

Abstract
The reversible addition fragmentation chain transfer (RAFT) polymerization method was adopted here to prepare a series of thermo-sensitive copolymers, poly (N,N-diethyl- acrylamide-b-N-vinylpyrrolidone). Their structures, molecular weight distribution and temperature sensitivity performances were characterized by the nuclear magnetic resonance (1HNMR), the gel permeation chromatography (GPC) and the fluorescence spectrophotometer, respectively. It has been identified that the synthesis reaction of the block copolymer was living polymerization. The thermo-sensitivity study suggested that N-vinylpyrrolidone (NVP), played a key role on the lower critical solution temperature (LCST) performance.
Keywords: RAFT polymerization, block copolymer, thermo-sensitivity, synthesis, characterization
1. Introduction
Block copolymers, also known as mosaic copolymer, is a special polymer which linked two or more polymer segments on the main chain directly [1]. Besides containing related properties of each block polymer, block copolymers also provide some novel properties, such as self-assembly, micro-phase separation, et al. [2–5], and which make them show an extensive application prospect. Among block copolymers, the thermosensitive block copolymer has been developed rapidly in recent years because the thermosensitive segment enabled the sol/gel phase transfer in selective solvents at their special LCST (Specifically, as temperature is higher than LCST, polymer would gather together and separate from solvent, while it would dissolve to the solvent again reversibly as temperature is lower than LCST). What’s more, during the gelatin process, thermo-sensitive block polymers will self-assemble and form a structure with a hydrophobic core-hydrophilic shell, which have shown a broad application as a prospect in electronic engineering [6, 7], drug release [8, 9], separation [10], nanomaterials [11], tissue engineering [12, 13], and so on. Reports on poly (N-isopropylacrylamide) (PNIPAM) block copolymer have been carried out more systematic and in-depth [14], but the poor biocompatibility and low LCST have seriously affected its future application prospects in medicine fields.
Compared with PNIPAM, a later found out thermo-sensitive polymer, poly (N, N-diethylacrylamide) (PDEA), has shown better biocompatible [15] property and higher LCST [16], which make the application of thermo-sensitive in polymer drug become more practical. Just for this reason, study about PDEA has attracted widely attention all over the world, and many reports about it could be indexed [17–21]. In modification aspect, researchers are mainly focused on improving LCST of PDEA or appending some other special properties to make it more versatile in future application [22–24]. However, multifunctional modifications were mainly realized by random copolymerization [25–27], the method about block copolymer is relatively small [21, 28–33], and the block copolymer of poly (N, N-diethylacrylamide-b-N-vinylpyrrolidone) was even not indexed at present. According to report, poly N-vinylpyrrolidone (PVP) is a kind of non-ionic water-soluble polymer, which possesses desirable properties such as biocompatibility, film-forming ability, biodegradability, and so on. Therefore, the property of block copolymer containing PVP and other segments has also been reported extensively [34–39]. Based on the nearer LCST to body temperature of PDEA and the excellent biocompatibility of PVP, it could be deduced that if combining PDEA together with PVP by block polymerization, and if LCST of block copolymer increased to equal to the body temperature, the supper properties of this block copolymer would show a very promising prospect in pharmaceutical field.
Generally, a block copolymer with a well-designed structure is usually prepared by the living radical polymerization method because the living radical polymerization always exists a reactive end groups and can be used to polymerize different monomers. For example, common monomers have styrene, methacrylates, acrylonitrile, vinyl acetate, etc., functional monomers have acrylic acid, sodium styrene sulfonate, dimethylaminoethyl methacrylate, etc. [40]. What’s more, polymer prepared by the living polymerization possessed a relative definite molecular weight and narrow molecular weight distribution [41], which would facilitate the study about properties of polymers with different structures in detail. According to reports, living radical polymerization methods always adopted contain nitroxide-mediated radical polymerization (NMP) [42], atom transfer radical polymerization (ATRP) [43] and RAFT [44] etc. Among them, RAFT is more popular since it can control the polymerization process better for both non polar and polar monomers. Besides, the monomer can be selected widely, the operation condition is more mild and assessable, the monomer conversion rate of RAFT is higher [45, 46], and it can be well applied in polymerization system of solution, emulsion and suspension [47–51], At the moment, the composition, sequential structure and molecular weight of block copolymer prepared by RAFT can be understood and calculated, and the end-group can be changed to others using an easy and mild chemical reaction because the existence of thiol end group [52]. At present, RAFT has been used extensively for the synthesis of the multi-block copolymer which requires multiple isolations and dispersion of the polymer [53, 54].
Based on the above findings, the preparation of DV using RAFT method was carried out in this work, the process was completed as follows: DV was prepared by polymerized PDEA, DV, in turn, using RAFT polymerization technology, and the structure contained the content of each block, the molecule weight and polydispersity index(PDI) were characterized by 1HNMR and GPC, respectively. All results documented that the polymerization was living polymerization. Then, the thermo-sensitive copolymer of DV was characterized, and the result of thermo-sensitivity about NVP showed that increasing the molar ratio of NVP, LCST increased, while the thermo-sensitivity was weakened, and when the ratio of nDEA:nNVP was in the range of (1:0.04, 1:0.05), LCST approximated to 36 °C, which was very close to body temperature.
