

Abstract
Epithelial-mesenchymal transition (EMT) is critical in the progression of numerous types of carcinoma, and endows invasive and metastatic properties upon cancer cells. The tumor microenvironment facilitates tumor metastasis to distant organs. Various signaling pathways contribute to this process. In the present study, SW480 colon adenocarcinoma cells were treated with transforming growth factor-β1 (TGF-β1; 10 ng/ml) and tumor necrosis factor-α (TNF-α; 20 ng/ml), alone or in combination, for 72 h, and EMT was assessed using immunofluorescence, western blot analysis and migration assays. The functions of p38 mitogen-activated protein kinase, extracellular signal-regulated kinase (ERK) and nuclear factor-κB (NF-κB) pathways in EMT were examined. It was demonstrated that the cooperation of TGF-β1 and TNF-α signaling promoted the morphological conversion of the SW480 cells from an epithelial to a mesenchymal phenotype. Furthermore, simultaneous exposure to TNF-α and TGF-β1 downregulated the expression of E-cadherin (an epithelial marker) and increased the expression of N-cadherin and vimentin (mesenchymal markers). Additionally, the migratory capacity of the SW480 cells increased. The inhibition of p38 and ERK signaling exhibited no effect on EMT, whereas the inhibition of inhibitor of NF-κB kinase subunit β blocked the EMT induced by TGF-β1 and TNF-α. In conclusion, the results of the present study demonstrated that TNF-α and TGF-β1 synergistically promoted EMT in SW480 cells via the NF-κB pathway, independent of p38 activation and ERK1/2 signaling. These results suggest a novel function of TGF-β1 and TNF-α during EMT in colon carcinoma and, thus, provide insights into potential therapeutic interventions.
Keywords: tumor necrosis factor-α, transforming-growth factor-β1, epithelial-mesenchymal transition, nuclear-factor-κB, colon carcinoma
Introduction
The metastatic spread of tumor cells to vital organs is the principle cause of mortality in patients with colon cancer. The tumor microenvironment is important in regulating the metastatic capacity of numerous types of cancer (1). Interactions of cancer cells with the tumor microenvironment throughout the epithelial-mesenchymal transition (EMT) are important determinants of cancer progression toward metastasis (2). The EMT program has emerged as a crucial early event for promoting the invasive and metastatic behavior of various types of carcinoma (3). EMT is the process by which the epithelial phenotype of a cell is converted to a mesenchymal phenotype. Specifically, adherens junction proteins, including E-cadherin and cytokeratin, are downregulated, and the expression of mesenchymal markers, including vimentin and N-cadherin, are upregulated (4). Furthermore, morphologically, the cell-cell junctions are weakened and cell polarity is altered to reduce cell-cell interactions. The morphology of cells becomes more fibroblast-like and cells become more invasive.
The exact mechanisms underlying EMT have yet to be elucidated; however, several cytokines, including transforming growth factor-β (TGF-β), tumor necrosis factor-α (TNF-α), interleukin-6 and macrophage migration inhibitory factor, have previously been demonstrated to be associated with EMT in experimental models and human cancer. These factors may either promote or inhibit tumor development by modulating EMT (5). TGF-β1, a member of the TGF-β super-family, is involved in embryonic development and also contributes to tumor progression by inducing EMT (6). Several studies have demonstrated that TNF-α, which is produced by macrophages, upregulates various adhesion molecules on endothelial cells that contribute to tumor cell adhesion and migration (7,8). Furthermore, in primary bronchial epithelial cells, TNF-α can accentuate TGF-β1-induced EMT, resulting in the dysregulated wound repair of injured lung epithelium (9). TNF-α has also been demonstrated to increase the metastatic potential in human colonic epithelial organoid models of colon cancer by promoting EMT, indicating a potential link between EMT and inflammation (10). TGF-β1-induced EMT is promoted by TNF-α via signaling between the Smad and nuclear factor-κB (NF-κB) pathways in the A549 lung adenocarcinoma epithelial cell line (11). However, the signaling pathways involved in the synergistic effect of TGF-β1 and TNF-α in colon adenocarcinoma epithelial cells remain to be elucidated, and further investigation is required to identify potential therapeutic targets to disrupt or even reverse EMT. Thus, the present study was designed to determine whether TNF-α and TGF-β1, two stroma-derived factors, facilitated EMT in colon cancer cells, and to understand the underlying mechanisms.
Materials and methods
Cell culture
SW480 colon cancer cells were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) containing 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) at 37°C in an incubator with 5% CO2.
