

Abstract
The role of environmental stressors, particularly exposure to air pollution, in the development of neurodegenerative disease remains underappreciated. We examined the neurological effects of acute ozone (O3) exposure in aged mice, where increased blood–brain barrier (BBB) permeability may confer vulnerability to neuroinflammatory outcomes. C57BL/6 male mice, aged 8–10 weeks or 12–18 months were exposed to either filtered air or 1.0 ppm O3 for 4 h; animals received a single IP injection of sodium fluorescein (FSCN) 20 h postexposure. One-hour post-FSCN injection, animals were transcardially perfused for immunohistochemical analysis of BBB permeability. β-amyloid protein expression was assessed via ELISA. Flow cytometric characterization of infiltrating immune cells, including neutrophils, macrophages, and microglia populations was performed 20 h post-O3 exposure. Flow cytometry analysis of brains revealed increased microglia “activation” and presentation of CD11b, F4/80, and MHCII in aged animals relative to younger ones; these age-induced differences were potentiated by acute O3 exposure. Cortical and limbic regions in aged brains had increased reactive microgliosis and β-amyloid protein expression after O3 insult. The aged cerebellum was particularly vulnerable to acute O3 exposure with increased populations of infiltrating neutrophils, peripheral macrophages/monocytes, and Ly6C+ inflammatory monocytes after insult, which were not significantly increased in the young cerebellum. O3 exposure increased the penetration of FSCN beyond the BBB, the infiltration of peripheral immune cells, and reactive gliosis of microglia. Thus, the aged BBB is vulnerable to insult and becomes highly penetrable in response to O3 exposure, leading to greater neuroinflammatory outcomes.
Keywords: neuroinflammation, microglia, aging, reproductive & developmental toxicology, ozone, blood–brain barrier, inhalation toxicology, respiratory toxicology
Ozone (O3) is a tropospheric photochemical air pollutant with global public health implications due to its role in driving adverse respiratory and cardiovascular health outcomes. Numerous modeling studies predict significant increases in global O3 concentrations over the next 30 years principally affecting urbanized regions of the world (Chang et al., 2014; Phares et al., 2006; Post et al., 2012; Stowell et al., 2017). Recent discoveries suggest that O3 may drive neurological outcomes, such as reduced cognitive performance, memory impairments, and Alzheimer’s disease (AD) symptoms (Akhter et al., 2015; Calderon-Garciduenas and de la Monte, 2017; Calderon-Garciduenas et al., 2016). More importantly, numerous studies reveal a relationship between other air pollutants, especially particulate matter, and neuroinflammatory and neurodegenerative phenotypes (Ailshire and Crimmins, 2014; Block and Calderón-Garcidueñas, 2009; Calderon-Garciduenas et al., 2008, 2012; Gomez-Mejiba et al., 2009; Levesque et al., 2011 b); however, lack of information regarding the pathophysiological mechanisms underlying these effects hampers identification of vulnerable subpopulations.
In the past decade, much attention has been given to the potential translocation and accumulation of particulate matter to the systemic vasculature and central nervous system (CNS) following exposure to air pollution (Block and Calderón-Garcidueñas, 2009; Maher et al., 2016; Mercer et al., 2013; Miller et al., 2017; Oberdorster et al., 2004; Peters et al., 2006). Localization of particulate matter in the CNS has been demonstrated after high dose exposures but has only been experimentally linked to neuroinflammation and pathology in an associative manner. However, indirect inflammatory effects due to pulmonary damage may be carried into circulation, as demonstrated mechanistically in several recent studies of both particulate and gaseous inhaled toxicants (Aragon et al., 2017; Channell et al., 2012; Lucero et al., 2017; Robertson et al., 2013; Schisler et al., 2015). For exposures to lower doses of inhaled toxicants, this may be the likely mechanism of neuroinflammatory effects. Using inhaled O3, which is unable to penetrate unreacted farther than 0.1 μm into the lung surfactant (Postlethwait et al., 1994), Mumaw and colleagues observed increased microglia activation in the brains of exposed rats; additionally, serum obtained postexposure initiated an inflammatory cascade in cultured microglia (Mumaw et al., 2016). Pulmonary exposure to multiwalled carbon nanotubes similarly generated circulating factors that induced inflammatory responses in cultured cerebrovascular endothelial cells and inhibited vasodilation in ex vivo vascular preparations; indeed, activation of microglia and astrocytes postexposure was contingent upon depletion of blood–brain barrier (BBB) function (Aragon et al., 2017). More recently, Ganguly and colleagues compared inhalation of carbon black particulate matter with intravenous infusion and found that inhalation induced a greater systemic inflammatory response, again indicating that indirect responses arising from the lung may have greater biological consequences than direct effects of systemically translocated particulate matter (Ganguly et al., 2017).