2. Experimental
2.1. Materials
Dodecylmercaptan (AR), Sodium hydride(AR), Carbon disulfide (AR), Iodine (AR), Sodium thiosulfate (AR),and Sodium sulfate (AR) were provided by the YantaiShuangshuang Chemical Co. Ltd. Ethyl acetate (AR), Petroleum ether (AR), Ether (AR),n-hexane (AR),and n-decane(AR) were purchased by the Rionlon Bo hua pharmaceutical chemical company. Acenaphthylene(AR) was provided by the Shanghai SA Ren Limited Liability Company. Acetone (AR) was produced by Beijing Chemical Works. All above reagents were used directly. Azobisisobutyronitrile(AR) was provided by YantaiShuangshuang Chemical Co. Ltd and was recrystallizedby95% ethanol before using.
2.2. Preparation of trithiocododecanoic acid-2-cyanoisopropyl
The synthesis theory of trithiocododecanoicacid-2-cyanoisopropyl (CPDTC), a RAFT agent, was shown in Scheme 1, and the process was shown in Scheme 2. A solution of sodium hydride in anhydrous diethyl ether (1.58 g sodium hydride was dissolved into 75 mL anhydrous ether) was loaded into a 250 mL three necked flask which was assembled with a dropping funnel and a thermometer. Then the flask was fixed on a magnetic stirrer equipped with an ice-water bath. After the solution (1#) was agitated and cooled to room temperature, carbon dodecylmercaptan (7.7 g) was added to the system with the rate of one drop for each two seconds (1d/2s). After all carbon dodecylmercaptan was transferred to the reaction system, the reaction was kept for another 2 h under the ice-water environment. After that, the agitation system was changed with an electric stirrer, and carbon disulfide (3 g) was added dropwise slowly to the system, the all dripping time was controlled in 10 min. Keeping the system under electric stirring and reaction for 40 min, when the reaction was finished, the obtained crude product was dissolved in ether and separated the white block insoluble matter (dodecyl-mercaptan) by filtration, the filtrate was concentrated to a yellow viscous product (2#). After dissolving, the yellow viscous product in the ether under a magnetic stirrer, iodine (5.8 g) was added into the solution in batches. After reacting for 1 hat room temperature, sodium iodide was filtered off, and a dark brown filtrate was obtained. After the iodine in the system was removed by adding the aqueous solution of sodium thiosulfate, the mixture was separated by a separating funnel, and the oil phase was dried with anhydrous sodium sulfate until no block appeared as some sodium sulfate was added. When the filtration was stopped, the filtrate (3#) was evaporated till ether was removed completely. Fixing the system on an oil bath at 79 °C, AIBN (3.4 g) and ethyl acetate (80 mL) were added in turn. Keeping the reaction for 24 h under magnetic stirrer and reflux state, the mixture was concentrated and filtrated again. After that, n-hexane was added to the filtrate till no white needle-like precipitate was produced. Filtering the mixture and evaporating the filtrate again, the crude product of CPDTC was obtained. Finally, the crude product of CPDTC was refined by a silica gel column. The process was conducted as follows: silica gel (140 g) was dispersed into petroleum ether (500 mL), then the dispersion was injected into a silica gel column. Adjusting the silica gel column till the upper surface of silica was flat, and keeping the petroleum ether flowing until it reached a distance of about 0.5 cm from the upper surface of the silica gel, the piston was closed and the crude product of CPDTC was injected into the silica gel column with a pipette. After that, the piston was opened again. Introducing petroleum ether to the silica gel column persistently until the entire color belt diffused to the middle of the silica gel column and was washed off, the silica gel column was washed off with a mixture of petroleum ether and ethyl acetate (the volume ratio of petroleum ether and ethyl acetate was 12: 1) until the belt that existed on the upper surface of the silica gel column was washed completely. Collecting and evaporating the elution of petroleum ether and ethyl acetate, pure CPDTC with 99% was obtained.
Scheme 1.

The mechanism of CPDTC synthesis process.
Scheme 2.

CPDTC synthesis process.
2.3. Preparation of N, N- diethylacrylamide
N, N- diethylacrylamide and its precursor, acryloyl chloride, were prepared according to literature [55], and the product was characterized by1HNMR.