Antibodies and reagents
Recombinant human TNF-α and human TGF-β1 were purchased from PeproTech, Inc. (Rocky Hill, NJ, USA) and Prospec-Tany TechnoGene, Ltd. (East Brunswick, NJ, USA), respectively. Rabbit polyclonal anti-N-cadherin (cat. no. ab12221), mouse monoclonal anti-E-cadherin (cat. no. ab1416) and anti-vimentin (cat. no. ab8069) antibodies were all purchased from Abcam (Cambridge, UK). FITC AffiniPure Goat Anti-Mouse IgG (H+L) (cat. no. E031210-01) and Cy3 AffiniPure Goat Anti-Mouse IgG (cat. no. E031610-01) were obtained from EarthOx Life Sciences (Millbrae, CA, USA). The rabbit monoclonal anti-extracellular signal-regulated kinase (ERK) antibody (cat. no. 4695), phospho-ERK (cat. no. 4370) and phospho-p38 mitogen-activated protein kinase (MAPK) antibodies (cat. no. 2387) were obtained from Cell Signaling Technology, Inc. (Danvers, MA, USA) and the antibody against GAPDH was from Sigma-Aldrich (Merck KGaA). IKKβ4i (cat. no. CAS 507475-17-4), a small molecule inhibitor against inhibitor of NF-κB kinase subunit β (IKKβ), an ERK1/2 inhibitor (PD98059; cat. no. sc-3532) and a p38 MAPK inhibitor (SB203580; cat. no. sc-3533) were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Inhibitors were dissolved in dimethyl sulfoxide and aliquots were stored at −80°C prior to use.
Cytokine and inhibitor assays
A total of 5×104 SW480 cells were seeded in 24-well plates with either TNF-α (10 ng/ml) or TGF-β1 (20 ng/ml), or a combination of the two. The morphology of the colon cancer cells was assessed by light microscopy (Olympus Corporation, Tokyo, Japan). For inhibition assays, cells were pretreated for 20 min with PD98059 ERK inhibitor (20 µM) or for 1 h with SB203580 p38 MAPK inhibitor (40 µM), and then seeded in culture plates in the presence of cytokines. In certain experiments, cells were pretreated with the IKKβ inhibitor, IKKβ4i (10 µM), for 2 h prior to the addition of cytokines and subsequent culture in 24-well plates.
Immunofluorescence
Cells were fixed in 4% paraformaldehyde at room temperature for 10 min and incubated with anti-E cadherin (1:100) and anti-vimentin (1:100) at 4°C overnight followed by incubation with the secondary antibodies of FITC AffiniPureGoat Anti-Mouse IgG (1:100) or Cy3AffiniPure Goat Anti-Mouse IgG (1:200) at room temperature for 1 h. DAPI was used as a nuclear counterstain. Images were acquired using a Leica TCS-SP-2UV laser scanning confocal microscope (Leica Microsystems GmbH, Wetzlar, Germany).
Migration assay
The migratory capacity of SW480 cells was assessed using a Transwell chamber assay (8 µm pore size, polycarbonate filters, 6.5 mm diameter; Costar Corporation, Cambridge, MA, USA) in 24-well plates. Briefly, 4×104 cells were treated with TGF-β1 (10 ng/ml), TNF-α (20 ng/ml) or a combination of the two for 72 h, incubated overnight in 250 µl of serum-free DMEM, then added to the upper chamber; and DMEM medium with 10% FBS (Gibco; Thermo Fisher Scientific, Inc.) was used as a chemoattractant in the lower chamber. Following incubation at 37°C for 24 h, the nonmigrating cells were removed from the upper surface of each Transwell using a cotton swab. Transwell membranes were then stained with crystal violet at room temperature for 5–10 min. The cells that migrated through the membrane to the lower surface were counted by light microscopy.
Western blotting
Proteins were extracted from cells in a Triton-X lysis buffer (1% Triton-X, 50 mM Tris and 150 mM NaCl) containing protease inhibitors (pepstatin, phenylmethane sulfonyl fluoride, aprotinin and leupeptin) for 1 h in 4°C. The protein concentration in the samples was determined using a Bradford protein assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Equal amounts of cell lysates (30 µg/lane) were separated by 10% SDS-PAGE and transferred onto polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA, USA). The membranes were probed with antibodies against E-cadherin (1:1,000), vimentin (1:500), N-cadherin (1:500), ERK (1:1,000), phospho-ERK (1:500) and phospho-p38 MAPK (1:11,000) overnight at 4°C. Membranes were incubated with horseradish peroxidase-conjugated anti-mouse antibody (cat. no. NXA931; 1:5,000) and anti-rabbit antibody (cat. no. NA934; 1:5,000) (GE Healthcare Life Sciences, Little Chalfont, UK) for 1 h at room temperature. Immune complexes were detected by enhanced chemiluminescence (Cell Signaling Technology, Inc.). GAPDH (1:1,000) was detected as a reference protein for equal sample loading, and Image Lab 5.0 software (Bio-Rad Laboratories, Inc.) was used for quantitative analysis.
Statistical analysis
Data were expressed as the mean ± standard error of the mean. The significance of differences was assessed by a one-way analysis of variance followed by Tukey's post hoc test, using the computer software package GraphPad Prism (version 5.0; GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Effects of TNF-α and TGF-β1 on EMT in SW480 cells
For further confirmation that the SW480 cells had undergone EMT, the effect of TGF-β1 and TNF-α on the invasive behavior of cells was investigated individually and simultaneously. In contrast to previous findings in colon cancer cells (12), TGF-β1 or TNF-α alone did not alter SW480 morphology within 72 h compared with the untreated cells (Fig. 1A); the cancer cells maintained uniform cobblestone morphology with tight cell-cell contact (Fig. 1A-a-i). However, the combined addition of TGF-β1 and TNF-α to SW480 cells for 72 h resulted in a complete mesenchymal transition, with cells exhibiting fibroblastic morphology, with a bipolar, spindle-like shape (Fig. 1A-j-l). Consistent with this change, the results of immunofluorescent staining demonstrated that E-cadherin immunoreactivity was predominantly on the cell membrane, with some immunoreactivity in the plasma. The expression of E-cadherin was markedly decreased (Fig. 1B) and the induction of vimentin was increased with a weak trend in the cancer cells treated with both cytokines (Fig. 1C). By contrast, no significant effect on cell phenotype was observed when SW480 cells were treated with TNF-α and TGF-β1 individually compared with the untreated control cells.
