

SUMMARY
Environmental stress can induce adult reproductive diapause, a state of developmental arrest that temporarily suspends reproduction. Deficiency for C. elegans Piwi protein PRG-1 results in strains that reproduce for many generations but then become sterile. We found that sterile-generation prg-1/Piwi mutants typically displayed pronounced germ cell atrophy as L4 larvae matured into 1-day-old adults. Atrophied germlines spontaneously reproliferated across the first days of adulthood, and this was accompanied by fertility for day 2–4 adults. Sterile day 5 prg-1 mutant adults remained sterile indefinitely, but providing an alternative food source could restore their fertility. Our data imply that late-generation prg-1 mutants experience a dynamic form of adult reproductive diapause, promoted by stress response, cell death, and RNAi pathways, where delayed fertility and reproductive quiescence represent parallel adaptive developmental outcomes. This may occur in response to a form of “heritable stress” that is transmitted by gametes and epigenetic in nature.
In Brief
Heestand et al. examine a form of diapause that occurs in late-generation C. elegans Piwi/prg-1 mutants. They argue that the sterility of late-generation prg-1 mutants may be a consequence of “heritable stress.” Thus, an endogenous form of stress may be transmitted by germ cells to modulate heredity and fertility.
INTRODUCTION
Diapause is a state of developmental arrest that promotes survival and delays fertility in response to harsh environmental conditions such as seasonal change or limited nutrient availability (Nystul et al., 2003; Padilla and Ladage, 2012; Schiesari and O’Connor, 2013; Tatar and Yin, 2001). For example, many mammals exhibit some form of in utero embryonic arrest in response to exogenous stress until favorable environmental conditions are encountered, thereby increasing the chance of survival for their offspring. Adult reproductive diapause (ARD) occurs when adults temporarily suspend reproduction in response to environmental stress and has been documented for a variety of invertebrates (Nystul et al., 2003; Padilla and Ladage, 2012; Schiesari and O’Connor, 2013; Tatar and Yin, 2001). C. elegans that experience starvation during the final larval stage (L4) can mature into long-lived sterile adults that are in ARD, also termed reproductive quiescence, where germ cell numbers are reduced and reproduction is suspended until food is encountered (Angelo and Van Gilst, 2009). Entry and exit for starvation-induced ARD is influenced by a second environmental stress, population density, which also promotes larval arrest during development (Golden and Riddle, 1982, 1984). Vertebrates, possibly including humans, can also enter a state of suspended animation in response to anoxia (Blackstone et al., 2005), which is plausibly reminiscent of hypoxia-induced embryonic diapause in C. elegans (Miller and Roth, 2009). While environmental cues can suspend reproduction in adulthood by activating ARD, it is unclear whether this developmental program can occur in response to an endogenous form of stress.
Germ cells possess the ability to remain eternally youthful as they are transmitted from one generation to the next. Studies of germ cell immortality in C. elegans have focused on mortal germline mutants that initially display normal levels of fertility, but growth for additional generations results in drops in fertility and eventually in complete sterility (Smelick and Ahmed, 2005). Conceptually, mortal germline mutants may transmit a form of transgenerational stress that accumulates over the generations prior to triggering sterility. C. elegans telomerase mutants are deficient for de novo telomere repeat addition at chromosome ends, and these mortal germline mutants transmit telomeres that shorten progressively and ultimately elicit chromosome fusion and genome catastrophe (Meier et al., 2006). Consistently, deficiency for telomerase in mice and humans results in phenotypes that display genetic anticipation and are exacerbated in successive generations (Armanios and Blackburn, 2012). Moreover, telomerase is deficient in somatic cells of normal humans and this triggers senescence after growth for a number of cell divisions, which represents one mechanism to suppress tumor formation (Sharpless and DePinho, 2007). Senescence can also be activated by transcriptional induction of p16/Ink4a, which is a major tumor suppressor protein that responds to many forms of stress and whose expression increases by several orders of magnitude in human and mouse tissues with age, even though inbred mouse tissues have long telomeres and their somatic cells are positive for telomerase. Thus, non-telomeric forms of endogenous stress are relevant to aging (He and Sharpless, 2017; Sharpless and DePinho, 2007), and it is possible that analysis of germ cell immortality pathways could reveal novel forms of stress that are relevant to aging of human somatic cells.
C. elegans PRG-1/Piwi interacts with Piwi-interacting RNAs (piRNAs) in germ cells to promote transgenerational genomic silencing and germ cell immortality (Bagijn et al., 2012; Batista et al., 2008; Das et al., 2008; Simon et al., 2014). However, C. elegans prg-1 mutants are unlikely to become sterile as a consequence of telomere erosion, as they fail to display telomere fusions near sterility (Simon et al., 2014). This implies that deficiency for PRG-1/Piwi results in accumulation of a distinct form of transgenerational damage.
We previously showed that the transgenerational fertility defect of prg-1/Piwi mutants could be suppressed by intermittent or constitutive reduction of the insulin/IGF1 stress response pathway (Kenyon, 2010; Simon et al., 2014). This discovery created a tacit link between an intervention that represses somatic aging and promotes stress resistance in diverse organisms with the transgenerational fertility defect caused by piRNA deficiency (Kenyon, 2010; Simon et al., 2014). Both small RNAs and histone H3K4 demethylases were required for reduced insulin signaling to promote fertility of prg-1/Piwi mutants, implying that an epigenomic silencing process can alleviate the piRNA-mediated genome silencing defect of Piwi mutants (Simon et al., 2014). How the transgenerational epigenetic defect of prg-1 mutants elicits sterility, and whether the sterility phenotype might be relevant to stress or aging (Smelick and Ahmed, 2005), remains unclear. Here, we report that prg-1/Piwi mutants become sterile via a mechanism that resembles starvation-induced ARD, implying that they succumb to a form of heritable stress, and we provide evidence suggesting that this sterility can be reversed via a small RNA pathway that is promoted by DAF-16/Foxo.
RESULTS
Sterile-Generation prg-1 Mutants Display Delayed Fertility and Germ Cell Atrophy
The brood size of late-generation prg-1 mutants becomes small prior to sterility and is accompanied by a high frequency of males that are incapable of mating (0/25 crosses of single prg-1 mutant males with unc-13 hermaphrodites were successful, whereas 25/25 analogous crosses with N2 wild-type males were successful). Individual sterile prg-1 adults were isolated from late-generation populations of animals that had grown for 20–50 generations. When randomly singled, late-generation mothers gave rise to very few progeny; these progeny were often sterile on day 1 of adulthood. In contrast, wild-type controls or early generation prg-1 mutants (F4) always laid embryos on day 1 (Figure S1B). Interestingly, some late-generation prg-1 mutant animals that failed to lay embryos on day 1 of adulthood became fertile on day 2 (24%). Of those animals that remained sterile on day 2, 14% became fertile on day 3, 6% of sterile day 3 adults become fertile on day 4, and 4% of sterile day 4 adults became fertile on day 5. When sterile prg-1 mutant animals became fertile on days 2–5, they each laid only a few eggs, similar to their late-generation siblings that were fertile on day 1. However, after day 5 of adulthood, all sterile animals remained sterile (Figure 1A). We created a prg-1 mutant strain with a Neon Green transgene, cpIs36, which allows the germline to be visualized, and found that similar fractions of late-generation animals that were sterile on day 1 of adulthood became fertile on days 2, 3, 4, and 5, with all day 5 sterile animals remaining sterile (Figure S1A). These data indicate that late-generation prg-1 mutants consist of two populations of adults that fail to lay embryos on day 1 of adulthood: one in which reproduction eventually occurs during days 2–5 of adulthood and a second that remains completely sterile after day 5 of adulthood.
Figure 1. Sterile prg-1 Animals Exhibit Germline Remodeling.

(A) Sterile day 1 prg-1(n4357) animals were scored daily for continued sterility from days 2 through 6. Percent fertile are the amount that reversed sterility on each day. n represents total worms scored.
(B) Germline phenotypes of fertile, early-generation (F4) germlines in L4 larva and young adults. n represents total germline arms scored (B–F). Germline categories are defined in Experimental Procedures.
