconventional development and function. Trends Immunol 32:212–218. doi: 10.1016/j.it.2011.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Gold MC, Cerri S, Smyk-Pearson S, Cansler ME, Vogt TM, Delepine J, Winata E, Swarbrick GM, Chua WJ, Yu YY, Lantz O, Cook MS, Null MD, Jacoby DB, Harriff MJ, Lewinsohn DA, Hansen TH, Lewinsohn DM. 2010. Human mucosal associated invariant T cells detect bacterially infected cells. PLoS Biol 8:e1000407. doi: 10.1371/journal.pbio.1000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansen TH, Huang S, Arnold PL, Fremont DH. 2007. Patterns of nonclassical MHC antigen presentation. Nat Immunol 8:563–568. doi: 10.1038/ni1475. [DOI] [PubMed] [Google Scholar]
- 9.Tsukamoto K, Deakin JE, Graves JA, Hashimoto K. 2013. Exceptionally high conservation of the MHC class I-related gene, MR1, among mammals. Immunogenetics 65:115–124. doi: 10.1007/s00251-012-0666-5. [DOI] [PubMed] [Google Scholar]
- 10.Meierovics A, Yankelevich WJ, Cowley SC. 2013. MAIT cells are critical for optimal mucosal immune responses during in vivo pulmonary bacterial infection. Proc Natl Acad Sci U S A 110:E3119–E3128. doi: 10.1073/pnas.1302799110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Georgel P, Radosavljevic M, Macquin C, Bahram S. 2011. The non-conventional MHC class I MR1 molecule controls infection by Klebsiella pneumoniae in mice. Mol Immunol 48:769–775. doi: 10.1016/j.molimm.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Chua WJ, Hansen TH. 2010. Bacteria, mucosal-associated invariant T cells and MR1. Immunol Cell Biol 88:767–769. doi: 10.1038/icb.2010.104. [DOI] [PubMed] [Google Scholar]
- 13.Chua WJ, Truscott SM, Eickhoff CS, Blazevic A, Hoft DF, Hansen TH. 2012. Polyclonal mucosa-associated invariant T cells have unique innate functions in bacterial infection. Infect Immun 80:3256–3267. doi: 10.1128/IAI.00279-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Bourhis L, Martin E, Peguillet I, Guihot A, Froux N, Core M, Levy E, Dusseaux M, Meyssonnier V, Premel V, Ngo C, Riteau B, Duban L, Robert D, Huang S, Rottman M, Soudais C, Lantz O. 2010. Antimicrobial activity of mucosal-associated invariant T cells. Nat Immunol 11:701–708. doi: 10.1038/ni.1890. [DOI] [PubMed] [Google Scholar]
- 15.Cui Y, Franciszkiewicz K, Mburu YK, Mondot S, Le Bourhis L, Premel V, Martin E, Kachaner A, Duban L, Ingersoll MA, Rabot S, Jaubert J, De Villartay JP, Soudais C, Lantz O. 2015. Mucosal-associated invariant T cell-rich congenic mouse strain allows functional evaluation. J Clin Invest 125:4171–4185. doi: 10.1172/JCI82424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serriari NE, Eoche M, Lamotte L, Lion J, Fumery M, Marcelo P, Chatelain D, Barre A, Nguyen-Khac E, Lantz O, Dupas JL, Treiner E. 2014. Innate mucosal-associated invariant T (MAIT) cells are activated in inflammatory bowel diseases. Clin Exp Immunol 176:266–274. doi: 10.1111/cei.12277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rahimpour A, Koay HF, Enders A, Clanchy R, Eckle SB, Meehan B, Chen Z, Whittle B, Liu L, Fairlie DP, Goodnow CC, McCluskey J, Rossjohn J, Uldrich AP, Pellicci DG, Godfrey DI. 2015. Identification of phenotypically and functionally heterogeneous mouse mucosal-associated invariant T cells using MR1 tetramers. J Exp Med 212:1095–1108. doi: 10.1084/jem.20142110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corbett AJ, Eckle SB, Birkinshaw RW, Liu L, Patel O, Mahony J, Chen Z, Reantragoon R, Meehan B, Cao H, Williamson NA, Strugnell RA, Van Sinderen D, Mak JY, Fairlie DP, Kjer-Nielsen L, Rossjohn J, McCluskey J. 2014. T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature 509:361–365. doi: 10.1038/nature13160. [DOI] [PubMed] [Google Scholar]