Figure 1.

Effects of TGF-β1 and TNF-α on SW480 cells. (A) Morphological characteristics of EMT in SW480 cells treated with (a-c) TGF-β1 or (d-f) TNF-α alone, or in (g-i) combination. (a-f) The majority of TGF-β1 or TNF-α-treated, or (g-i) untreated SW480 cells demonstrated a pebble-like shape and tight cell-cell adhesion at 72 h. (j-l) Cotreatment with TGF-β1 and TNF-α decreased cell-cell contacts and resulted in a more elongated morphological shape compared with individual treatments. (l) TGF-β1 and TNF-α treatment resulted in EMT of SW480 cells, with cells exhibiting a bipolar, spindle-like shape, characteristic of fibroblasts, by 72 h. Magnification, ×200. (B) Immunofluorescent images of SW480 cells treated with TGF-β1, TNF-α or a combination of both for 48 h. Cells were stained with an antibody against E-cadherin (green), and the nucleus was counterstained with DAPI (blue). Subpanels are representative images of the nuclear staining, E-cadherin staining, and a merge of the two, respectively, in SW480 cells. There was a decrease in the an immunostaining of E-cadherin in TGF-β1-and TNF-α-treated SW480 cells compared with cells treated with TNF-α, TGF-β1 alone or untreated cells group. Magnification, ×200. (C) Cells were stained with antibody against vimentin (red), and the nucleus was counterstained with DAPI. Subpanels are representative images of nuclear staining, vimentin staining and a merge of the two, respectively, in SW480 cells. There was an increase in immunostaining of vimentin in TGF-β1- and TNF-α-treated SW480 cells compared with cells treated with TNF-α or TGF-β1 alone, or untreated. Magnification, ×200. (D) Western blotting of the treatment of SW480 cells with TGF-β1 and TNF-α. SW480 cells were incubated with TGF-β1 and TNF-α for 0–72 h and E-cadherin expression was visibly depleted at 72 h. N-cadherin and vimentin expression levels were increased from 24 h. TGF-β1, transforming growth factor-β1; TNF-α, tumor necrosis factor-α; EMT, epithelial-mesenchymal transition.
E-cadherin expression was almost undetectable at 72 h in the presence of the two cytokines based on a western blotting analysis, which indicated that simultaneous stimulation with TNF-α and TGF-β1 promoted EMT (Fig. 1D). By contrast, E-cadherin expression remained discernible following a 72-h treatment with TGF-β1 or TNF-α alone (Fig. 1D). Additionally, E-cadherin protein levels were downregulated and the N-cadherin expression was upregulated following incubation with TNF-α or TGF-β1 alone compared with the control cells (n=3; P<0.05) (data not shown). Vimentin expression levels were marginally increased compared with the control; however, the difference was not significant (n=3; P>0.05).
The expression levels of the mesenchymal markers vimentin and N-cadherin were upregulated following combined treatment with TNF-α and TGF-β1 (Fig. 2A-D), indicating the occurrence of EMT. To measure cell migration, the ability of SW480 cells to penetrate Transwell membranes was examined (Fig. 2E). Few non-treated cells spontaneously penetrated the filters, and the addition of either TNF-α or TGF-β1 alone for 72 h did not observably initiate migration (Fig. 2E-a and -c) compared with the untreated control (Fig. 2E-d). The addition of the two cytokines together increased the rate of migration compared with the control (P<0.05; n=3; Fig. 2E-b and -F), which corroborated the morphological observations of EMT, as demonstrated in Fig. 2E-b. Collectively, the data of the present study suggested that TNF-α and TGF-β1 cooperated to reduce the level of E-cadherin, induce mesenchymal phenotypes and, thus, promote EMT.
Figure 2.