(C) Germline phenotypes of sterile L4 larva and young adults. Sterile day 1 adult prg-1 germline profiles were significantly different than wild-type controls. ***p < 0.0001, chi-square test with Bonferroni correction.
(D) Effects of starvation-induced ARD genes and stress response transcription factors on sterile germline phenotypes of prg-1. p values determined using pairwise chi-square tests with Bonferroni correction (all comparisons in Table S1).
(E) Germline phenotypes of sterile prg-1 mutants at day 1, day 2, day 3, and day 4. Scored using fixed DAPI stain. ***p < 0.0001, chi-square test with Bonferroni correction between day 1 and all other day germline profiles.
(F) Live score of germline phenotypes of sterile prg-1 in day 1, day 2, day 3, and day 4 prg-1:cpIs36 mutants. ***p < 0.0001, chi-square test with Bonferroni correction between day 1 and all other day germline profiles. For all panels, data were combined from a minimum of three experiments.
To study the germlines of sterile-generation prg-1 mutants, we identified late-generation prg-1 mutant populations laying very few progeny, and individually isolated hundreds of candidate animals. From the progeny of some of these animals, we could identify sterile day 1 adults along with younger sibling animals likely to become sterile that were available for L4 larval-stage analysis. In both early-generation (F4) and sterile-generation prg-1 mutant animals, we found that the early L4 larval stage, which immediately precedes adulthood, possessed germlines whose size and developmental stage were generally comparable to those of control wild-type L4 larvae (Figures 1B, 1C, 2A, 2B, and S2A). The germlines of both fertile and sterile late-generation prg-1 mutant adults were smaller in total length in comparison to wild-type or for early-generation prg-1 mutants, such that their distal tip cells rarely crossed and their germline bends were premature, which may be consistent with the small brood sizes of fertile late-generation prg-1 mutant strains (Figures 2, 6A, S2H, and S2I). Some sterile day 1 prg-1 mutant adults contained large germline arms whose sizes were comparable to those of fertile late-generation siblings and whose developmental transitions were comparable to wild-type (Figure S2F). However, there was a significant change in germline profiles of sterile prg-1 mutants compared to wild-type (Figure 1C), with most sterile 1-day-old prg-1 mutant adults displaying a germ cell atrophy phenotype and possessing only a small population of mitotic germ cells near the distal tip cell (Figures 1E, 1F, 2G, 2H, and 2L). Notably, animals that displayed germ cell atrophy were devoid of germ cells at various stages of meiosis (Dernburg et al., 1998), which were abundant in sterile-generation prg-1 mutant L4 larvae. Two additional minor fractions of sterile prg-1 mutant adults either lacked germ cells altogether (empty) or had short germlines with both mitotic and meiotic germ cells (Figures 1C, 1E, 1F, 2E, 2F, 2I, 2M, and 2N). These results imply that a dramatic germ cell atrophy phenotype typically occurs during the L4-to-adult transition for most sterile prg-1 mutant 1-day-old adults.
Figure 2. Germline Atrophy in Sterile prg-1 Animals.

(A–O) DAPI-stained photos of (A) N2 wild-type L4, (B) sterile-generation prg-1 L4, (C) N2 wild-type adult, (D) fertile prg-1 mutant, (E and F) short germline from sterile adult prg-1 mutants, (G and H) atrophied germlines from sterile adult prg-1 mutants, (I) empty germline from sterile adult prg-1 mutant lacking germ cells, (J) distal mitotic region of a fertile N2 wild-type adult, (K) meiotic region of a fertile N2 adult containing a transition zone where chromosomes pair during initiation of meiosis as well as meiotic pachytene cells undergoing homologous recombination, (L) arrested cells in an atrophied germline of a sterile adult prg-1 mutant, (M) distal region of a short germline in a sterile adult prg-1 mutant, (N) meiotic cells near the germline bend in a short germline from a sterile prg-1 adult, and (O) meiotic cells in a sterile prg-1 daf-16 adult. Dashed lines indicate germline. Scale bars, 50 μm in (A–I) and 10 μm in (J–O). (J) and (L) show examples of mitotic cells, and (K) and (O) arrows show examples of meiotic cells.
Figure 6. Insulin Signaling and Downstream Small RNA Pathway Are Necessary for Reversal of prg-1-Induced ARD.

(A) Average brood size of day 2 wild-type worms or fertile, late-generation prg-1 mutants on empty vector (EV) or RNAi clone targeting daf-2. Error bars indicate SEM.
(B) Mortality assay for 30 generations of prg-1; daf-2 mutants homozygous at F2 and prg-1; daf-2 mutants that were made homozygous for daf-2 at F18.
(C) Sterile day 1 prg-1(n4357) daf-16 animals were scored for continued sterility from days 2 through 6. n represents total worms scored on that day that were previously sterile.
(D) Sterile day 1 prg-1(n4357) mut-7 (pk204) animals were scored for continued sterility from days 2 through 6. n represents total worms scored on that day that were previously sterile.
(E) Sterile day 5 prg-1 daf-16 or prg-1 cep-1 or prg-1; mut-7 mutants on either standard laboratory NGM plates seeded with OP50 E. coli (OP50:NGM) or standard RNAi plates seeded with HT115 E. coli (HT115:RNAi) scored for percent fertility 48 hr after treatment (day 7). ***p < 0.0001, Fisher’s exact test.
(F) Sterile day 5 prg-1 mutants were treated with total RNA isolated from bacteria on standard laboratory plate conditions (NGM) or standard RNAi plate conditions (RNAi) and scored for fertility. p = n.s., Fisher’s exact test.
(G) Percent fertile sterile day 5 prg-1 mutants on either standard OP50:NGM plate conditions or OP50:NGM with ampicillin (AMP) or tetracycline (TET) or IPTG. Above data combined from minimum of three independent experiments; n represents total worms scored.
We next studied the structure of the germlines of sterile prg-1 mutants using the transgene cpIs36, which expresses Pmex-5::mNeonGreen brightly throughout the germline. We found that we could enrich for late-generation prg-1; cpIs36 mutants that give rise to sterile progeny by isolating late-generation parents with small germlines. Sterile day 1 prg-1; cpIs36 mutant animals were singled and blindly scored for fertility daily and for anterior and posterior germline arm size based on fluorescence detected on agar plates using a stereomicroscope until sterility was reversed or remained completely sterile for 5 days. As a control, we mounted a number of sterile day 1 adults on slides and observed them using a 40× objective. We found that a fraction of germline arms were entirely devoid of germ cells and another fraction contained only a few germ cells, which would be difficult to see at low resolution on the stereomicroscope. Most germ-line arms that were scored as empty on day 1 remained empty, although some reproliferated and these presumably correspond to sterile day 1 prg-1 mutant germline arms that were not entirely devoid of germ cells. Most day 1 animals that had atrophied germlines displayed reproliferation of the germline arm. Of those animals in which both germline arms were either atrophied or empty on day 1 of adulthood, 16% became fertile on days 2–5 of adulthood, indicating that germ cell atrophy represents a state of transient sterility. Additionally, the germline arms of all 35 animals that became fertile displayed germ cell proliferation such that none possessed atrophied germlines in both anterior and posterior germline arms on day 2 of adulthood (Figure 3). Moreover, of 35 worms that became fertile, only 4 arms out of 70 scored were in atrophy on day 2, indicating that germ cell proliferation on day 2 was an excellent predictor of reversibility of sterility. Regrowth did not guarantee reversal of sterility since we found numerous examples of worms that regrew their germ-lines yet remained sterile. Furthermore, while empty germlines generally remained empty, germlines that were either atrophied or short on day 1 of adulthood were dynamic and regrew across the first 5 days of adulthood. When scored either by staining with DAPI or by observing a Neon Green germline reporter, germline arms of sterile day 2, day 3, and day 4 prg-1 mutant adults displayed a significant change in germline size when compared to sterile day 1 adults, with an increased frequency of short germ-lines that contained cells in various stages of meiosis in comparison to the number of day 1 atrophied germline arms that were devoid of meiotic germ cells (Figures 1E and 1F). Thus, the germ-line arms of most sterile-generation prg-1 mutant adults shrink to a state of atrophy as L4 larvae mature to day 1 adults but then reproliferate. These observations are consistent with the activation of a mechanism to delay the onset of fertility in late-generation prg-1 mutants.