The BBB consists of the nonfenestrated brain endothelial cells contouring the vasculature of the CNS, the astrocytic end feet surrounding this vasculature, neurons, pericytes, and a host of proteins all of which control the flow of nutrients, hormones, and toxins between the brain and circulation (Haddad-Tóvolli et al., 2017). Trafficking of leukocytes is heavily regulated by the BBB, yet despite the “immune privilege” of the brain, surveillance by peripheral immune cells occurs, particularly during disease (Engelhardt et al., 2017). Systemic immune activation correlates with neuroinflammation and can perpetuate chronic inflammation in the brain. Conversely, neurodegenerative outcomes, including the accumulation of β-amyloid plaques (Aβ) and tau proteins, induce innate immune activation. This diverts microglia, the resident immune cells of the brain, from their beneficial physiological function toward an adverse phenotype, better known as reactive gliosis, resulting in proneuroinflammatory niches (Burda and Sofroniew, 2014). Studies reveal that chronic neuroinflammation itself may underlie, or at least potentiate, neurodegenerative outcomes (Ransohoff, 2016 b). The potential role of environmental stressors in this relationship remains an underappreciated contributor to neurodegenerative disease states (Jayaraj et al., 2017).
As the population ages, the prevalence of neurodegenerative disorders increases (Niccoli and Partridge, 2012); yet the extent to which the aging population is more vulnerable to neural insult and induction of neuroinflammation upon exposure to environmental pollutants is relatively unexplored. A recent meta-analysis revealed that in all assessed studies, save one, a correlation was found between increased exposure to at least one type of air pollutant and decreased cognition or a dementia-related outcome (Power et al., 2016). As the brain ages, the permeability of the BBB increases, likely due to reduced endothelial cell integrity and vasculature remodeling (Lee et al., 2012; Montagne et al., 2015); indeed, normal changes in vasculature during the aging process are exaggerated upon exposure to air pollutants. This exposure also induces neuroinflammation that appears dependent on an initial insult to the BBB (Aragon et al., 2017; Mumaw et al., 2016; Tyler et al., 2016). As such, the elderly population may be at greater risk for neurological outcomes induced by neuroinflammation in response to air pollution exposure, simply because they have increased baseline BBB permeability.
The purpose of this study was to evaluate the effect of acute exposure to air pollution on brain permeability, neuroinflammation, and neurodegenerative outcomes in aged animals. We hypothesized that aged animals, with increased BBB permeability compared with young animals, would have a greater neuroinflammatory response, even to one acute dose of environmentally relevant O3 exposure.
MATERIALS AND METHODS
O3 exposure paradigm
All procedures were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of New Mexico. Animals were maintained in a 22°C, AAALAC-approved vivarium on a 12-h light/dark cycle with ad libitum access to chow and water. Adult male C57BL/6 mice (age-matched from Taconic Labs and Jackson Labs) were group housed and allowed to age either 8–10 weeks or 12–18 months. The resulting average age for assessment of the “aged” cohort was 431 ± 66 days. Animals derived from the 2 different vendors were randomized throughout the study design into groups for exposure to either filtered air (FA) or 1 ppm O3 via whole body inhalation for 4 h from 08:00 to 12:00 h (Robertson et al., 2013). O3 was generated using an OREC silent arc discharge O3 generator (Osmonics, Phoenix, AZ) and monitored using a photometric O3 analyzer (TG-501, GrayWolf, Shelton, CT). Food was withheld during the 4 h exposure to preclude ingestion of O3-derived byproducts, and mice were exposed without bedding to ensure optimal delivery of O3. Animals were euthanized 20 h postexposure at 08:00 h the following day.