2.4. Preparation of block copolymer
2.4.1. Preparation of PDEA
Synthesis of PDEA was shown in Scheme 3, the product was 3# of Scheme 3: A solution of ethyl acetate (24 g), acenaphthylene (0.0661 g) and DEA (24 g) was added into a three necked flask, then CPDTC and AIBN were added in turn with the molar ratio of AIBN: CPDTC: DEA = 1: 5: 1000, and the mass ratio of DEA to acenaphthylene was 1.2700: 0.0035. After a nitrogen purge for 30 min, the flask was placed on an oil bath of 70 °C. Keeping the reaction under a persistent nitrogen purge and stirring for 24 h, the reaction was stopped, and the product solution was diluted with about 3 times the volume of ethyl acetate. After introduced, the solution dropwise slowly to about 10 times volume of n-decane under quickly stirring, the mixture was filtrated. Repeating the dissolution and precipitation process for three times, the final obtained precipitate was dried in a 40 °C vacuum oven under the vacuum degree of 0.08 to constant, and PDEA was obtained.
Scheme 3.

Synthesis of block copolymer DV.
2.4.2. Preparation of DV
Synthesis of DV was shown in Scheme 3, the product was 4# of Scheme 3. The synthesis process of DV was similar to that of PDEA except for PDEA here was conscripted as a RAFT reagent, NVP was used as a monomer, and the solvent was changed to acetone, the reaction time was different. After the final obtained precipitate was dried in a 40 °C vacuum oven under the vacuum degree of 0.08 to constant, different molar ratios of DV were obtained.
2.5. Structure characterization of RAFT reagents, monomers and polymers
Deuterated chloroform (CDCl3) solution (10 mg/mL) of CPDTC, PDEA and DV were prepared, respectively. And then1HNMR for each compound was determined by nuclear magnetic resonance spectroscopy (AV-400, Bruker, U.S.A.), and the molecular structure was characterized according to the chemical shift of hydrogen in each compound.
2.6. Characterization of molecular weight distribution
Tetrahydrofuran (THF, 5 mg/mL) solution of PDEA and DV were prepared, respectively, and the molecular weight distribution for each compound was detected by a gel permeation chromatograph (GPC, Waters 1525/2414/2487, Fairburn industrial development co. LTD, Shanghai, China) which was calibrated with narrowly distributed polymethyl methacrylate. During this process, DMF in which 0.05 mmol/L lithium bromide was contained was adopted as the mobile phase, and the injection volume was 10 μL at 1 mL/min flow rate and 25 °C.
2.7. Characterization of thermo-sensitive performance of copolymer
The thermo-sensitive performance of DV was determined by static fluorescence spectroscopy technology using a fluorescence spectrometer (LS-55, Perkin-Elmer Cetus Corporation, USA). During this process, 0.01 mg/mL of DV aqueous solution was prepared, and operation conditions were set as follows: the excitation wavelength was 290 nm, the scanning speed was 240 nm/min, the excitation and emission slit were10 nm, the scanning emission wavelength was 330–430 nm, and the fluorescent intensity determination wavelength was 380 nm.
3. Results and discussion
3.1. Structure of monomer and polymer
3.1.1. Structure of CPDTC
Figure 1 shows 1HNMR (400 MHz, CDCl3, δ, ppm) spectra of CPDTC: 2.84 (t, 2H, –CH2–), 1.88 (s, 6H, –CH3), 1.72–1.65 (m, 2H, –CH2–), 1.26 (s, 16H, –CH2–) and 0.87 (t, 3H, –CH3).
Figure 1.

1HNMR spectra of the CPDTC.
3.1.2. Structure of DEA and NVP
Figure 2 showed 1HNMR (400 MHz, CDCl3, δ, ppm) spectra of DEA:1.16 (t, 6H, –CH3), 3.40 (q, 4H, –CH2–), 5.65 (dd, 1H, CH2=), 6.32 (dd, 1H, CH2=), 6.53 (m, 1H, CH2=CH–).
Figure 2.

1HNMR spectra of the DEA.
Figure 3 showed 1HNMR (400 MHz, CDCl3, δ, ppm) spectra of NVP:2.10 (dt, 2H, –CH2–), 2.48 (t, 2H, –CH2–), 3.50 (t, 2H, –CH2–), 4.40 (q, 1H, CH2=), 7.08 (m, 1H, CH2=CH–)
Figure 3.

1HNMR spectra of NVP.
3.1.3. Structure of PDEA
Figure 4 shows the 1HNMR spectra of PDEA. It could be found in a polymer, except for peak at δ = 2.96 in CPDTC and δ = 3.34 in DEA, almost all peaks for both CPDTC and DEA were submersed and could not be identified. The reason could be ascribed to large molecule of PDEA, because of the polymerization, large amounts of DEA were bonded together, and the quantities of hydrogen for a given group would increase hundreds of times. Due to the superposition of each group, and the influence of other groups adjacent to them both in link and in space, peaks related to a given group could not be distinguished clearly, naturally. However, due to the far δ distance of q and x from the others, the influence they suffered was relatively small, and could be conscripted as reference to calculate molecular weight of PDEA as Equation (1):
| (1) |
Figure 4.