Figure 3. Regrowth of Sterile Germline Arms.

Anterior (top of pair) and posterior (bottom of pair indicated with black connector bar) germline arms in day 1 sterile prg-1; cpIs36 were blindly scored live daily for germline phenotype along with the presence of F1 progeny. Each sequential block represents a score for particular day, and the color indicates the germline phenotype observed. Each gonad arm was tracked daily until fertility returned (F, fertile; left column) or remained completely sterile for 5 days (S, sterile; right column). In many cases, only one gonad arm regained fertility while the other remained sterile, such that embryos were observed only on one side of the uterus or the other germline arm remained in state of atrophy or empty.
Apoptosis and Necrosis Promote Germ Cell Atrophy in Sterile prg-1 Mutants
A pattern of germline development that is grossly normal until the L4 larval stage followed by strong germline atrophy was reminiscent of a germline degeneration phenotype that was proposed to occur when C. elegans L4 larvae are starved, some of which emerge as adults that are in a stress-resistant state of reproductive quiescence known as ARD (Angelo and Van Gilst, 2009). The apoptosis gene ced-3 and the nuclear hormone receptor nhr-49, which is an NHF4-alpha homolog responsible for mediating fatty acid oxidation and gluconeogenesis during starvation (Van Gilst et al., 2005a, 2005b), promoted germline remodeling in starvation-induced ARD (Angelo and Van Gilst, 2009). We found that ced-3 and ced-4 mutations, which eliminate apoptotic cell death (Metzstein et al., 1998), led to a significant change to the germline profiles of day 1 sterile prg-1 mutant adults (Table S1), and overall reduced levels of germline atrophy, as did deficiency for nhr-49 (Figure 1D). Thus, the sterility of late-generation prg-1 mutants is accompanied by a germ cell atrophy phenotype that requires the genetic constituents of starvation-induced ARD.
Mammalian p53 as well as the C. elegans homolog of p53, CEP-1, can promote apoptosis or cell cycle arrest in response to DNA damage, heat shock, anoxia, and oxidative or osmotic stresses (Gartner et al., 2008). The germline profile of sterile prg-1 cep-1 double mutant day 1 adults was significantly altered (Figure 1D; Table S1), with germline atrophy being strongly inhibited but not abolished. Sterile prg-1 cep-1 double mutants became fertile on day 2 through day 5, similar to prg-1 single mutants (Figure 5A), indicating that germ cell atrophy is not required for delayed fertility of late-generation prg-1 mutants. In starvation-induced ARD, mutation to ced-3 did not impair ARD entry, but the reduction in germ cell number was impaired and, upon addition of food, the level of ARD exit dropped significantly but was not eliminated (Angelo and Van Gilst, 2009). We constructed prg-1 cep-1; ced-3 and prg-1 cep-1; ced-4 triple mutants, which are deficient for both CEP-1/p53 and core apoptosis genes. prg-1 cep-1; ced-4 had a similar percentage of atrophied germ-lines in comparison to prg-1 cep-1 and prg-1; ced-4 controls, although their germline profiles were significantly different (Figure 1D; Table S1). prg-1 cep-1; ced-3 triple mutants displayed the lowest level of germ cell atrophy and the strongest return of large germlines of any strain tested, whereas prg-1 cep-1; ced-4 triple mutants generally displayed short rather than large germline arms (Figures 1D and 5C). The simplest interpretation of the cep-1 and ced double- and triple-mutant data is that CEP-1 functions in the classical apoptosis pathway to promote germ cell atrophy in sterile prg-1 animals (Schumacher et al., 2001). The short germlines observed in prg-1 cep-1; ced-4 triple mutants could reflect a caspase-independent function of CED-4, for example as previously shown for regulation of cell size (Chen et al., 2008).
Figure 5. prg-1 Germline Arm Phenotypes and Sterility Can Be Suppressed.

(A) Sterile day 1 prg-1(n4357) cep-1(gk138) animals were scored for continued sterility from days 2 through 6. n represents total worms scored on that day that were previously sterile.
(B) Germline phenotypes of sterile germlines in day 1, day 2, and day 3 prg-1 daf-16 mutants.
(C) Effects of apoptosis and necrotic cell death pathways on sterile germline phenotypes of prg-1. For comparison, we include wild-type, prg-1, and prg-1 cep-1 data that are the same data as shown in Figure 1D. p values were determined using pairwise chi-square tests with Bonferroni correction that can be found in Table S1.
(D) Sterile day 5 prg-1 mutants on either standard laboratory plate NGM plates with OP50 E. coli (OP50:NGM) or on RNAi plates seeded with various bacteria relevant to generating dsRNA (HT115:RNAi clone; EV is empty vector) scored for percent fertility 48 hr after treatment (day 7) *p < 0.05, Fisher’s exact test with Bonferroni correction.
(E) Sterile day 5 prg-1 mutants on OP50 bacteria on standard RNAi plate condition (OP50:RNAi) scored for percent fertility 48 hr after treatment (day 7). Data combined from minimum of three independent experiments; n represents total worms (A, D, and E) or germline arms (B and C) scored.
Apoptotic corpses occur in the pachytene region of the adult C. elegans germline and can be visualized as discrete rings using GFP-tagged CED-1, a transmembrane protein that promotes engulfment of apoptotic corpses (Zhou et al., 2001). Consistently, we observed apoptotic corpses in adult wild-type control germlines (Figure 4A), but CED-1::GFP activity was not observed for wild-type L4 larvae (Figures 4D, 4G, and 4J). We constructed prg-1; ced-1::GFP strains and observed that day 1 adult early-generation (F4), late-generation, and sterile-generation 1-day-old adults displayed elevated apoptotic corpses with a trend toward increased occurrence in later generations (Figures 4B, 4C, and 4M), consistent with the concept that transposon-induced DNA damage may promote the fertility defect of prg-1 mutants (McMurchy et al., 2017). Sterile-generation but not early-generation prg-1 mutant L4 larvae displayed pronounced CED-1::GFP activity in both proximal and distal regions of the germline (Figures 4E, 4F, 4H, 4I, 4K, and 4L). A developmental time course of sterile-generation L4 larvae revealed strong CED-1::GFP activity in germlines of mid-L4 larvae (Figures 4I and 4N). Together, our data imply that an apoptotic program can act throughout the germline of L4 larvae to promote germ cell atrophy as sterile-generation prg-1 mutants mature into day 1 adults.
Figure 4. Increased Apoptosis in Early- and Late-Generation prg-1 Animals.

(A and B) Compared to wild-type (A), prg-1 animals (B) display and increase number of apoptotic cells, indicated by white arrows.
(C) Sterile-generation animals with atrophied germline.
(D, G, and J) Wild-type L4 germlines observed at early (L4 0 hr) (J), mid (L4 +4 hr) (G), and late (L4 +8 hr) (D) time courses.
(E, H, and K) Fertile, early-generation prg-1 L4 germlines observed at early (L4 0 hr) (K), mid (L4 +4 hr) (H), and late (L4 +8 hr) (E) time courses.
(F, I, and L) Sterile-generation prg-1 L4 germlines observed at early (L4 0 hr) (L), mid (L4 +4 hr) (I), and late (L4 +8 hr) (F) time courses, indicating an increase in the number of apoptotic cells.
(M) Average number of apoptotic cells in early-generation adult germlines (n > 30 for all samples).
(N) Sterile-generation animals displayed CED-1::GFP activity throughout L4 germline development. Arrows indicate apoptotic cells. Error bars indicate SEM.