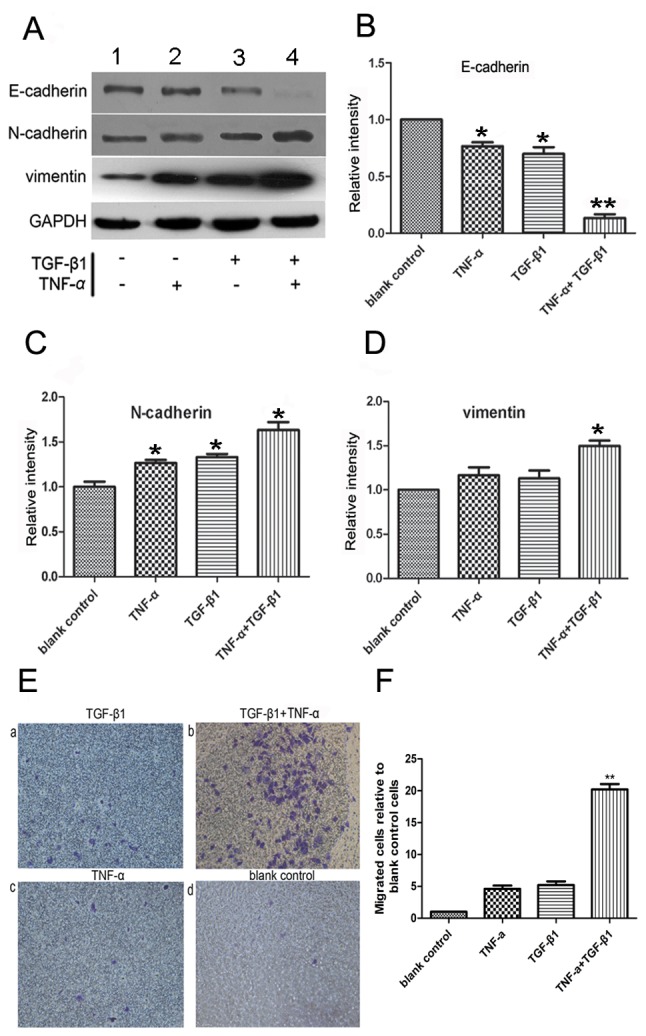
Analysis of EMT activity of SW480 cells by TGF-β1 and TNF-α. (A) Western blot analysis of the EMT activity of SW480 cells. The relative expression of (B) E-cadherin, (C) N-cadherin and (D) vimentin were examined. TGF-β1 or TNF-α alone decreased E-cadherin expression and increased the expression level of the EMT marker, N-cadherin; *P<0.05 vs. blank control (n=3). Cotreatment with TGF-β1 and TNF-α significantly altered the protein expression levels of E-cadherin, vimentin and N-cadherin; *P<0.05 vs. TGF-β1 or TNF-α alone (n=3). TGF-β1 or TNF-α alone exerted no significant effect on vimentin expression. The results were normalized to the level of GAPDH, used as a loading control. (E) Modulation of the mesenchymal-like properties of SW480 cells by TGF-β1 and TNF-α, (×200 magnification) (a and c) SW480 cells were incubated with TGF-β1 or TNF-α for 72 h, and the number of migratory cells was determined. (b) Cotreatment with TGF-β1 + TNF-α induced an increased number of migratory cells compared with TGF-β1 or TNF-α treatment alone. The majority of cells exhibited a bipolar phenotype characteristic of fibroblasts. (d) Untreated cells demonstrated few or no migrated cells. (F) Cotreatment with TGF-β1 + TNF-α induced an increased number of migratory cells compared with TGF-β1 or TNF-α treatment alone. **P<0.01 vs. TGF-β1 or TNF-α treatment alone (n=3). Values are presented as the mean ± standard error of the mean of triplicate samples. EMT, epithelial-mesenchymal transition; TGF-β1, transforming growth factor-β1; TNF-α, tumor necrosis factor-α.
TNF-α promotes ERK activation, with no effect on EMT
To elucidate the cytokine-stimulated signaling pathways that induce EMT, the present study investigated the ERK1/2 signaling pathway. This signaling cascade can be stimulated by TNF-α (13) and has previously been demonstrated to regulate epithelial cell plasticity and EMT induced by TGF-β1 (14,15). Western blotting for phospho-ERK1/2 demonstrated that ERK1/2 was activated in SW480 cells exposed to TNF-α or TGF-β1. Furthermore, TGF-β1 and TNF-α treatment demonstrated an additive effect on phospho-ERK1/2 activation (P<0.05; n=3). A negligible effect of ERK isoform was detected in control cells (Fig. 3A). To identify the relative contribution of ERK1/2 signaling to EMT, selective inhibitors were used. Pretreatment of SW480 cells with PD98059 eliminated ERK1/2 phosphorylation induced by the co-treatment with TGF-β1 and TNF-α (Fig. 3B). However, pre-incubation with the ERK1/2 inhibitor exerted no significant effect on E-cadherin and vimentin expression examined by western blotting (Fig. 3C) or morphological phenotype analysis (Fig. 3D) of the cells in which EMT was induced by TGF-β1 alone or TGF-β1 plus TNF-α (P>0.05; n=3).
Figure 3.

Analysis of ERK activation following TGF-β1 + TNF-α treatment. (A) Western blot analysis of ERK activation in response to cytokine stimulation. Cells were treated with TNF-α, TGF-β1 or a combination of the two. ERK1/2 activity was determined by western blotting with a phospho-specific ERK antibody. ERK protein expression was confirmed using an ERK antibody. Cotreatment of the cells with TGF-β1 and TNF-α increased ERK1/2 phosphorylation compared with TGF-β1 or TNF-α treatment alone; **P<0.01 (n=3). (B) Pretreatment of SW480 cells with an ERK1/2 inhibitor (PD98059) inhibited ERK1/2 phosphorylation induced by co-treatment with TGF-β1 and TNF-α. Untreated SW480 cells were the control. ERK1/2 activation was determined using the phospho-specific ERK antibody. ERK protein expression was confirmed using an ERK antibody. However, no significant effects on (C) E-cadherin and vimentin protein expression or (D) cell phenotype were observed following pre-incubation with PD98059. ERK, extracellular signal-regulated kinase; TGF-β1, transforming growth factor-β1; TNF-α, tumor necrosis factor-α.