1HNMR spectra of the PDEA.
where, and represent the total hydrogen quantities of hydrogen at chemical shifts 2.96 and 3.34, respectively. There are 2 mol H at δ(2.96) in 1 mol CPDTC, but 4 mol H at δ(3.34) in DEA, so as 1 mol CPDTC was combined with 1 mol DEA, the integral of H at δ(3.34)would be 2 times that at δ(2.96), 127 was the molecular weight of DEA.
For example, from Figure 3, it could be found the peak area ratio of q and x was 1: 43.70, so the molecular weight of PDEA should be 43.70/2 × 127 = 2774.95 g/mol (±0.2%).
Besides, GPC characterization showed a polydispersity index (PDI) of PDEA as 1.05, which was far smaller than the general free radical polymerization, and it indicated that the process of preparing PDEA was living polymerization.
3.1.4. Structure of DV
Figures 5–9 showed 1HNMRspectra of DV, the molar ratio between DEA and NVP segment could also be calculated according to the specific δ of DEA and NVP as Equation (2):
| (2) |
Figure 5.

1HNMR spectra of the DV1.
Figure 6.

1HNMR spectra of the DV2.
Figure 7.

1HNMR spectra of the DV3.
Figure 8.

1HNMR spectra of the DV4.
Figure 9.

1HNMR spectra of the DV5.
where, and represented the total hydrogen quantities of hydrogen at chemical shift 3.68 and 3.31, respectively. There are 2 mol H at δ(3.68) in 1 mol NVP, but 4 mol H at δ(3.31) in DEA, so as 1 mol NVP combined with 1 mol DEA, the integral of H at δ(3.31) would be 2 times that at δ(3.68), 127 was the molecular weight of DEA.
As the molar ratio of DEA and NVP was obtained, the molecular weight of PVP could also be calculated using Equation (3):
| (3) |
where, M(DEA), M(NVP) and represent the molecular weight of DEA, NVP, the molecular weight of PDEA, respectively.
After was obtained, the molecular weight of DV could be calculated easily. For example, from Figure 8, it could be found the peak area ratio of x and o was 1: 0.01, so the molecular weight of PDEA could be calculated as Equation (4):
| (4) |
where and represent the molecular weight of PDEA and PVP, respectively.
The 1HNMR spectra of the DV2, DV3, DV4 and DV5 were shown as Figure 5–9. Similarly, , , , and could be obtained as the way of .
3.2. The relationship among the molecular weight, polydispersity index of DV and polymerization time
Figure 10 displayed the relationships among the molecular weights and poly dispersity index (PDI) of DV at different reaction times. It was shown that the molecular weight was linearly increased with the polymerization time, and PDI was (1.20, 1.35). The drift off of experimental results to their related theoretical could be ascribed to the variation of viscosity. With prolonged reaction time, the molecular weight of the polymer increased, and the viscosity augmented correspondently [56]. Besides, the concentration of the monomer would decrease with increase of time. Both of which would decrease the polymerization probability, while increase the chain termination probability. As a result, PDI became wide gradually with the prolonging of polymerization time.
Figure 10.
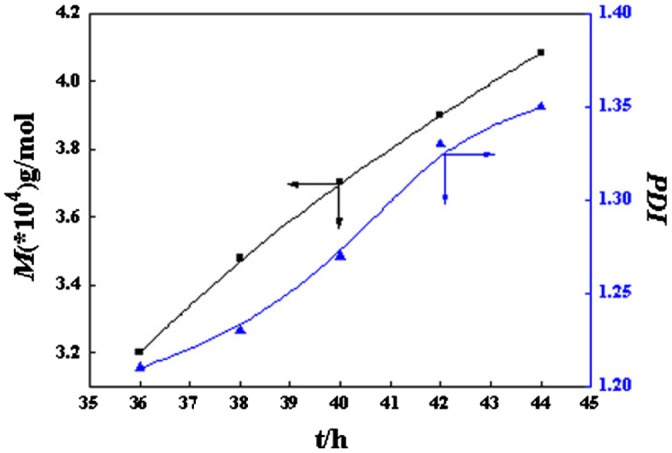
The relationship between relative molecular weight and PDI of PDEA-b-PVP and polymerization time.
3.3. Thermo-sensitive performance of DV
Figure 11 showed the relationship between the molar fraction of DV and LCST in block copolymer. It was shown obviously that the fluorescence intensity (FI) of DV decreased with the increase of the temperature (T). Besides, with the increase of the molar ratio of monomer NVP, the abrupt amplitude of FI was reduced, while LCST increased. Table 1 showed the LCST of DVs.
Figure 11.