Germ cell atrophy still occurred for some sterile-generation prg-1 mutant adults that were deficient for the core cell death proteins ced-3 or ced-4 (Figure 1D). We asked whether necrotic cell death contributed to germ cell atrophy by inactivating the C. elegans calreticulin ortholog crt-1 (Syntichaki et al., 2002). Sterile-generation prg-1; crt-1 double mutants displayed reduced levels of germ cell atrophy, and a significant change to their germline profile (Figure 5C; Table S1). We constructed prg-1 cep-1; crt-1 triple mutants to eliminate both necrosis and apoptosis. This resulted in a significant change in the germline profile compared to prg-1 cep-1 (Figure 5C; Table S1) but surprisingly failed to remove the germ cell atrophy phenotype. However, an increased frequency of germ cell tumors was observed (Figure 5C). These results imply that CEP-1/p53 promotes germ cell atrophy by activating necrotic cell death, which is consistent with an established role for Mammalian p53 in promoting necrotic cell death (Baumann, 2012). However, the increase in germ cell tumors for prg-1; crt-1 double mutants and a further increase for prg-1 cep-1; crt-1 triple mutants, suggests a non-redundant role for CEP-1/p53 and the necrotic cell factor CRT-1 in suppression of germ cell tumor formation.
Insulin/IGF1 Signaling Promotes Germ Cell Atrophy in Sterile prg-1 Mutants
The C. elegans DAF-16/Foxo transcription factor can promote resistance to diverse stresses including starvation, heat shock, proteotoxicity, oxygen radicals, pathogenic bacteria, and high metal concentrations (Zhou et al., 2011), and even promotes germline apoptosis in a manner that can repress germ cell tumors (Pinkston-Gosse and Kenyon, 2007). We found that sterile late-generation prg-1 daf-16 double-mutant day 1 adults had a significant change in their germline profile compared to day 1 sterile prg-1 mutant adults (Figure 1D; Table S1), with strongly reduced frequencies of germline atrophy. In agreement with these data, when prg-1 daf-16; daf-2 mutants were analyzed for their germline profiles there was a strong reduction in germ-line atrophy and a profile that was similar overall to that of prg-1 daf-16 double mutants (Figure S1F). This demonstrates that daf-2 mutation does not rescue the germline phenotypes of prg-1 daf-16 double mutants back to wild-type or otherwise alter them. Strongly reduced frequencies of germline atrophy were also observed in sterile late-generation day 2 and day 3 prg-1 daf-16 double-mutant adults (Figure 5B), which instead possessed an increased number of “short” germline arms that displayed hallmarks of normal meiosis including a transition zone and synapsed homologous chromosomes (Dernburg et al., 1998), as well as mature oocytes at the proximal end of the germline (Figures 1D, 2O, and S2G). DAF-16 can function downstream of C. elegans PTEN tumor suppressor homolog DAF-18 to promote longevity in response to reduced DAF-2 signaling but can also respond to distinct signals from other pathways (Ogg and Ruvkun, 1998; Vowels and Thomas, 1992). We found that sterile late-generation prg-1; daf-18 double-mutant day 1 adults had a significant change in their germline profiles (Table S1) and displayed reduced germ cell atrophy and an increased frequency of short germlines with differentiated germ cells, in comparison to prg-1 single-mutant controls (Figure 1D). While it is possible that these genes could function in parallel pathways, we suggest that DAF-16/FOXO functions downstream of DAF-18/PTEN to promote germline remodeling in late-generation prg-1 mutant L4 larvae that are poised to become sterile, and suggest that DAF-16 responds to reduced daf-2 insulin/IGF1-signaling cues in this context.
DAF-16 interacts with the heat shock transcription factor HSF-1 to promote longevity (Hsu et al., 2003). DAF-16 and HSF-1 can respond to common forms of stress, and they influence the expression of common transcriptional targets (Hsu et al., 2003). Sterile day 1 prg-1 hsf-1 double-mutant adults displayed reduced germline atrophy and an increase in small germlines at frequencies similar to prg-1 daf-16 double mutants, resulting in no significant difference between their germline pro-files (Figure 1D; Table S1). This suggests that HSF-1 and DAF-16 interact to promote germ cell atrophy in sterile prg-1 adults.
To investigate the relationship between CEP-1/CED-3/CED-4 apoptosis and DAF-16/DAF-18/HSF-1/NHR-49 stress response pathways (Kenyon, 2010; Metzstein et al., 1998; Ratnappan et al., 2014), we generated triple mutants involving prg-1, apoptosis, and longevity mutations. prg-1 daf-16; ced-4 mutants and prg-1 nhr-49; ced-4 mutants both favored short rather than large germline arms, comparable to prg-1 daf-16 and prg-1 nhr-49 double mutants at day 1 (Figure 1D). Furthermore, when comparing their germline profiles, they were either weakly significant or not significantly different (Table S1). The lack of additivity observed for these triple mutants, in terms of a further reduction of germ cell atrophy on day 1 of adulthood, suggests that these genes act in a single pathway to promote germ cell atrophy in sterile prg-1 mutants.
prg-1 Mutant Sterility Is a Reversible State of Reproductive Quiescence
For starvation-induced ARD, addition of food can restore fertility to a small fraction of sterile animals, demonstrating that ARD is a facultative stress response that suspends reproduction in response to starvation until favorable nutritional cues are encountered (Angelo and Van Gilst, 2009). We previously found that reduced daf-2 signaling can abolish the progressive sterility phenotype of prg-1 mutants (Simon et al., 2014). Consistently, when we removed daf-2 from early-generation prg-1 mutants, this resulted in robust fertility for many generations (Figure 6B). We also asked whether a daf-2 mutation could rescue the progressive sterility of late-generation prg-1 mutants by propagating sibling strains that were heterozygous for a recessive daf-2 mutation but homozygous mutant for prg-1 until late generations (F18) and then isolated prg-1 −/−; daf-2 −/− homozygotes. However, mutation of daf-2 failed to ameliorate the fertility defect of late-generation prg-1 mutants. In parallel, we asked whether RNAi knockdown of daf-2 in fertile late-generation prg-1 mutants would ameliorate the very low levels of fertility that they experience prior to the onset of sterility. However, daf-2 RNAi failed to improve the weak fecundity of fertile late-generation prg-1 mutants (Figure 6A).
As a negative control, we next asked whether sterile prg-1 adults would remain sterile when daf-2 signaling is reduced. Wild-type worms can reproduce until day 8 of adulthood (Hughes et al., 2007), so we identified 10-day-old sterile prg-1 adults with atrophied germlines and surprisingly found that a small but significant population became fertile upon treatment with daf-2 RNAi (Figure S1C). We next re-tested these results under our present laboratory conditions for prg-1 mutant animals that were sterile through day 5 of adulthood. These animals remained sterile on nematode growth medium (NGM) plates with standard OP50 bacteria (Figure 1A), but fertility was restored to a small yet significant fraction of animals placed on RNAi plates seeded with HT115 bacteria expressing double-stranded RNA (dsRNA) targeting daf-2 or gfp and also for the L4440 empty vector RNAi control (Figure 5D). Additionally, crossing day 7 sterile prg-1 mutant adults that were transferred to RNAi plates with wild-type males did not markedly improve the rescue from sterility (Figure S1E). Furthermore, treatment of HT115 bacteria expressing dsRNA for L4440 empty vector or daf-2 did not enhance the restoration of fertility for late-generation sterile day 2 prg-1 mutants (Figure S1D). We next asked whether HT115 E. coli used for RNAi feeding was responsible for this effect, but found that RNAi plates seeded with OP50 E. coli that lack an antibiotic resistance plasmid resulted in restoration of fertility to sterile prg-1 mutant adults (Figure 5E). These results imply that the sterility of 5-day-old sterile prg-1 adults represents a state of reversible reproductive diapause. We conclude that bacteria grown on RNAi plates can reverse the sterility of a fraction of sterile prg-1 mutants. We tried restoring fertility to day 5 prg-1 mutant adults using bacteria grown on plates containing single compounds found in RNAi plates, which contain ampicillin, tetracycline, and isopropyl β-D-1-thiogalactopyranoside (IPTG) but found that no single compound was sufficient to restore fertility (Figure 6G). These results also indicate that deficiency for daf-2, either by RNAi or by mutation, does not specifically suppress the fertility defects of late-generation prg-1 mutants.