- 19.Soudais C, Samassa F, Sarkis M, Le Bourhis L, Bessoles S, Blanot D, Herve M, Schmidt F, Mengin-Lecreulx D, Lantz O. 2015. In vitro and in vivo analysis of the Gram-negative bacteria-derived riboflavin precursor derivatives activating mouse MAIT cells. J Immunol 194:4641–4649. doi: 10.4049/jimmunol.1403224. [DOI] [PubMed] [Google Scholar]
- 20.Chen Z, Wang H, D'Souza C, Sun S, Kostenko L, Eckle SB, Meehan BS, Jackson DC, Strugnell RA, Cao H, Wang N, Fairlie DP, Liu L, Godfrey DI, Rossjohn J, McCluskey J, Corbett AJ. 2017. Mucosal-associated invariant T-cell activation and accumulation after in vivo infection depends on microbial riboflavin synthesis and co-stimulatory signals. Mucosal Immunol 10:58–68. doi: 10.1038/mi.2016.39. [DOI] [PubMed] [Google Scholar]
- 21.Ussher JE, Bilton M, Attwod E, Shadwell J, Richardson R, de Lara C, Mettke E, Kurioka A, Hansen TH, Klenerman P, Willberg CB. 2014. CD161++ CD8+ T cells, including the MAIT cell subset, are specifically activated by IL-12+IL-18 in a TCR-independent manner. Eur J Immunol 44:195–203. doi: 10.1002/eji.201343509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakala IG, Kjer-Nielsen L, Eickhoff CS, Wang X, Blazevic A, Liu L, Fairlie DP, Rossjohn J, McCluskey J, Fremont DH, Hansen TH, Hoft DF. 2015. Functional heterogeneity and antimycobacterial effects of mouse mucosal-associated invariant T cells specific for riboflavin metabolites. J Immunol 195:587–601. doi: 10.4049/jimmunol.1402545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Latz E, Xiao TS, Stutz A. 2013. Activation and regulation of the inflammasomes. Nat Rev Immunol 13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.von Moltke J, Ayres JS, Kofoed EM, Chavarria-Smith J, Vance RE. 2013. Recognition of bacteria by inflammasomes. Annu Rev Immunol 31:73–106. doi: 10.1146/annurev-immunol-032712-095944. [DOI] [PubMed] [Google Scholar]
- 25.Bergsbaken T, Fink SL, Cookson BT. 2009. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7:99–109. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin S, Brodsky IE. 2015. The inflammasome: learning from bacterial evasion strategies. Semin Immunol 27:102–110. doi: 10.1016/j.smim.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 27.Ulland TK, Ferguson PJ, Sutterwala FS. 2015. Evasion of inflammasome activation by microbial pathogens. J Clin Invest 125:469–477. doi: 10.1172/JCI75254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dotson RJ, Rabadi SM, Westcott EL, Bradley S, Catlett SV, Banik S, Harton JA, Bakshi CS, Malik M. 2013. Repression of inflammasome by Francisella tularensis during early stages of infection. J Biol Chem 288:23844–23857. doi: 10.1074/jbc.M113.490086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma J, Li Q, Mishra BB, Pena C, Teale JM. 2009. Lethal pulmonary infection with Francisella novicida is associated with severe sepsis. J Leukoc Biol 86:491–504. doi: 10.1189/jlb.1208728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kingry LC, Petersen JM. 2014. Comparative review of Francisella tularensis and Francisella novicida. Front Cell Infect Microbiol 4:35. doi: 10.3389/fcimb.2014.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barker JR, Chong A, Wehrly TD, Yu JJ, Rodriguez SA, Liu J, Celli J, Arulanandam BP, Klose KE. 2009. The Francisella tularensis pathogenicity island encodes a secretion system that is required for phagosome escape and virulence. Mol Microbiol 74:1459–1470. doi: 10.1111/j.1365-2958.2009.06947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Golovliov I, Baranov V, Krocova Z, Kovarova H, Sjostedt A. 2003. An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect Immun 71:5940–5950. doi: 10.1128/IAI.71.10.5940-5950.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O'Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, Dixit VM, Monack DM. 2010. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci U S A 107:9771–9776. doi: 10.1073/pnas.1003738107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mahawar M, Atianand MK, Dotson RJ, Mora V, Rabadi SM, Metzger DW, Huntley JF, Harton JA, Malik M, Bakshi CS. 2012. Identification of a novel Francisella tularensis factor required for intramacrophage survival and subversion of innate immune response. J Biol Chem 287:25216–25229. doi: 10.1074/jbc.M112.367672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schmerk CL, Duplantis BN, Howard PL, Nano FE. 2009. A Francisella novicida pdpA mutant exhibits limited intracellular replication and remains associated with the lysosomal marker LAMP-1. Microbiology 155:1498–1504. doi: 10.1099/mic.0.025445-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mares CA, Ojeda SS, Morris EG, Li Q, Teale JM. 2008. Initial delay in the immune response to Francisella tularensis is followed by hypercytokinemia characteristic of severe sepsis and correlating with upregulation and release of damage-associated molecular patterns. Infect Immun 76:3001–3010. doi: 10.1128/IAI.00215-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turtle CJ, Delrow J, Joslyn RC, Swanson HM, Basom R, Tabellini L, Delaney C, Heimfeld S, Hansen JA, Riddell SR. 2011. Innate signals overcome acquired TCR signaling pathway regulation and govern the fate of human CD161(hi) CD8alpha(+) semi-invariant T cells. Blood 118:2752–2762. doi: 10.1182/blood-2011-02-334698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohno K, Kataoka J, Ohtsuki T, Suemoto Y, Okamoto I, Usui M, Ikeda M, Kurimoto M. 1997. IFN-gamma-inducing factor (IGIF) is a costimulatory factor on the activation of Th1 but not Th2 cells and exerts its effect independently of IL-12. J Immunol 158:1541–1550. [PubMed] [Google Scholar]
- 39.Freeman BE, Hammarlund E, Raue HP, Slifka MK. 2012. Regulation of innate CD8+ T-cell activation mediated by cytokines. Proc Natl Acad Sci U S A 109:9971–9976. doi: 10.1073/pnas.1203543109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kawachi I, Maldonado J, Strader C, Gilfillan S. 2006. MR1-restricted V alpha 19i mucosal-associated invariant T cells are innate T cells in the gut lamina propria that provide a rapid and diverse cytokine response. J Immunol 176:1618–1627. doi: 10.4049/jimmunol.176.3.1618. [DOI] [PubMed] [Google Scholar]
- 41.Gherardin NA, Keller AN, Woolley RE, Le Nours J, Ritchie DS, Neeson PJ, Birkinshaw RW, Eckle SB, Waddington JN, Liu L, Fairlie DP, Uldrich AP, Pellicci DG, McCluskey J, Godfrey DI, Rossjohn J. 2016. Diversity of T cells restricted by the MHC class I-related molecule MR1 facilitates differential antigen recognition. Immunity 44:32–45. doi: 10.1016/j.immuni.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 42.Reantragoon R, Kjer-Nielsen L, Patel O, Chen Z, Illing PT, Bhati M, Kostenko L, Bharadwaj M, Meehan B, Hansen TH, Godfrey DI, Rossjohn J, McCluskey J. 2012. Structural insight into MR1-mediated recognition of the mucosal associated invariant T cell receptor. J Exp Med 209:761–774. doi: 10.1084/jem.20112095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lopez-Sagaseta J, Dulberger CL, Crooks JE, Parks CD, Luoma AM, McFedries A, Van Rhijn I, Saghatelian A, Adams EJ. 2013. The molecular basis for mucosal-associated invariant T cell recognition of MR1 proteins. Proc Natl Acad Sci U S A 110:E1771–E1778. doi: 10.1073/pnas.1222678110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lopez-Sagaseta J, Dulberger CL, McFedries A, Cushman M, Saghatelian A, Adams EJ. 2013. MAIT recognition of a stimulatory bacterial antigen bound to MR1. J Immunol 191:5268–5277. doi: 10.4049/jimmunol.1301958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Young MH, U'Ren L, Huang S, Mallevaey T, Scott-Browne J, Crawford F, Lantz O, Hansen TH, Kappler J, Marrack P, Gapin L. 2013. MAIT cell recognition of MR1 on bacterially infected and uninfected cells. PLoS One 8:e53789. doi: 10.1371/journal.pone.0053789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patel O, Kjer-Nielsen L, Le Nours J, Eckle SB, Birkinshaw R, Beddoe T, Corbett AJ, Liu L, Miles JJ, Meehan B, Reantragoon R, Sandoval-Romero ML, Sullivan LC, Brooks AG, Chen Z, Fairlie DP, McCluskey J, Rossjohn J. 2013. Recognition of vitamin B metabolites by mucosal-associated invariant T cells. Nat Commun 4:2142. doi: 10.1038/ncomms3142. [DOI] [PubMed] [Google Scholar]