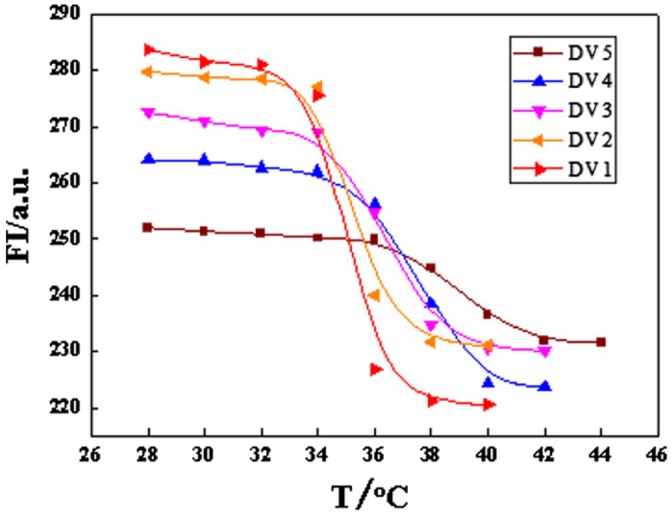
The relationship between fluorescence intensity and temperature of PDEA-b-PVP.
Table 1. The relationship between Mn and LCST.
| PDEA-b-PVP | Mna/g⋅mol−1 | LCST/oC |
|---|---|---|
| DV1 | 3413 | 33.3 |
| DV2 | 3524 | 33.8 |
| DV3 | 3746 | 34.0 |
| DV4 | 3857 | 35.7 |
| DV5 | 3968 | 36.0 |
And the molar ratios of NVP to DEA in DV1 to DV5 was 0.038, 0.077, 0.154, 0.192, 0.231, respectively.
meant the deviation of the Mn of DVs was ±(0.2–0.4)%.
The FI of DV decreased with the increase of temperature, which could be ascribed to the variation of the interaction force between the polymer and solvent. When the temperature was lower than LCST, polymer chains would interact mainly with the solvent by the hydrogen bond and Van der Waals force (VDW) [57]. At this state, solvated molecules would be around macromolecular chains through the hydrogen bond, and a higher ordering solvation shell will form correspondingly, which would lead the polymer chain to an extended random coil [56], the rotation of the molecular chain was relative free, which would be conducive to the fluorescent label, acenaphthylene, absorption energy and emission fluorescence spectrum. As a result, the FI of the polymer was large. As temperature reached LCST, the hydrophobic interaction among polymer chains gradually strengthened, polymer would shrink, and the phase separation would take place slowly, and the rotation of the molecular chain became gradually difficult. At this state, the energy absorption ability of acenaphthylene immobilized on the polymer chain would become difficult, and result in the excitation and fluorescence emission increasing in difficulty, the fluorescence intensity decreased accordingly. As the solvated molecular chain dehydrated, and the hydrophobic part in polymer chains collected together completely. At this stage, the phase separation would finish, and the conformation of polymer chains would change to a stable tight global [58], which would result in the hard rotation of polymer chain. As a result, acenaphthylene immobilized on the polymer chain could not absorb energy and also could not be excited, the fluorescence emission would be less, FI would display a smaller value and keep as a constant, accordingly.
As NVP was introduced, the interaction of the hydrogen bond between NVP and solvent would be strengthened. Thus, the phase separation would become difficult because the process would need to destroy more hydrogen bonds, which would increase LCST of the polymer naturally. With the introduction of NVP, the content of acenaphthylene would decrease the abrupt amplitude of FI reduced naturally.
4. Conclusions
This work synthesized DV successfully, and compared the relationship between polymerization time and molecular weight distribution, which verified the synthesis reaction of a block copolymer, which was identified as living polymerization. The thermo-sensitivity study of NVP found that LCST increased with the increase of NVP, while the thermo-sensitivity was weakened, and when the ratio of nDEA:nNVP was in the range of (1:0.04, 1:0.05), LCST approximated to 36 °C, which was very close to body temperature.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This work was supported by the National Natural Science Foundation of China [grant numbers 51563015, 21762027].