TNF-α activates p38 MAPK signaling but does not promote EMT
Another pathway activated by TNF-α which has previously been proposed to be important in EMT is the p38 MAPK signaling pathway, which is known to be a convergence point for TNF-α and TGF-β signaling (16). Therefore, it was hypothesized that elevated p38 MAPK activity, induced by TNF-α, may provide a mechanism by which the cytokine promoted EMT. Western blotting with phospho-specific antibody demonstrated that p38 MAPK activity was increased following treatment with TGF-β1 or TNF-α alone compared with the control. Notably, cotreatment of the cells with TGF-β1 and TNF-α exerted an additive effect (Fig. 4A and B) compared with TGF-β1 or TNF-α treatment alone.
Figure 4.

Western blot analysis of phospho-p38 in response to cytokine stimulation. (A) Cells were treated with TNF-α, TGF-β1 or combination of both cytokines. p38 activity was determined by western blotting with a phospho-specific p38 antibody. p38 protein expression was confirmed using a p38 antibody. Cotreatment of the cells with TGF-β1 + TNF-α increased p38 phosphorylation compared with TGF-β1 or TNF-α treatment alone. (B) Densitometry of phospho-p38 protein expression in response to cytokine stimulation; *P<0.05 (n=3). (C) Treatment with a p38 inhibitor (SB203580, 10 µM) prevented p38 activation following stimulation with TNF-α and TGF-β1. (D) Densitometry of groups untreated (lanes 1–3) and treated (lanes 4–6) with a p38 inhibitor. **P<0.01 vs. lanes 1–3 (n=3). (E) Effect of SB203580 on SW480 cell morphology. Cells were examined following treatment with TGF-β1 and TNF-α. (a-c) Compared to the control groups from 24 h to 72 h, treatment with SB203580 reduced the early transitional effects and (d-f) prevented cellular elongation and spreading. (e) The SB203580-treated cells remained similar to cells at 48 h after cytokine treatment (f) though underwent epithelial-mesenchymal transition over a longer time course (72 h). Values are presented as the mean ± standard error of triplicate samples. p38, p38 mitogen-activated protein kinase; TNF-α, tumor necrosis factor-α; TGF-β1, transforming growth factor-β1.
To elucidate the effect of p38 MAPK activation on EMT, a specific p38 MAPK inhibitor, SB203580, was used. As demonstrated in Fig. 4C and D, the expression of phospho-p38 MAPK was inhibited by SB203580 following treatment with TNF-α/TGF-β1 for 1 h, and a significant difference was observed between groups with or without treatment of p38 MAPK inhibitor (P<0.01). Subsequently, the effects of SB203580 on morphology were examined at 48 h following addition of the cytokines (the initial discernible effects of accelerated EMT could be observed at this time-point). At 48 h, the morphology of the SW480 cells changed from epithelial to fibroblast-like following treatment with TNF-α plus TGF-β1. However, treatment with SB203580 appeared to reduce the early transitional effects, completely inhibiting cellular elongation and spreading. Over a longer time period (72 h), the SB203580-treated cells underwent EMT, similar to those examined at 48 h after the addition of the cytokines (Fig. 4E).
TNF-α activates NF-κB signaling to promote EMT
As inhibition of the p38 MAPK pathway demonstrated no effect on EMT, alternative pathways initiated by TNF-α were investigated. An apparent candidate was the NF-κB pathway, which is important for TNF-α-induced tumor initiation and promotion (8).
SW480 cells were incubated with IKKβ4i and the effect on EMT was examined. Pretreatment with IKKβ4i significantly inhibited EMT marker expression in cells incubated with TGF-β1 plus TNF-α compared with the control (E-cadherin, 62±15%; vimentin, 75±15%; P<0.05; n=3; Fig. 5A). Additionally, IKKβ4i inhibited the change in cell phenotype induced by TGF-β1 and TNF-α. It also reduced the extent of EMT stimulated by TGF-β1 and TNF-α (Fig. 5B). This suggested that NF-κB signaling is a critical pathway involved in TGF-β1 and TNF-α-induced EMT.
Figure 5.