- 47.Loh L, Wang Z, Sant S, Koutsakos M, Jegaskanda S, Corbett AJ, Liu L, Fairlie DP, Crowe J, Rossjohn J, Xu J, Doherty PC, McCluskey J, Kedzierska K. 2016. Human mucosal-associated invariant T cells contribute to antiviral influenza immunity via IL-18-dependent activation. Proc Natl Acad Sci U S A 113:10133–10138. doi: 10.1073/pnas.1610750113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jo J, Tan AT, Ussher JE, Sandalova E, Tang XZ, Tan-Garcia A, To N, Hong M, Chia A, Gill US, Kennedy PT, Tan KC, Lee KH, De Libero G, Gehring AJ, Willberg CB, Klenerman P, Bertoletti A. 2014. Toll-like receptor 8 agonist and bacteria trigger potent activation of innate immune cells in human liver. PLoS Pathog 10:e1004210. doi: 10.1371/journal.ppat.1004210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cayrol C, Girard JP. 2014. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol 31:31–37. doi: 10.1016/j.coi.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 50.Sattler A, Dang-Heine C, Reinke P, Babel N. 2015. IL-15 dependent induction of IL-18 secretion as a feedback mechanism controlling human MAIT-cell effector functions. Eur J Immunol 45:2286–2298. doi: 10.1002/eji.201445313. [DOI] [PubMed] [Google Scholar]
- 51.Smithgall MD, Comeau MR, Yoon BR, Kaufman D, Armitage R, Smith DE. 2008. IL-33 amplifies both Th1- and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol 20:1019–1030. doi: 10.1093/intimm/dxn060. [DOI] [PubMed] [Google Scholar]
- 52.Bourgeois E, Van LP, Samson M, Diem S, Barra A, Roga S, Gombert JM, Schneider E, Dy M, Gourdy P, Girard JP, Herbelin A. 2009. The pro-Th2 cytokine IL-33 directly interacts with invariant NKT and NK cells to induce IFN-gamma production. Eur J Immunol 39:1046–1055. doi: 10.1002/eji.200838575. [DOI] [PubMed] [Google Scholar]
- 53.Rider P, Voronov E, Dinarello CA, Apte RN, Cohen I. 2017. Alarmins: feel the stress. J Immunol 198:1395–1402. doi: 10.4049/jimmunol.1601342. [DOI] [PubMed] [Google Scholar]
- 54.Hinks TS. 2016. Mucosal-associated invariant T cells in autoimmunity, immune-mediated diseases and airways disease. Immunology 148:1–12. doi: 10.1111/imm.12582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reantragoon R, Boonpattanaporn N, Corbett AJ, McCluskey J. 2016. Mucosal-associated invariant T cells in clinical diseases. Asian Pac J Allergy Immunol 34:3–10. [PubMed] [Google Scholar]
- 56.Fortier AH, Slayter MV, Ziemba R, Meltzer MS, Nacy CA. 1991. Live vaccine strain of Francisella tularensis: infection and immunity in mice. Infect Immun 59:2922–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, Affaticati P, Gilfillan S, Lantz O. 2003. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature 422:164–169. doi: 10.1038/nature01433. [DOI] [PubMed] [Google Scholar]
- 58.Bosio CM, Elkins KL. 2001. Susceptibility to secondary Francisella tularensis live vaccine strain infection in B-cell-deficient mice is associated with neutrophilia but not with defects in specific T-cell-mediated immunity. Infect Immun 69:194–203. doi: 10.1128/IAI.69.1.194-203.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cowley SC, Elkins KL. 2003. Multiple T cell subsets control Francisella tularensis LVS intracellular growth without stimulation through macrophage interferon gamma receptors. J Exp Med 198:379–389. doi: 10.1084/jem.20030687. [DOI] [PMC free article] [PubMed] [Google Scholar]