References
- [1].Ghamkhari A, Massoumi B, Jaymand M. Novel ‘schizophrenic’diblock copolymer synthesized via RAFT polymerization: poly (2-succinyloxyethyl methacrylate)-b-poly [(N-4-vinylbenzyl), N. Des Monomers Polym. 2017;20(1):190–200. 10.1080/15685551.2016.1239165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cong H, Li L, Zheng S. Formation of nanostructures in thermosets containing block copolymers: from self-assembly to reaction-induced microphase separation mechanism. Polymer. 2014;55(5):1190–1201. 10.1016/j.polymer.2014.01.049 [DOI] [Google Scholar]
- [3].Wen GH, Zhang B, Xie HL, et al. Microphase separation facilitating and stabilizing hierarchical segment self-assembly of combined main-chain/side-chain liquid crystalline polymer in diblock copolymer. Macromolecules. 2013;46(13):5249–5259. 10.1021/ma400325g [DOI] [Google Scholar]
- [4].Borah D, Rasappa S, Senthamaraikannan R, et al. Tuning PDMS brush chemistry by UV–O3 exposure for PS-b-PDMS microphase separation and directed self-assembly. Langmuir. 2013;29(28):8959. 10.1021/la401561k [DOI] [PubMed] [Google Scholar]
- [5].Wang Z, Li Y, Huang Y, et al. Enzyme-regulated topology of a cyclic peptide brush polymer for tuning assembly. Chem Commun. 2015;51:17108–17111. 10.1039/C5CC05653E [DOI] [PubMed] [Google Scholar]
- [6].Bass PS, Zhang L, Cheng ZY. Time-dependence of the electromechanical bending actuation observed in ionic-electroactive polymers. J Adv Dielect. 2017:1720002. [Google Scholar]
- [7].Zhang L, Liu Z, Lu X, et al. Nano-clip based composites with a low percolation threshold and high dielectric constant. Nano Energy. 2016;26:550–557. [Google Scholar]
- [8].Mali F, Zhang T, Li Yao, et al. Preparation and characterization of the PBLA-POLO-PBLA nanoparticles. J Sichuan Univ (Eng Sci Ed), 2016, 48(3):202–208. [Google Scholar]
- [9].Ayman M, Allohedan HA, Hamad A. Insulin release behavior of;poly(N-isopropylacrylamide-co-N-vinyl-2-pyrrolidone) hydrogel based on modified melamine crosslinkers. Polymer Sci. 2013;55(3–4):233–239. [Google Scholar]
- [10].Yu Y, Wang X-H, Lang W-C, et al. Microstructures and transformation of linear triblock copolymer films induced by surface field. Acta Polymerica Sinica, 2016(7):955–962. [Google Scholar]
- [11].Jing D, Wei T, Yang B, et al. Progress of nanoporous templates from self-assembly of block copolymers. Chemistry. 2013;76(10):909–914. [Google Scholar]
- [12].Ge J-H, Wang Y-J, Zheng Y-D. Degradable performance and bio-mineralization function of PLA-PEG-PLA/PLA tissue engineering scaffold in SBF. Polymer Mater Sci Eng. 2011, 27(11):126–128. [PubMed] [Google Scholar]
- [13].Zou Y, Zhang L, Yang L, Zhu F, et al. “Click” chemistry in polymeric scaffolds: bioactive materials for tissue engineering. J Controlled Release. 2018;273:160–179. 10.1016/j.jconrel.2018.01.023 [DOI] [PubMed] [Google Scholar]
- [14].Xia YJ, Huang YW, Yi GB, et al. Reversible aggregation kinetics of poly(N-isopropylacrylamide-co-N-vinylpyrrolidone) in aqueous solutions revealed by elastic light scattering spectroscopy. J Wuhan Univ Technology-Mater Sci Ed. 2013;28(4):766–772. 10.1007/s11595-013-0766-6 [DOI] [Google Scholar]
- [15].Perera RH, Wu H, Peiris P, et al. Improving performance of nano-scale ultrasound contrast agents using N. Nanomed Nanotechnol Biol Med. 2017;13(1):59–67. 10.1016/j.nano.2016.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ahmadkhani L, Abbasian M, Akbarzadeh A. Synthesis of sharply thermo and PH responsive PMA-b-PNIPAM-b-PEG-b-PNIPAM-b-PMA by RAFT radical polymerization and its schizophrenic micellization in aqueous solutions. Des Monomers Polym. 2017;20(1):406–418. 10.1080/15685551.2017.1314654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lessard DG, Ousalem M, Zhu XX, et al. Study of the phase transition of poly(N, N-diethylacrylamide) in water by rheology and dynamic light scattering. J Polymer Sci Part B Polymer Phys. 2003;41(14):1627–1637. 10.1002/(ISSN)1099-0488 [DOI] [Google Scholar]
- [18].André X. New double-responsive micelles of block copolymers based on N, N-diethylacrylamide: synthesis, kinetics, micellization, and application as emulsion stabilizers. Bibliogr; 2005. [Google Scholar]
- [19].Winnik FM, Ottaviani MF, Bossman SH, et al. Phase separation of poly(N-isopropylacrylamide) in water: a spectroscopic study of a polymer tagged with a fluorescent dye and a spin label. J Phys Chem. 1993;97(49):12998–13005. 10.1021/j100151a058 [DOI] [Google Scholar]