A DAF-16-Mediated Small RNA Pathway Promotes Fertility of Sterile prg-1 Adults
DAF-16 is a FOXO transcription factor that acts downstream of the insulin/IGF1-like receptor daf-2 and is required for the immortality of prg-1 daf-2 double mutants (Simon et al., 2014). When we passaged prg-1 daf-16 double mutants until the sterile generation, we found that similar to the prg-1 single mutants, 23% of prg-1 daf-16 double-mutant worms that were sterile on day 1 became fertile on day 2. Likewise, 18% of sterile day 2 adults, 13% of sterile day 3 adults, and 2% of sterile day 4 adults became fertile (Figure 6C). However, no prg-1 daf-16 adults that were sterile through day 5 exited ARD to become fertile when placed on RNAi plates seeded with HT115 bacteria (Figure 6E). As mutation of daf-16 suppressed germ cell atrophy in sterile prg-1 mutants (Figure 1D), we asked whether cep-1, mutation of which more strongly suppressed prg-1 mutant germ cell atrophy (Figures 1D and 5C), affected the restoration of fertility to sterile 5-day-old prg-1 mutants. However, cep-1 was not required for ARD exit (Figure 6E).
Constitutive activation of DAF-16 in early-generation prg-1 mutants indefinitely promotes the fertility of prg-1 mutants by activating an endogenous small RNAi silencing pathway that depends on mut-7, which promotes secondary small interfering RNA (siRNA) biogenesis (Simon et al., 2014). We therefore asked whether MUT-7 might function downstream of DAF-16 in reversing the sterility of 5-day-old sterile prg-1 mutants. Although significant fractions of sterile prg-1; mut-7 day 1 double mutants became fertile on days 2–5, we found that no 5-day-old sterile prg-1; mut-7 double-mutant adults became fertile when placed on E. coli grown on RNAi plates (Figures 6D and 6E). This indicates that MUT-7 and a small RNAi silencing pathway downstream of DAF-16 are required for exit of prg-1-induced ARD but are not required for spontaneous reversal of fertility that occurs on days 2–5.
As MUT-7 promotes secondary siRNA biogenesis and therefore promotes many types of small RNA responses in C. elegans, we asked whether RNA generated by bacteria grown on RNAi plates might be responsible for restoring fertility to sterile late-generation prg-1 mutants. Total RNA was isolated from bacteria grown on NGM or RNAi plates and was either injected or soaked into sterile day 5 prg-1 mutant adults. However, RNA from bacteria grown on RNAi plates failed to specifically restore fertility to sterile 5-day-old prg-1 mutant adults (Figure 6F). Taken together, these results suggest that E. coli grown on RNAi plates but not RNA from these E. coli cells can activate a DAF-16-mediated endogenous RNAi pathway that can restore fertility to a small fraction of sterile prg-1 mutant adults. Together, our results indicate that although activation of DAF-16 was not capable of suppressing the fertility defect of late-generation prg-1 mutants (Figure 6A), that activation of DAF-16 in early generations abolishes the prg-1 mutant fertility defect (Figure 6B) (Simon et al., 2014) and that an endogenous DAF-16-mediated RNAi pathway can restore fertility to sterile 5-day-old prg-1 mutant adults in response to bacteria grown on RNAi plates.
DISCUSSION
Here, we demonstrate that sterile-generation prg-1 mutants activate two mechanisms to delay fertility. The first mechanism delays fertility from 1 to 4 days (Figure 1) and involves a large-scale germ cell apoptotic program during the final larval stage (Figures 1 and 4). On day 1 of sterility, the majority of germlines were atrophied (Figures 1 and 3). Germline regrowth can occur for day 2, 3, and 4 adults, accompanied by restoration of fertility for some animals (Figures 1 and 3). Defects in insulin signaling and cell death pathways suppressed but did not eliminate germ cell atrophy (Figures 1 and 5). Apoptosis, insulin signaling pathways, and necrosis pathways appear to function in a single pathway to promotes germ cell atrophy whose wiring remains to be elucidated (Figure 7). Although germ cell atrophy is a pronounced feature of the delayed fertility mechanism that we describe, it is not necessary for it.
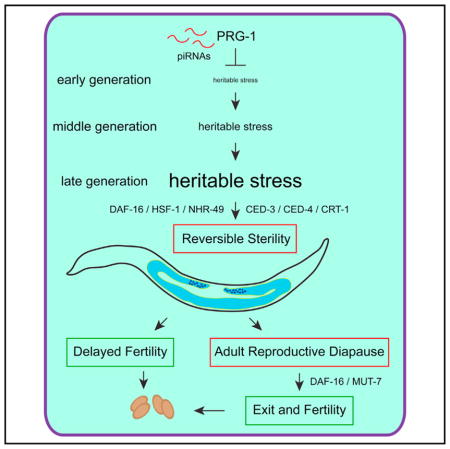
Figure 7. Model of PRG-1.

PRG-1 suppresses a heritable form of stress that triggers adult germ cell remodeling and drives a form of arrest. Delayed fertility pathway (left) is prg-1 mutants that spontaneously reverse from loss of fertility to fertile state. Reproductive diapause pathway (right) is prg-1 mutants that remain sterile throughout adulthood and only return to fertility when given an undetermined stimulus. Blue represents germ cells. Brown represents embryos.
prg-1 mutant animals that remained sterile until day 5 of adulthood revealed a second mechanism to delay fertility, where they can remain sterile indefinitely but can become fertile if provided with a cue from a food source (Figure 5). This second response of prg-1 mutants to delay reproduction represents a form of ARD (Roy et al., 2016; Seidel and Kimble, 2011, 2015). Thus, sterile-generation prg-1 mutants undergo a dynamic form of ARD that delays sterility in two ways: first, by delaying the onset of fertility for 1–4 days, a phenotype that may be masked by the highly crowded conditions of starvation-induced ARD (Angelo and Van Gilst, 2009), and second by inducing a form of reproductive quiescence in day 5 adults that can be reversed when an alternative food source is provided.
Given this parallel with starvation-induced ARD, we propose that germ cells of prg-1 mutants transmit an escalating form of “heritable stress” that ultimately becomes comparable to severe environmental stresses such as starvation (Figure 7). A heritable stress that can be transmitted by gametes could be broadly relevant to the concept of inheritance and related epigenetic phenomena, and possibly to human adult-onset disorders that are affected by exogenous stresses. The delayed fertility and reproductive quiescence responses of sterile-generation prg-1 adults reflect a dynamic developmental program that delays fertility in a non-uniform manner in populations of sterile-generation prg-1 mutant adults, which may represent a concerted effort to outlast or adapt to the heritable stress prior to reproduction. Loss of Piwi in Drosophila, zebrafish, and mice results in immediate sterility that correlates with transposon-induced genome instability (Juliano et al., 2011), and expression of the Mirage transposon has been demonstrated to contribute to the fertility defects of late-generation prg-1 mutants (McMurchy et al., 2017). Our results suggest that the sterility of Piwi mutants in other species could be due to an epigenetic stress response that immediately triggers ARD. If so, it might be possible to restore fertility to these Piwi mutants.
What is the nature of this heritable stress? We considered that homologous recombination defects might promote sterility. Sterile prg-1 mutants at 20°C displayed six DAPI bodies in their oocytes (Simon et al., 2014), which correspond to homologous chromosomes that are held together by recombination crossovers. Small RNA mutants that become sterile only if grown at 25°C show an increase in these DAPI bodies (Sakaguchi et al., 2014); we examined sterile prg-1 mutants that were grown at 25°C but these did not show more than six DAPI bodies in their oocytes (Figure S1G). This implies that homologous recombination is nominally fine in sterile prg-1 mutants, although minor meiotic or mitotic chromosome segregations defects may occur, as a weakly penetrant X chromosome loss phenotype is observed in mid- and late-generation prg-1 mutant strains. Previous work revealed that immediate sterility occurred if prg-1/piRNA mutants that are devoid of secondary siRNAs are crossed in a manner that restores secondary siRNAs but leaves prg-1 mutant (de Albuquerque et al., 2015; Phillips et al., 2015). In this scenario, Ketting and colleagues observed that sterile prg-1 mutants displayed a range of adult germline phenotypes that are consistent with those that we observed for prg-1 single mutants after many generations of growth. The sterility observed by the Ketting and Ruvkun groups, which was attributed to secondary siRNA dysfunction in the context of the pro- and anti-small RNA silencing pathways of C. elegans, could be relevant to the sterility in prg-1 mutants.