Flow cytometry analysis of BBB permeability and immune activation
All flow cytometry analyses were performed on tissues isolated 20 h post-O3 exposure. Animals were transcardially perfused with ice-cold phosphate-buffered saline (PBS; pH 7.4) to remove circulating immune cells from blood vessels. The cerebellum was separated from the rest of the brain and the spleen was removed. Tissues were digested according to the manufacturer’s instructions using the Miltenyi gentleMACS protocol to obtain a single-cell suspension as previously described (Noor et al., 2017). Briefly, tissue was minced and digested with 10 mg/ml deoxyribonuclease-1, DNase-1 (Sigma-Aldrich, St. Louis, MO), and 100 mg/ml collagenase (Roche Diagnostics, Indianapolis, IN) in C-tubes (Miltenyi Biotec, San Diego, CA) at 37°C under continuous rotation. Samples were dissociated using a gentleMACS Dissociator (Miltenyi Biotec); following dissociation, cells were passed through a 40 μm cell strainer (Corning sterile cell strainers, Fisher Scientific) and incubated with magnetic myelin removal beads (Miltenyi Biotec) for 15 min at 4°C. Cells were then passed through MACS LS columns adjacent to MACS Separator magnets (Miltenyi Biotec) to remove myelin and enrich the population of microglia and infiltrating immune cells. After counting, between 0.2 and 1 × 106 cells were pelleted in a FACS tube (BD Falcon, MA) and stained with Viability Dye eFluor 450 (eBioscience, San Diego, CA) for 30 min at 4°C. After washing with FACS buffer (PBS containing 1.0% bovine serum albumin and 1 mM EDTA), cells were preincubated with Fc block (BD Biosciences, San Jose, CA) for 10 min and then incubated with fluorochrome-conjugated antimouse antibodies for 30 min at 4°C. Approximately 0.125–0.5 μg per 106 cells of the following antibodies were used: CD45, F4/80, 1A8, MHCII, Ly6C (eBioscience), and CD11b (BD Biosciences). After staining, cells were washed and resuspended in FACS buffer prior to analysis. Approximately 50 000 events were collected for each sample on a BD LSR Fortessa Cytometer (BD Biosciences). Single stain and isotype controls were used for laser compensation. Data were analyzed using FlowJo software v.8.7.4. The same gating was applied to all samples regardless of age or condition.
Flow cytometry gating strategy
Figures 1A–G demonstrate the gating strategy for determining different cell subsets in the brain tissues. Briefly, the live cells were identified using forward scatter (FSC), side scatter (SSC), and negative viability dye staining (Figs. 1A–D) in combination with positive CD45 expression (Figure 1E). The subpopulation of neutrophils within the leukocyte population was determined using 1A8 expression (neutrophil marker) and CD11b expression (Figure 1F). The population of 1A8−CD11b+ cells (all leukocytes that are not neutrophils) was analyzed for F4/80 and CD45 expression. Identification of infiltrating monocytes/macrophages was determined based on 1A8−CD11b+F4/80+CD45high expression as distinguished from microglia with 1A8−CD11b+F4/80+CD45low expression (Figure 1G). Median fluorescence intensities (MFIs) were determined for CD11b, F4/80, and MHCII expression on microglia in either the cerebellum or rest of brain (ROB) where n = 3 mice per group. Additionally, infiltrating monocytes/macrophages were further analyzed for positive Ly6C expression to identify inflammatory monocytes (1A8−CD11b+F4/80+CD45high Ly6C+).
Figure 1.

Gating strategy for determining the population of infiltrating, live leukocytes. Representative flow cytometry plots demonstrate the gating strategy used for analysis of immune cell percentages and median fluorescence intensities (MFIs). A, Potential live cells were gated based on their light scatter properties: side scatter-area (SSC-A) versus forward scatter-area (FSC-A). Doublets were removed from this population by gating on (B) forward scatter-width (FSC-W) versus forward scatter-height (FSC-H) and (C) side scatter-width versus side scatter-height (SSC-W versus SSC-H). Dead cells were excluded from the single-cell population (C), as demonstrated in (D) showing FSC-A versus viability dye staining. The cell population identified from A–D gating was then assessed for (E) positive CD45 staining, representing live leukocytes. This subset was used to compare (F) 1A8 versus CD11b expression, with CD45+CD11b+1A8+ denoting the neutrophil population and CD45+1A8C−CD11b+ denoting the CD11b+ population (subset, F). This population was then analyzed for (G) F4/80 versus CD45 expression, where 1A8−CD11b+F4/80+CD45low denotes microglia and 1A8−CD11b+F4/80+CD45low denotes macrophage/monocytes. MFIs were determined for CD11b, F4/80, and MHCII expression on microglia in either the cerebellum or brain where n = 3 mice per group.