- [20].Chan JW, Yu B, Hoyle CE, et al. Convergent synthesis of 3-arm star polymers from RAFT-prepared poly(N, N-diethylacrylamide) via a thiol-ene click reaction. Chem Commun. 2008;40(40):4959–4961. 10.1039/b813438c [DOI] [PubMed] [Google Scholar]
- [21].Marcelo G, Prazeres TJV, Charreyre MT, et al. Thermoresponsive micelles of phenanthrene-α-end-labeled poly( N -decylacrylamide-b-N, N-diethylacrylamide) in Water. Macromolecules. 2010;43(1):501–510. 10.1021/ma902103q [DOI] [Google Scholar]
- [22].Zhang X, Monge S, In M, et al. Thermo-and pH-sensitive aggregation behavior of PDEAm-b-P(l-lysine) double hydrophilic block copolymers in aqueous solution. Soft Matter. 2012;9(4):1301–1309. [Google Scholar]
- [23].André X, Zhang M, Müller AHE. Thermo- and pH-Responsive Micelles of Poly(acrylic acid)-block-Poly(N, N-diethylacrylamide). Macromol Rapid Commun. 2010;26(7):558–563. [Google Scholar]
- [24].Maeda Y, Nakamura TA, Ikeda I. Change in solvation of poly(N, N-diethylacrylamide) during phase transition in aqueous solutions as observed by IR spectroscopy. Macromolecules. 2002;35(27):10172–10177. 10.1021/ma020945w [DOI] [Google Scholar]
- [25].Maeda Y, Yamabe M. A unique phase behavior of random copolymer of -isopropylacrylamide and -diethylacrylamide in water. Polymer. 2009;50(2):519–523. 10.1016/j.polymer.2008.11.032 [DOI] [Google Scholar]
- [26].Chen ZB, Liu DL, Zhao YY, et al. Synthesis and characterization of copolymer of poly(N,N-diethylacrylamide-co-acrylic acid). J Lanzhou Univ Technol. 2017;02(43):30–34. [Google Scholar]
- [27].Savoji MT, Strandman S, Zhu XX. Block random copolymers of N-Alkyl-substituted acrylamides with double thermosensitivity. Macromolecules. 2012;45(4):2001–2006. 10.1021/ma2027269 [DOI] [Google Scholar]
- [28].Angelopoulos SA, Tsitsilianis C. Thermo-reversible hydrogels based on Poly(N, N-diethylacrylamide)-block-poly(acrylic acid)-block-poly(N, N-diethylacrylamide) double hydrophilic triblock copolymer. Macromol Chem Phys. 2006;207(23):2188–2194. 10.1002/(ISSN)1521-3935 [DOI] [Google Scholar]
- [29].Beija M, Fedorov A, Charreyre MT, et al. Fluorescence anisotropy of hydrophobic probes in poly(N-decylacrylamide)-block-poly(N, N-diethylacrylamide) block copolymer aqueous solutions: evidence of premicellar aggregates. J Phys Chem B. 2010;114(31):9977–9986. 10.1021/jp101613y [DOI] [PubMed] [Google Scholar]
- [30].Telmo JV. Prazeres, Mariana Beija, Marie-Thérèse Charreyre. RAFT polymerization and self-assembly of thermoresponsive poly(-decylacrylamide–,-diethylacrylamide) block copolymers bearing a phenanthrene fluorescent α-end group. Polymer. 2010;51(2):355–367. [Google Scholar]
- [31].Amélia MPS, da Silva Gonçalves, Lopes SIC, Brogueira P, et al. Thermo-responsiveness of poly(N, N N, N mathContainer Loading Mathjax -diethylacrylamide) polymers at the air–water interface: The effect of a hydrophobic block. J Colloid Interface Sci. 2008;327(1):129–137. [DOI] [PubMed] [Google Scholar]
- [32].Fuchise K. Facile synthesis of thermoresponsive block copolymers bearing poly(N,N-diethylacrylamide) segment through group transfer polymerization. Macromol Rapid Commun. 2014;61–77. 10.1007/978-4-431-55046-4 [DOI] [Google Scholar]
- [33].Ren C, Liu X, Jiang X, et al. Polyisobutylene-b-Poly(N, N-diethylacrylamide) well-defined amphiphilic diblock copolymer: synthesis and thermo-responsive phase behavior. J Polym Sci Part A Polym Chem. 2015;53(9):1143–1150. 10.1002/pola.v53.9 [DOI] [Google Scholar]
- [34].Feng Q, Yaxuan Z, Zhiwei X, et al. Frontal polymerization and characterization of poly (N-Isopropylacrylamide-co-N-Vinyi-2-Pyrrolidone) hydrogels. Polym Mater Sci Eng. 2015;31(4):37–41. [Google Scholar]
- [35].Shen J, Jiang T, Feng J. Preparation and characterization of PAM-b-PVP and its hydrolysate by RAFT polymerization. Polym Mater Sci Eng. 2012;28(9):124–128. [Google Scholar]
- [36].Chen L-F, Bian X-K, Hou Z-C, et al. Preparation and characterization of the antifouling porous membranes from poly(vinylidene fluoride)-graft-poly(N-vinyl pyrrolidone) powders. Nucle Sci Techn. 2014;25(5):39–46. [Google Scholar]
- [37].Gao J, Zhao H, Li L, et al. Preparation and properties of poly(lactide)-poly(ethylene glycol)-poly(lactide)/poly(N-vinylpyrrolidone) crosslinked copolymer films. J Polym Mater Sci Eng. 2017;33(1):18–22. [Google Scholar]