We previously suggested that desilencing of repetitive segments of the genome may explain the sterility phenotype of prg-1 mutants (Simon et al., 2014). Two recent studies support this possibility, as acute desilencing of heterochromatin and perturbation of a variety of genes that promote heterochromatin formation can elicit fertility defects that are consistent with a form of DNA damage evoked by desilencing of heterochromatin (McMurchy et al., 2017; Zeller et al., 2016). Consistently, the Kennedy group recently identified additional mortal germline mutants that are defective for RNA inheritance and likely become sterile due to epigenetic deregulation in germ cells (Spracklin et al., 2017).
Transgenerational fertility defects that result in heterochromatin dysfunction may provide insight into forms of stress that accumulate as human somatic cells proliferate during aging and senesce. Links between aging and the epigenome have been noted, such as desilencing of retrotransposons in senescent human cells, and epigenetic silencing of retrotransposons by the anti-aging protein SIRT6 in the mouse (De Cecco et al., 2013; Gorbunova et al., 2014; Van Meter et al., 2014). Moreover, repetitive elements play major roles in genome evolution. For example, rapid evolution of tandem repeats that specify vertebrate centromeres promotes genomic conflict and speciation (Malik and Henikoff, 2002). Given that harsh environmental conditions can trigger ARD (Angelo and Van Gilst, 2009; Ragland et al., 2010), we propose that ARD can also occur in response to endogenous epigenetic stress, such as that encountered in the context of genomic conflict, or when transposons or retrovi-ruses enter the germline in horizontal transfer events and create bursts of genomic stress. Perinuclear P granules, or germ granules, that contain components of anti-viral and anti-transposon response systems may represent a focal point for the interface between epigenomic regulation and germ cell development (Strome and Updike, 2015).
When starvation-induced ARD worms are returned to food, a small percentage of worms are able to regrow their germlines and become fertile again (Angelo and Van Gilst, 2009). We observed a similar reproductive quiescence phenotype in sterile 5-day-old prg-1 mutant adults, a small percentage of which can respond to a signal from bacteria grown on RNAi plates to exit reproductive diapause. Although DAF-16 played a less pronounced role in germ cell atrophy than CEP-1/p53, we found that DAF-16 and MUT-7, which promotes a DAF-16-dependent small RNA-mediated genomic silencing pathway that can eliminate the transgenerational fertility defect of prg-1 mutants, but not cep-1/p53, were required for ARD exit of day 5 prg-1 mutant adults in reproductive diapause. Further insight into the restoration of fertility to sterile prg-1 mutants is an important future goal, but one that may be technically challenging given the low frequency of such events. Given that HSF-1 and DAF-16 can act at the same promoters in the context of longevity (Hsu et al., 2003), it is plausible that CEP-1/p53 shares transcriptional targets with these proteins, or that insulin signaling in the soma could function upstream of cell death/necrosis in the germline. We suggest that the DAF-16 stress response transcription factor plays distinct roles in responding to a heritable stress transmitted by prg-1 mutants, where it promotes germ cell atrophy in L4 larvae and ARD exit for day 5 sterile worms. For ARD exit, DAF-16 and the small RNA pathway protein MUT-7 are likely to promote an epigenomic silencing process that is capable of restoring fertility to prg-1 mutants (Simon et al., 2014).
We conclude that ARD may represent a biological checkpoint that can integrate development with genome evolution, transiently suspending reproduction until the germ cells of an organism can respond or adapt to a genomic incident. Environmental stress has the potential to induce either ARD (Angelo and Van Gilst, 2009; Ragland et al., 2010) or Lamarckian inheritance (Choi and Mango, 2014), and our data suggest that Piwi/prg-1 mutants may transmit a form of heritable stress that may accumulate to engage the ARD stress response. Our study therefore suggests a possible interface between heritable and environmental forms of stress, which in conjunction with ARD could represent a cornerstone of the evolutionary biology of aging in metazoans. Our study lays the foundation for future work to investigate how cell death, insulin signaling, and small RNA pathways interface with germline development to modulate germ cell immortality and ARD.
EXPERIMENTAL PROCEDURES
Strains
Unless noted otherwise, all strains were cultured at 20°C on NGM plates seeded with E. coli OP50. Strains used are listed in Supplemental Experimental Procedures.
prg-1 mutations were outcrossed versus an outcrossed stock of dpy-5(e61) unc-13(e450), and freshly isolated homozygous F2 lines were established for analysis. dpy-5 unc-55; daf-2 triple mutants were crossed with prg-1/dpy-5(e61) unc-13(e450) males, which were then selected based on a Dauer phenotype at 25°C and on loss of dpy-5 unc-55 to create prg-1; daf-2. Analogous crosses using marker mutations dpy-17 for ced-4, or dpy-9 for daf-18, or unc-24 for ced-3, as balancers to create double-mutant strains. To create the linked prg-1 daf-16 double mutant, prg-1 dpy-24 and unc-13 daf-16 double mutants were first created, and then progeny of prg-1 dpy-24/unc-13 daf-16 heterozygotes that lost unc-13 were identified, and the resulting putative prg-1 daf-16 recombinant chromosomes were made homozygous and PCR genotyped to verify the presence of prg-1 or daf-16 deletions. prg-1 daf-16 double mutants were crossed with unc-13 dpy-24; daf-2/+ +; + heterozygous males and then selected for Daf and against Dpy Unc phenotypes to create prg-1 daf-16; daf-2 triples.
Sterility Assays
Due to the heterogeneity of the Mortal Germline phenotype (Mrt) in the prg-1 population, it is technically difficult to obtain large populations of sterile late-generation animals. prg-1 worms were grown by transferring six L1s to fresh NGM plates every other generation until small brood sizes were observed in later generations, and then many individual animals were singled and scored for sterility. At L4 (day 0), individual worms were tracked for sterility and scored each day for the presence of progeny. In brood size assays, total progeny were counted.
Germline Mortality Assay
Worms were assessed for the Mrt phenotype using the assay previously described (Ahmed and Hodgkin, 2000). L1 or L2 larvae were transferred for all assays.
Germline Visualization
DAPI staining was performed as previously described (Ahmed and Hodgkin, 2000). L4 larvae were selected from sibling plates, and sterile adults were singled as late L4s and observed every 24 hr to monitor sterility. Sterile worms were stained, and each germline arm scored using immunofluorescence on Nikon Eclipse E800 microscope using NIS Elements software. Germline arms were qualitatively characterized into five categories based on comparisons to wild-type (WT) controls. Large germlines contained both mitotic and meiotic cells, a bend in the gonad arm, and overall size and architecture comparable to a wild-type worm. Tumorous germlines resembled proximal proliferative tumors (Korta et al., 2012). Short germlines were characterized as germline arms that had both mitotic and meiotic cells, typically a bend in the gonad arm, but were far shorter and under proliferated compared to both large germ-lines and wild-type controls. Atrophy germlines were characterized as a small population of germ cells (50–100) near the distal tip cell that were typically uniformly mitotic and devoid of meiotic germ cells. Empty germlines were devoid of all or almost all germ cells.
cpIs36 was crossed into prg-1 mutants and used to visualize the germline in live animals. Germlines were blindly qualitatively scored by eye, comparing size and architecture to cpIs36 controls, and sorted into five categories. Large germ-lines were similar in size and architecture to cpIs36, tumorous germlines typically displayed more intense fluorescence but could be difficult to distinguish from large germlines at this resolution, shorts were shorter than control germlines, atrophy was scored as a small pocket of fluorescence near the distal tip of the gonad, and empty was missing Neon Green expression. L4 larvae were selected from sibling sterile plates, singled, and each germline arm was blindly scored daily for germline size on a Leica M205 FA fluorescence microscope.
RNAi Rescue
Late-generation L4 mutants were individually cloned and confirmed to be sterile through day 5. Sterile day 5 worms were then selected and singled to NGM plates with 50 mg/mL ampicillin, 12.5 mg/mL tetracycline, and 1 mM IPTG (RNAi plate condition). These plates were seeded with E. coli HT115 expressing dsRNA targeting gene of interest, or an empty vector (L4440), or E. coli OP50 (Kamath et al., 2001). The worms were then scored daily for progeny at 20°C. For individual drug reversal experiments, the same drug concentrations found in RNAi plates were added to NGM plates, which were then seeded with OP50 E. coli.