Immunohistochemistry for analysis of the BBB
BBB permeability was assessed using the injectable tracer, sodium fluorescein (FSCN), as previously described (Aragon et al., 2017). Briefly, animals were intraperitoneally injected with 100 μl of 2% (wt/vol) FSCN (Sigma Aldrich) 20-h post-O3 exposure. One hour following injection, animals were euthanized and transcardially perfused with 0.9% (wt/vol) NaCl (saline solution), followed by 4% paraformaldehyde. Brains were removed and fixed at 4°C in 4% paraformaldehyde for 48 h and transferred to a 30% (wt/vol) sucrose solution. Following cryoprotection in sucrose, brains were rinsed in 1× PBS and frozen in OCT (optimal cutting temperature compound). Sagittal sections (25 μm, 50 μm, or 100 μm) were collected with a cryostat and stored at −20°C until use. For BBB analysis, sections were washed with 1× PBS and stained with DAPI (4′,6-diamidino-2-phenylindole; 1:1000; Life Technologies, D3571) to label cell nuclei. Sections were imaged on a Zeiss AxioPlan2 Microscope with Nuance Multi-Spectral Camera using 20× objective, as previously described (Aragon et al., 2017). Briefly, in 3 separate tissue sections, 2–3 images per region, including the brainstem, cerebellum, frontal cortex, dentate gyrus, hippocampus (CA1 area), and hypothalamus, were obtained per animal. Images were analyzed using ImageJ software for total fluorescence of FSCN as a measure of BBB permeability. For analysis of reactive gliosis of microglia, 25 μm sections were permeabilized with PBS solution containing 1% BSA, 5% (vol/vol) serum, and 0.2% Triton X-100 for 2 h at room temperature (RT). Sections were incubated with anti-calcium binding adapter molecule 1 (Iba-1; 1:500, WAKO, 019-19741) overnight at 4°C. Sections were washed and treated with a secondary antibody, Alexa Fluor 555 (1:1000; Life Technologies), for 2 h at RT, and nuclei were stained with DAPI. Imaging analysis was performed for Iba-1 as for FSCN, with total fluorescence quantification of Iba-1 to indicate the presence of microglia in ImageJ software. Using sections stained with Iba-1, microglia morphology was assessed as previously described (Aragon et al., 2017). Briefly, the soma size and territory occupied by an individual cell, as determined by the perimeter created from the microglia processes, was assessed using ImageJ software. Data are presented as an activation index, or the ratio of soma size over total territory occupied. This ratio should increase as microglia become reactive: soma size increases while territory decreases. The number of microglia was assessed by counting the number of soma present in representative images of Iba-1 stained microglia in each brain region.
For confocal microscopic visualization of the neurovascular units, 50 μm sections were permeabilized and stained overnight at 4°C with rabbit anti-platelet endothelial cell adhesion molecule-1 (PECAM-1; 1:100; Santa Cruz, sc-1506-R) to visualize vasculature. The following day, sections were stained with the secondary antibody, Alexa Fluor 555 (1:1000; Life Technologies) and the conjugated glial fibrillary acidic protein (GFAP)-Alexa Fluor 647 antibody (1:500, Biolegend, 644706) to visualize astrocytes for 2 h at RT. DAPI was applied to label cell nuclei. Images were captured using a Zeiss LSM800 Airyscan confocal microscope at 63× with appropriate compensations for visualizing FSCN, PECAM, GFAP, and DAPI simultaneously.
Preparation of brain tissue for protein expression analysis
Frozen cortical/limbic tissue was weighed and manually homogenized in tissue homogenization buffer containing 2 mM Tris (pH 7.4), 250 mM sucrose, 0.5 mM EDTA, and 0.5 mM EGTA with 1:100 protease inhibitor cocktail (Sigma, P8340) using the Biomasher II disposable microhomogenizer (Kimble Chase, Rochester, NY). Homogenates were mixed 1:1 with 0.4% diethylamine (DEA) solution and centrifuged at 150 000 × g for 1 h at 4°C. The supernatant was neutralized 1:10 with 0.5 M Tris–HCl (pH 6.8). Protein concentrations were determined using spectrophotometric absorbance at 280 nm. Homogenates were snap frozen in liquid nitrogen and stored at –80°C until use.