- [38].Tamami B, Farjadian F, Ghasemi S, et al. Palladium nanoparticles supported on poly (N-vinylpyrrolidone)-grafted silica as an efficient catalyst for copper-free sonogashira and suzuki cross-coupling reactions. J Braz Chem Soc. 2015;26(8):1591–1598. [Google Scholar]
- [39].Shi J, Wang W, Wang J, et al. Synergetic effects of multiwalled carbon nanotubes and polyvinyl pyrrolidone on structure and properties of polysulfone microporous membranes. Polym Mater Sci Eng. 2015;31(12):63–66. [Google Scholar]
- [40].Liu Y, Jin YZ. Research progress of application of reversible addition-fragmentation chain transfer living radical polymerization. Polym Bull. 2009;23(6):59–67. [Google Scholar]
- [41].Fuxi H, Gang T, Xiaoyan M, et al. The active/controlled radical polymerization of N-vinylcaprolactam. Prog Chem. 2016(z2):328–336. [Google Scholar]
- [42].Hawker CJ, Bosman AW, Harth E. New polymer synthesis by nitroxide mediated living radical polymerizations. Chem Rev. 2001;101(12):3661–3688. 10.1021/cr990119u [DOI] [PubMed] [Google Scholar]
- [43].Matyjaszewski K, Xia J. Atom transfer radical polymerization. Chem Rev. 2001;101(9):866–868. [DOI] [PubMed] [Google Scholar]
- [44].Ren R, Wang Y, Liu D, et al. Facile preparation of a novel nickel-containing metallopolymer via RAFT polymerization. Des Monomers Polym. 2017;20(1):300–307. 10.1080/15685551.2016.1257378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Loiseau J, Doërr N, Suau JM, et al. Synthesis and characterization of poly(acrylic acid) produced by RAFT polymerization. application as a very efficient dispersant of CaCO3, kaolin, and TiO2. Macromolecules. 2003;36(9):3066–3077. 10.1021/ma0256744 [DOI] [Google Scholar]
- [46].Ray B, Isobe Y, Morioka K, et al. Synthesis of isotactic poly(N-isopropylacrylamide) by RAFT polymerization in the presence of lewis acid. Macromolecules. 2003;36(3):543–545. 10.1021/ma0257595 [DOI] [Google Scholar]
- [47].Xiao-hui LIU. Investigation of RAFT-mediated Controlled/Living Radical Polymerization of Acrylonitrile. Changchun Institute of Applied Chemistry, Academia Sinica Academia Sinica; 2007. p. 20–25. [Google Scholar]
- [48].Shen X-L, Zhou J-H, Ma J-Z. Synthesis and properties of PAA-b-PBA based on RAFT polymerization in water. Fine Chemicals. 2010;27(3). [Google Scholar]
- [49].Chen Z, Geng L, Yu Y, et al. Emulsion/suspension polymerization of MMA RAFT polymerization in the presence of trithiocarbonate. National Polymer Academic Paper Report; 2007. [Google Scholar]
- [50].Wi Y, Lee K, Lee BH, et al. Soap-free emulsion polymerization of styrene using poly(methacrylic acid) macro-RAFT agent. J Polym. 2008;49(26):5626–5635. [Google Scholar]
- [51].Charleux B, D’Agosto F, Delaittre G. Preparation of hybrid latex particles and core–shell particles through the use of controlled radical polymerization techniques in aqueous media. J Adv Polym Sci. 2010;233(9):125–183. [Google Scholar]
- [52].Zhang Y. Progress of azo polymer prepared by living free radical polymerization. Chem Indus Times. 2008;22(6):62–67. [Google Scholar]
- [53].Wang L-P, Tao X-Q, Ge X-C. RATRP/RAFT polymerization. Polym Mater Sci Eng. 2009;25(6):32–34. [Google Scholar]
- [54].Zhang H, Gong Y-K. Research progress of RAFT polymerization on surface modification. Polym Bull. 2011(12):17–32. [Google Scholar]
- [55].Chen Y. The research on mechanism of the conformational transition of poly (N, N-diethylacrylamide) in extremely dilute aqueous solution. Lanzhou: Lanzhou University; 2006. [Google Scholar]
- [56].Arrigo R, Morici E, Cammarata M, et al. Rheological percolation threshold in high-viscosity polymer/CNTs nanocomposites. J Eng Mech. 2017;143(5):D4016006. 10.1061/(ASCE)EM.1943-7889.0001096 [DOI] [Google Scholar]
- [57].Lee SH, Ouchi M, Sawamoto M. Functionalization at the central position of vinyl polymer chains: highly associable multipoint hydrogen bonds for complementary self-assemblies. Macromol Rapid Commun. 2014;35(4):431–436. 10.1002/marc.v35.4 [DOI] [PubMed] [Google Scholar]
- [58].Hirosawa K, Fujii K, Ueki T, et al. SANS study on the solvated structure and molecular interactions of a thermo-responsive polymer in a room temperature ionic liquid. Phys Chem Chem Phys. 2016;18(27):17881–17889. 10.1039/C6CP02254E [DOI] [PubMed] [Google Scholar]