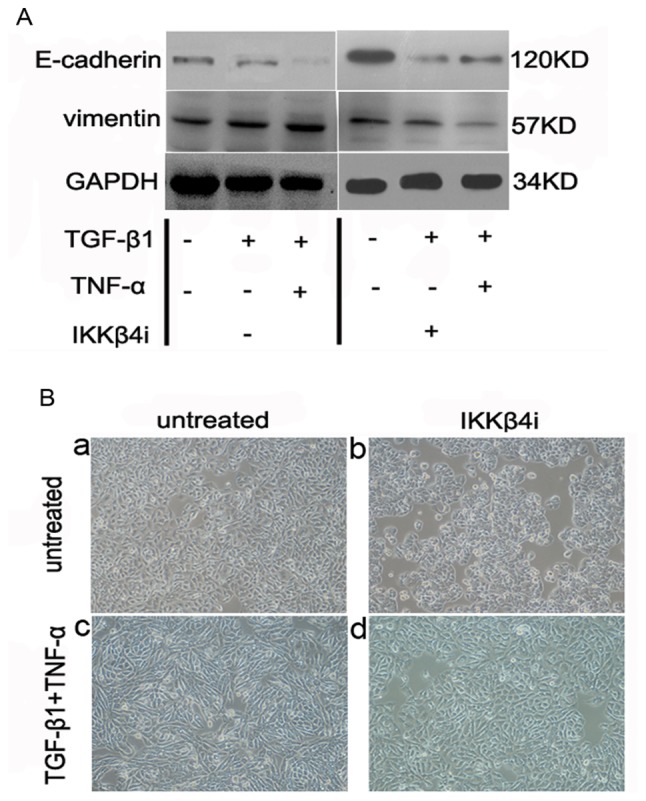
Analysis of the activation of the NF-κB pathway with TGF-β1 and TNF-α. (A) Western blot analysis of the effect of IKKβ4i on the response to cytokine stimulation. Pretreatment of the cells with IKKβ4i (10 µM) significantly attenuated TGF-β1- and TNF-α-induced EMT; P<0.05 (n=3). All results were normalized to GAPDH. (B) Effects of IKKβ4i on SW480 cell morphology. (a-c) Cotreatment of the cells with TGF-β1 + TNF-α enhanced the changes in phenotype compared with untreated cells and IKKβ4i-treated cells. (d) The change in phenotype was inhibited by pretreatment with IKKβ4i. The results are representative of three independent experiments. Magnification, ×100. NF-κB, nuclear-factor-κB; TGF-β1, transforming growth factor-β1; TNF-α, tumor necrosis factor-α; IKKβ4i, small molecule inhibitor against inhibitor of NF-κB kinase subunit β; EMT, epithelial-mesenchymal transition.
Discussion
Despite advances in clinical treatment, recurrence occurs in ~50% of all patients with colorectal cancer, including ~25% of patients who are theoretically curable during the early stages of disease development (17). A potential mechanism to explain metastatic progression is EMT, a complex cellular reprogramming process in which epithelial cells acquire a mesenchymal phenotype; EMT is crucial for tumor invasive and metastatic behaviour (18), and facilitates metastasis to distant organs (2). EMT is promoted by stimulatory signals (19). The results of the present study demonstrated that TNF-α or TGF-β1 alone exerted almost no effect on SW480 cell morphology. By contrast, the addition of both cytokines resulted in a complete mesenchymal transition, upon which cells changed to exhibit a spindle-shaped, fibroblast-like morphology. Previous research has indicated that TGF-β1 stimulation alters endothelial morphology; however, this is a rare occurrence (20). Furthermore, downregulation of E-cadherin expression is a crucial event in EMT (21). The findings of the present study demonstrated that combined treatment with TGF-β1 and TNF-α resulted in the reduced expression of E-cadherin and the increased expression of vimentin and N-cadherin.
In the present study, the migration of SW480 cells treated with both the cytokines was increased, which is characteristic of an invasive phenotype. By contrast, the addition of either TNF-α or TGF-β1 alone did not significantly induce migration. It was also demonstrated that TNF-α promoted phenotypic transition and increased the expression of invasive markers of EMT compared with TGF-β1 alone, which suggested that a pro-inflammatory tumor microenvironment enriched in TNF-α may accentuate TGF-β1-induced EMT. Notably, it is well established that levels of pro-inflammatory cytokines are strongly associated with tumor progression in patients with colorectal cancer and positive lymph node metastasis (22). Furthermore, the levels of TGF-β1 and TNF-α have previously been demonstrated to be increased in the serum of patients with colorectal cancer, which also indicates an association with tumor progression of colon adenocarcinomas (6,23). Potentially, an increased level of TNF-α, coupled with the increased levels of TGF-β1, may initiate EMT and contribute to disease pathogenesis. The results of the present study indicated that TNF-α and TGF-β1 are crucial for the initiation of EMT, and suggested that an inflammatory microenvironment rich in TNF-α is important for the regulation of EMT in colon adenocarcinoma.
Signaling cascades involved in the effect of TNF-α- and TGF-β1-induced EMT were investigated in the present study. Several studies have previously demonstrated that the ERK1/2 pathway activity is required for TGF-β1-induced EMT in non-transformed cells (24–26). To further elucidate the signaling pathways involved in EMT, the present study examined the effect of TGF-β1 and TNF-α on ERK signaling. However, inhibition of ERK exerted no effect on TGF-β1- and TNF-α-induced EMT. An alternative candidate pathway is the p38 MAPK signaling pathway, which is a point of convergence in TNF-α and TGF-β1 signaling and is responsive to both cytokines (27). The results of the present study indicated that p38 MAPK activation decreased E-cadherin levels, but did not promote mesenchymal transition.