RNA Injection/Soaking
RNA was isolated from E. coli OP50 seeded on either standard NGM plate or RNAi plate condition using TRIzol Max Bacterial RNA Isolation Kit (Ambion by Life Technologies). RNA presence and quality were confirmed using gel electrophoresis and a Nano Drop spectrophotometer. Total RNA was either injected or soaked into sterile day 5 worms according to published protocols (Fire et al., 1998; Maeda et al., 2001).
Statistical Analysis
Statistical analysis was performed using the R statistical environment (R Core Team, 2017). For prg-1 sterile-mutant statistical analysis, due to the limitations of obtaining large experimental pools of sterile prg-1 mutants, worms with low brood sizes were expanded and sterile progeny were collected. Each worm was treated as an individual sterility event, and all data from various pools were aggregated and analyzed. For analysis of germline phenotypes, contingency tables were constructed from the observed frequencies of the different phenotypes. Pairwise chi-square tests or Fisher’s exact test were performed to test for significant differences in the distribution of germline phenotypes between different strains/conditions. Bonferroni correction was used to control the familywise error rate when multiple comparisons were made.
Supplementary Material
Highlights.
Sterile prg-1/Piwi mutants display germ cell atrophy in adult germlines
Indefinitely sterile prg-1 mutants become fertile when fed alternate food source
Sterility of prg-1 mutants is a form of adult reproductive diapause
prg-1 mutant sterility may therefore be the consequence of “heritable stress”
Acknowledgments
We thank J. Mitchell for insight into germline remodeling, D. Reiner for suggestions on experimental design and strains, L. Leopold for assistance with statistical analysis, P. Hu for daf-2 and age-1 RNAi clones, M. Jarstfer and members of the Ahmed lab for critical reading of the manuscript, J. Powell-Coffman for lucid comments on ARD exit, and D. Dickinson for the strain cpIs36. Some strains used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440). B.H., M.S., S.F., D.T., and S.A. were supported by NIH Grant GM083048 to S.A.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, two figures, and one table and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.03.015.
AUTHOR CONTRIBUTIONS
B.H., M.S., D.T., and S.A. designed and performed experiments. S.A. supervised the research. B.H., M.S., S.F., and S.A. analyzed data and wrote the paper.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Ahmed S, Hodgkin J. MRT-2 checkpoint protein is required for germline immortality and telomere replication in C. elegans. Nature. 2000;403:159–164. doi: 10.1038/35003120. [DOI] [PubMed] [Google Scholar]
- Angelo G, Van Gilst MR. Starvation protects germline stem cells and extends reproductive longevity in C. elegans. Science. 2009;326:954–958. doi: 10.1126/science.1178343. [DOI] [PubMed] [Google Scholar]
- Armanios M, Blackburn EH. The telomere syndromes. Nat Rev Genet. 2012;13:693–704. doi: 10.1038/nrg3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagijn MP, Goldstein LD, Sapetschnig A, Weick EM, Bouasker S, Lehrbach NJ, Simard MJ, Miska EA. Function, targets, and evolution of Caenorhabditis elegans piRNAs. Science. 2012;337:574–578. doi: 10.1126/science.1220952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Ruby JG, Claycomb JM, Chiang R, Fahlgren N, Kasschau KD, Chaves DA, Gu W, Vasale JJ, Duan S, et al. PRG-1 and 21U-RNAs interact to form the piRNA complex required for fertility in C. elegans. Mol Cell. 2008;31:67–78. doi: 10.1016/j.molcel.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann K. Cell death: multitasking p53 promotes necrosis. Nat Rev Mol Cell Biol. 2012;13:480–481. doi: 10.1038/nrm3401. [DOI] [PubMed] [Google Scholar]
- Blackstone E, Morrison M, Roth MB. H2S induces a suspended animation-like state in mice. Science. 2005;308:518. doi: 10.1126/science.1108581. [DOI] [PubMed] [Google Scholar]
- Chen L, McCloskey T, Joshi PM, Rothman JH. ced-4 and proto-oncogene tfg-1 antagonistically regulate cell size and apoptosis in C. elegans. Curr Biol. 2008;18:1025–1033. doi: 10.1016/j.cub.2008.06.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Mango SE. Hunting for Darwin’s gemmules and Lamarck’s fluid: transgenerational signaling and histone methylation. Biochim Biophys Acta. 2014;1839:1440–1453. doi: 10.1016/j.bbagrm.2014.05.011. [DOI] [PubMed] [Google Scholar]
- Das PP, Bagijn MP, Goldstein LD, Woolford JR, Lehrbach NJ, Sapetschnig A, Buhecha HR, Gilchrist MJ, Howe KL, Stark R, et al. Piwi and piRNAs act upstream of an endogenous siRNA pathway to suppress Tc3 transposon mobility in the Caenorhabditis elegans germline. Mol Cell. 2008;31:79–90. doi: 10.1016/j.molcel.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Albuquerque BF, Placentino M, Ketting RF. Maternal piRNAs are essential for germline development following de novo establishment of endo-siRNAs in Caenorhabditis elegans. Dev Cell. 2015;34:448–456. doi: 10.1016/j.devcel.2015.07.010. [DOI] [PubMed] [Google Scholar]
- De Cecco M, Criscione SW, Peckham EJ, Hillenmeyer S, Hamm EA, Manivannan J, Peterson AL, Kreiling JA, Neretti N, Sedivy JM. Genomes of replicatively senescent cells undergo global epigenetic changes leading to gene silencing and activation of transposable elements. Aging Cell. 2013;12:247–256. doi: 10.1111/acel.12047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dernburg AF, McDonald K, Moulder G, Barstead R, Dresser M, Villeneuve AM. Meiotic recombination in C. elegans initiates by a conserved mechanism and is dispensable for homologous chromosome synapsis. Cell. 1998;94:387–398. doi: 10.1016/s0092-8674(00)81481-6. [DOI] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Gartner A, Boag PR, Blackwell TK. Germline survival and apoptosis. WormBook: The Online Review of C. elegans Biology (WormBook) 2008 doi: 10.1895/wormbook.1.145.1. https://www.ncbi.nlm.nih.gov/books/NBK116071/ [DOI] [PMC free article] [PubMed]
- Golden JW, Riddle DL. A pheromone influences larval development in the nematode Caenorhabditis elegans. Science. 1982;218:578–580. doi: 10.1126/science.6896933. [DOI] [PubMed] [Google Scholar]
- Golden JW, Riddle DL. The Caenorhabditis elegans dauer larva: developmental effects of pheromone, food, and temperature. Dev Biol. 1984;102:368–378. doi: 10.1016/0012-1606(84)90201-x. [DOI] [PubMed] [Google Scholar]
- Gorbunova V, Boeke JD, Helfand SL, Sedivy JM. Human Genomics. Sleeping dogs of the genome. Science. 2014;346:1187–1188. doi: 10.1126/science.aaa3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Sharpless NE. Senescence in health and disease. Cell. 2017;169:1000–1011. doi: 10.1016/j.cell.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu AL, Murphy CT, Kenyon C. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142–1145. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- Hughes SE, Evason K, Xiong C, Kornfeld K. Genetic and pharmacological factors that influence reproductive aging in nematodes. PLoS Genet. 2007;3:e25. doi: 10.1371/journal.pgen.0030025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano C, Wang J, Lin H. Uniting germline and stem cells: the function of Piwi proteins and the piRNA pathway in diverse organisms. Annu Rev Genet. 2011;45:447–469. doi: 10.1146/annurev-genet-110410-132541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol. 2001;2:RESEARCH0002. doi: 10.1186/gb-2000-2-1-research0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Korta DZ, Tuck S, Hubbard EJ. S6K links cell fate, cell cycle and nutrient response in C. elegans germline stem/progenitor cells. Development. 2012;139:859–870. doi: 10.1242/dev.074047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda I, Kohara Y, Yamamoto M, Sugimoto A. Large-scale analysis of gene function in Caenorhabditis elegans by high-throughput RNAi. Curr Biol. 2001;11:171–176. doi: 10.1016/s0960-9822(01)00052-5. [DOI] [PubMed] [Google Scholar]