NF-κB is a heterodimeric transcription factor composed of members of the Rel family of transcription factors, including RelA (p65) (28), and is involved in numerous biological processes. In resting cells, NF-κB protein is retained in an inactive state in the cytoplasm, complexed with inhibitor of κB proteins (IκBs). When cells are exposed to cytokines, including TNF-α, interleukin-1 or epidermal growth factor, NF-κB dissociates from IκBs, and is subsequently translocated to the nucleus to activate the transcription of target genes. Activated NF-κB promotes the expression of various proteins, including proteins that facilitate metastasis in the tumor microenvironment (29,30). In the present study, pretreatment of SW480 cells with IKKβ4i significantly reduced EMT stimulated by the combination of TGF-β1 and TNF-α. This suggested that inflammation-induced EMT is closely associated with NF-κB activation. Notably, NF-κB activation has been previously indicated to be involved in inflammation-induced cancer or metastatic progression (31). NF-κB has been previously identified as an important regulator of EMT during breast cancer progression (32), which highlights NF-κB as a potential therapeutic target for suppressing EMT in cancer. In addition, it is notable that the results of the present study show IKK inhibitor enhanced E-cadherin expression without TGF-β1 and TNF-α treatment, though the expression of vimentin was not altered. To a certain degree, NF-κB constitutive activation in the cell line used in the present study may account for this phenomenon. The alternative pathway may be involved, including WNT/β-catenin-mediated target gene expression, which participates in NF-κB activation pathway and eventually regulates EMT (3).
In conclusion, the present study investigated the ability of TNF-α to promote TGF-β1-induced EMT and the potential pathways involved in this crucial transition in SW480 colon cancer cells. The results indicated that the effects of TNF-α and TGF-β1 on EMT are not associated with p38 activation and ERK1/2 signaling; however, the effects may be mediated by activation of the NF-κB pathway. These findings provide potential therapeutic targets for the treatment of metastatic colon cancer. However, further investigation is required.
Acknowledgements
The present study was supported by the Shan'xi Provincial Health Department of Science and Technology Research Projects (grant no. 201301062). The authors thank Professor Shao Lee (Chinese University of Hong Kong, China) for critically reviewing the manuscript in advance of submission.
Glossary
Abbreviations
- EMT
epithelial-mesenchymal transition
- ERK
extracellular regulated protein kinases
- FBS
fetal bovine serum
- IKKβ
inhibitor of nuclear factor-κB kinase
- MAPK
mitogen-activated protein kinase
- NF-κB
nuclear-factor κB
- SEM
standard error of the mean
- TNF
tumor necrosis factor
- TGF
transforming growth factor
Competing interests
The authors declare that they have no competing interests.
References
- 1.Candido J, Hagemann T. Cancer-related inflammation. J Clin Immunol. 2013;33(Suppl 1):S79–S84. doi: 10.1007/s10875-012-9847-0. [DOI] [PubMed] [Google Scholar]
- 2.Gao D, Vahdat LT, Wong S, Chang JC, Mittal V. Microenvironmental regulation of epithelial-mesenchymal transitions in cancer. Cancer Res. 2012;72:4883–4889. doi: 10.1158/0008-5472.CAN-12-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Göktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G, et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152:25–38. doi: 10.1016/j.cell.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 4.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: Acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 5.Jung HY, Fattet L, Yang J. Molecular pathways: Linking tumor microenvironment to epithelial-mesenchymal transition in metastasis. Clin Cancer Res. 2015;21:962–968. doi: 10.1158/1078-0432.CCR-13-3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hawinkels LJ, Verspaget HW, van der Reijden JJ, van der Zon JM, Verheijen JH, Hommes DW, Lamers CB, Sier CF. Active TGF-beta1 correlates with myofibroblasts and malignancy in the colorectal adenoma-carcinoma sequence. Cancer Sci. 2009;100:663–670. doi: 10.1111/j.1349-7006.2009.01100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9:361–371. doi: 10.1038/nrc2628. [DOI] [PubMed] [Google Scholar]
- 8.Wu Y, Zhou BP. TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. Br J Cancer. 2010;102:639–644. doi: 10.1038/sj.bjc.6605530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borthwick LA, McIlroy EI, Gorowiec MR, Brodlie M, Johnson GE, Ward C, Lordan JL, Corris PA, Kirby JA, Fisher AJ. Inflammation and epithelial to mesenchymal transition in lung transplant recipients: Role in dysregulated epithelial wound repair. Am J Transplant. 2010;10:498–509. doi: 10.1111/j.1600-6143.2009.02953.x. [DOI] [PubMed] [Google Scholar]
- 10.Bates RC, Mercurio AM. Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol Biol Cell. 2003;14:1790–1800. doi: 10.1091/mbc.E02-09-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borthwick LA, Gardner A, De Soyza A, Mann DA, Fisher AJ. Transforming Growth Factor-β1 (TGF-β1) driven epithelial to mesenchymal transition (EMT) is accentuated by tumour necrosis factor α (TNFα) via crosstalk between the SMAD and NF-κB pathways. Cancer Microenviron. 2012;5:45–57. doi: 10.1007/s12307-011-0080-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pino MS, Kikuchi H, Zeng M, Herraiz MT, Sperduti I, Berger D, Park DY, Iafrate AJ, Zukerberg LR, Chung DC. Epithelial to mesenchymal transition is impaired in colon cancer cells with microsatellite instability. Gastroenterology. 2010;138:1406–1417. doi: 10.1053/j.gastro.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kyriakis JM, Avruch J. Protein kinase cascades activated by stress and inflammatory cytokines. Bioessays. 1996;18:567–577. doi: 10.1002/bies.950180708. [DOI] [PubMed] [Google Scholar]
- 14.Ellenrieder V, Hendler SF, Boeck W, Seufferlein T, Menke A, Ruhland C, Adler G, Gress TM. Transforming growth factor beta1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation. Cancer Res. 2001;61:4222–4228. [PubMed] [Google Scholar]
- 15.Zavadil J, Bitzer M, Liang D, Yang YC, Massimi A, Kneitz S, Piek E, Bottinger EP. Genetic programs of epithelial cell plasticity directed by transforming growth factor-beta. Proc Natl Acad Sci USA. 2001;98:6686–6691. doi: 10.1073/pnas.111614398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Obata T, Brown GE, Yaffe MB. MAP kinase pathways activated by stress: The p38 MAPK pathway. Crit Care Med. 2000;28(4 Suppl):N67–N77. doi: 10.1097/00003246-200004001-00008. [DOI] [PubMed] [Google Scholar]
- 17.Young PE, Womeldorph CM, Johnson EK, Maykel JA, Brucher B, Stojadinovic A, Avital I, Nissan A, Steele SR. Early detection of colorectal cancer recurrence in patients undergoing surgery with curative intent: Current status and challenges. J Cancer. 2014;5:262–271. doi: 10.7150/jca.7988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 19.Fuxe J, Karlsson MC. TGF-β-induced epithelial-mesenchymal transition: A link between cancer and inflammation. Semin Cancer Biol. 2012;22:455–461. doi: 10.1016/j.semcancer.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 20.Brown KA, Aakre ME, Gorska AE, Price JO, Eltom SE, Pietenpol JA, Moses HL. Induction by transforming growth factor-beta1 of epithelial to mesenchymal transition is a rare event in vitro. Breast Cancer Res. 2004;6:R215–R231. doi: 10.1186/bcr778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–746. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 22.Hamadien MA, Khan Z, Vaali-Mohammed MA, Zubaidi A, Al-Khayal K, McKerrow J, Al-Obeed O. Polymorphisms of tumor necrosis factor alpha in Middle Eastern population with colorectal cancer. Tumour Biol. 2016;37:5529–5537. doi: 10.1007/s13277-015-4421-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Al Obeed OA, Alkhayal KA, Al Sheikh A, Zubaidi AM, Vaali-Mohammed MA, Boushey R, Mckerrow JH, Abdulla MH. Increased expression of tumor necrosis factor-α is associated with advanced colorectal cancer stages. World J Gastroenterol. 2014;20:18390–18396. doi: 10.3748/wjg.v20.i48.18390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buonato JM, Lazzara MJ. ERK1/2 blockade prevents epithelial-mesenchymal transition in lung cancer cells and promotes their sensitivity to EGFR inhibition. Cancer Res. 2014;74:309–319. doi: 10.1158/0008-5472.CAN-12-4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen X, Xiao W, Wang W, Luo L, Ye S, Liu Y. The complex interplay between ERK1/2, TGFβ/Smad, and Jagged/Notch signaling pathways in the regulation of epithelial-mesenchymal transition in retinal pigment epithelium cells. PLoS One. 2014;9:e96365. doi: 10.1371/journal.pone.0096365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL. Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia. 2004;6:603–610. doi: 10.1593/neo.04241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Itoigawa Y, Harada N, Harada S, Katsura Y, Makino F, Ito J, Nurwidya F, Kato M, Takahashi F, Atsuta R, Takahashi K. TWEAK enhances TGF-β-induced epithelial-mesenchymal transition in human bronchial epithelial cells. Respir Res. 2015;16:48. doi: 10.1186/s12931-015-0207-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell. 2002;9:625–636. doi: 10.1016/S1097-2765(02)00477-X. [DOI] [PubMed] [Google Scholar]
- 29.Xia Y, Shen S, Verma IM. NF-κB, an active player in human cancers. Cancer Immunol Res. 2014;2:823–830. doi: 10.1158/2326-6066.CIR-14-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bradford JW, Baldwin AS. IKK/nuclear factor-kappaB and oncogenesis: Roles in tumor-initiating cells and in the tumor microenvironment. Adv Cancer Res. 2014;121:125–145. doi: 10.1016/B978-0-12-800249-0.00003-2. [DOI] [PubMed] [Google Scholar]
- 31.Epanchintsev A, Shyamsunder P, Verma RS, Lyakhovich A. IL-6, IL-8, MMP-2, MMP-9 are overexpressed in Fanconi anemia cells through a NF-κB/TNF-α dependent mechanism. Mol Carcinog. 2015;54:1686–1699. doi: 10.1002/mc.22240. [DOI] [PubMed] [Google Scholar]
- 32.Chua HL, Bhat-Nakshatri P, Clare SE, Morimiya A, Badve S, Nakshatri H. NF-kappaB represses E-cadherin expression and enhances epithelial to mesenchymal transition of mammary epithelial cells: Potential involvement of ZEB-1 and ZEB-2. Oncogene. 2007;26:711–724. doi: 10.1038/sj.onc.1209808. [DOI] [PubMed] [Google Scholar]