- Malik HS, Henikoff S. Conflict begets complexity: the evolution of centromeres. Curr Opin Genet Dev. 2002;12:711–718. doi: 10.1016/s0959-437x(02)00351-9. [DOI] [PubMed] [Google Scholar]
- McMurchy AN, Stempor P, Gaarenstroom T, Wysolmerski B, Dong Y, Aussianikava D, Appert A, Huang N, Kolasinska-Zwierz P, Sapetschnig A, et al. A team of heterochromatin factors collaborates with small RNA pathways to combat repetitive elements and germline stress. eLife. 2017;6:e21666. doi: 10.7554/eLife.21666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier B, Clejan I, Liu Y, Lowden M, Gartner A, Hodgkin J, Ahmed S. trt-1 is the Caenorhabditis elegans catalytic subunit of telomerase. PLoS Genet. 2006;2:e18. doi: 10.1371/journal.pgen.0020018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzstein MM, Stanfield GM, Horvitz HR. Genetics of programmed cell death in C. elegans: past, present and future. Trends Genet. 1998;14:410–416. doi: 10.1016/s0168-9525(98)01573-x. [DOI] [PubMed] [Google Scholar]
- Miller DL, Roth MB. C. elegans are protected from lethal hypoxia by an embryonic diapause. Curr Biol. 2009;19:1233–1237. doi: 10.1016/j.cub.2009.05.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nystul TG, Goldmark JP, Padilla PA, Roth MB. Suspended animation in C. elegans requires the spindle checkpoint. Science. 2003;302:1038–1041. doi: 10.1126/science.1089705. [DOI] [PubMed] [Google Scholar]
- Ogg S, Ruvkun G. The C. elegans PTEN homolog, DAF-18, acts in the insulin receptor-like metabolic signaling pathway. Mol Cell. 1998;2:887–893. doi: 10.1016/s1097-2765(00)80303-2. [DOI] [PubMed] [Google Scholar]
- Padilla PA, Ladage ML. Suspended animation, diapause and quiescence: arresting the cell cycle in C. elegans. Cell Cycle. 2012;11:1672–1679. doi: 10.4161/cc.19444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips CM, Brown KC, Montgomery BE, Ruvkun G, Montgomery TA. piRNAs and piRNA-dependent siRNAs protect conserved and essential C. elegans genes from misrouting into the RNAi pathway. Dev Cell. 2015;34:457–465. doi: 10.1016/j.devcel.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkston-Gosse J, Kenyon C. DAF-16/FOXO targets genes that regulate tumor growth in Caenorhabditis elegans. Nat Genet. 2007;39:1403–1409. doi: 10.1038/ng.2007.1. [DOI] [PubMed] [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2017. https://www.R-project.org/ [Google Scholar]
- Ragland GJ, Denlinger DL, Hahn DA. Mechanisms of suspended animation are revealed by transcript profiling of diapause in the flesh fly. Proc Natl Acad Sci USA. 2010;107:14909–14914. doi: 10.1073/pnas.1007075107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnappan R, Amrit FR, Chen SW, Gill H, Holden K, Ward J, Yamamoto KR, Olsen CP, Ghazi A. Germline signals deploy NHR-49 to modulate fatty-acid β-oxidation and desaturation in somatic tissues of C. elegans. PLoS Genet. 2014;10:e1004829. doi: 10.1371/journal.pgen.1004829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy D, Michaelson D, Hochman T, Santella A, Bao Z, Goldberg JD, Hubbard EJ. Cell cycle features of C. elegans germline stem/progenitor cells vary temporally and spatially. Dev Biol. 2016;409:261–271. doi: 10.1016/j.ydbio.2015.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi A, Sarkies P, Simon M, Doebley AL, Goldstein LD, Hedges A, Ikegami K, Alvares SM, Yang L, LaRocque JR, et al. Caenorhabditis elegans RSD-2 and RSD-6 promote germ cell immortality by maintaining small interfering RNA populations. Proc Natl Acad Sci USA. 2014;111:E4323–E4331. doi: 10.1073/pnas.1406131111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiesari L, O’Connor MB. Diapause: delaying the developmental clock in response to a changing environment. Curr Top Dev Biol. 2013;105:213–246. doi: 10.1016/B978-0-12-396968-2.00008-7. [DOI] [PubMed] [Google Scholar]
- Schumacher B, Hofmann K, Boulton S, Gartner A. The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr Biol. 2001;11:1722–1727. doi: 10.1016/s0960-9822(01)00534-6. [DOI] [PubMed] [Google Scholar]
- Seidel HS, Kimble J. The oogenic germline starvation response in C. elegans. PLoS One. 2011;6:e28074. doi: 10.1371/journal.pone.0028074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel HS, Kimble J. Cell-cycle quiescence maintains Caenorhabditis elegans germline stem cells independent of GLP-1/Notch. eLife. 2015;4:e10832. doi: 10.7554/eLife.10832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007;8:703–713. doi: 10.1038/nrm2241. [DOI] [PubMed] [Google Scholar]
- Simon M, Sarkies P, Ikegami K, Doebley AL, Goldstein LD, Mitchell J, Sakaguchi A, Miska EA, Ahmed S. Reduced insulin/IGF-1 signaling restores germ cell immortality to Caenorhabditis elegans Piwi mutants. Cell Rep. 2014;7:762–773. doi: 10.1016/j.celrep.2014.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smelick C, Ahmed S. Achieving immortality in the C. elegans germline. Ageing Res Rev. 2005;4:67–82. doi: 10.1016/j.arr.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Spracklin G, Fields B, Wan G, Becker D, Wallig A, Shukla A, Kennedy S. The RNAi inheritance machinery of Caenorhabditis elegans. Genetics. 2017;206:1403–1416. doi: 10.1534/genetics.116.198812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strome S, Updike D. Specifying and protecting germ cell fate. Nat Rev Mol Cell Biol. 2015;16:406–416. doi: 10.1038/nrm4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syntichaki P, Xu K, Driscoll M, Tavernarakis N. Specific aspartyl and calpain proteases are required for neurodegeneration in C. elegans. Nature. 2002;419:939–944. doi: 10.1038/nature01108. [DOI] [PubMed] [Google Scholar]
- Tatar M, Yin C. Slow aging during insect reproductive diapause: why butterflies, grasshoppers and flies are like worms. Exp Gerontol. 2001;36:723–738. doi: 10.1016/s0531-5565(00)00238-2. [DOI] [PubMed] [Google Scholar]
- Van Gilst MR, Hadjivassiliou H, Jolly A, Yamamoto KR. Nuclear hormone receptor NHR-49 controls fat consumption and fatty acid composition in C. elegans. PLoS Biol. 2005a;3:e53. doi: 10.1371/journal.pbio.0030053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gilst MR, Hadjivassiliou H, Yamamoto KR. A Caenorhabditis elegans nutrient response system partially dependent on nuclear receptor NHR-49. Proc Natl Acad Sci USA. 2005b;102:13496–13501. doi: 10.1073/pnas.0506234102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Meter M, Kashyap M, Rezazadeh S, Geneva AJ, Morello TD, Seluanov A, Gorbunova V. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat Commun. 2014;5:5011. doi: 10.1038/ncomms6011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vowels JJ, Thomas JH. Genetic analysis of chemosensory control of dauer formation in Caenorhabditis elegans. Genetics. 1992;130:105–123. doi: 10.1093/genetics/130.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller P, Padeken J, van Schendel R, Kalck V, Tijsterman M, Gasser SM. Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat Genet. 2016;48:1385–1395. doi: 10.1038/ng.3672. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Hartwieg E, Horvitz HR. CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell. 2001;104:43–56. doi: 10.1016/s0092-8674(01)00190-8. [DOI] [PubMed] [Google Scholar]
- Zhou KI, Pincus Z, Slack FJ. Longevity and stress in Caenorhabditis elegans. Aging (Albany NY) 2011;3:733–753. doi: 10.18632/aging.100367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.