β-Amyloid protein expression analysis in brain via ELISA
Soluble toxic Aβ42 was assessed in cortical tissue samples via ELISA with the human/rat β-amyloid (42) ELISA kit (Wako Pure Chemical Industries, 290-62601, Richmond, VA), per manufacturer instructions. Briefly, after overnight incubation of tissue samples and standards, the microplate was washed ×5 and subsequently incubated with horseradish peroxidase (HRP)-conjugated antibody for 1 h at 4°C. Wells were washed ×5 again and 3,3′,5,5′-tetramethylbenzidine (TMB) solution was added to initiate the HRP reaction. The reaction was terminated after 30 min and absorbance at 450 nm for each well was determined using a Tecan plate reader. A standard curve from known Aβ42 protein concentrations was generated; concentrations for each sample were determined and converted to pg Aβ42/mg cortical tissue loaded by correcting for dilution factors and variable protein concentrations per sample.
Statistics
Statistical analyses were conducted on GraphPad Prism 6 Software, version 6.03 (GraphPad Software, San Diego, CA). For assays where O3 exposure effects were generated in both young and old mice, a 2-factor analysis of variance (ANOVA) with Bonferroni correction for group differences was applied. For certain assays where only aged mice were used, a Student’s t test was used. p values less than .05 were considered significant.
RESULTS
Acute O3 Exposure Induces Peripheral Leukocyte Infiltration in the Brain in an Age-Dependent and Region-Specific Manner
Assessment of the infiltrating population of immune cells was separated into 3 categories: neutrophils, macrophages/monocytes, and Ly6C+ inflammatory monocytes. For these 3 subcategories, we found a main effect of acute O3 exposure on the population of infiltrating peripheral immune cells but not resident immune cells in the aged cerebellum, with no significant effect observed in the young cerebellum. Figures 2A–2C demonstrate a 333% increase in the percentage of neutrophils from 0.68% to 2.82% of the total CD11b+ subset of live leukocytes with 1A8high expression for the aged cerebellum after acute O3 exposure. Two-way ANOVA analysis of the neutrophil population demonstrates a main effect of O3 exposure (p < .01) but no main effect of age and no interaction. Multiple comparison analyses with Bonferroni correction revealed a significant difference (p < .05) between the aged FA and aged O3 exposure groups (Figure 2C). A slightly different pattern was observed for the population of infiltrating macrophages/monocytes in the cerebellum: 2-way ANOVA statistical analysis suggests a significant main effect of age (p < .01) and O3 exposure (p < .05), though no significant interaction (p = .2483). Figures 2D and 2E demonstrate a 72% increase in the percentage of macrophages/monocytes, from 5.24% to 9.03% of the total CD11b+ subset of live leukocytes with 1A8−CD45high expression for aged animals with acute O3 exposure. Multiple comparison analyses with Bonferroni correction revealed a significant difference (p < .05) in infiltrating macrophages/monocytes between the aged FA and aged O3 exposure groups (Figure 2F) and a significant difference (p < .01) between young and aged animals with acute O3 exposure (Table 1).
Figure 2.

Acute ozone (O3) exposure induces peripheral leukocyte infiltration in the cerebellum in aged animals. A, Representative flow cytometry dot plots comparing 1A8 (neutrophil marker) and CD11b expression in the aged cerebellum; the neutrophil population was determined from the live leukocyte population and was denoted as CD45+1A8+CD11b+. B, Effects of O3 exposure on the neutrophil population in the aged cerebellum compared with filtered air (FA) aged controls. C, Acute O3 exposure significantly increased the neutrophil population only in the aged cerebellum. D, Expression of F4/80 and CD45 in the aged cerebellum; the macrophage/monocyte population was denoted as CD45high1A8−CD11b+F4/80+ distinct from the microglia population, which was denoted as CD45low1A8−CD11b+F4/80+. E, Effects of acute O3 exposure on the peripheral macrophage/monocyte population in the aged cerebellum. F, O3 exposure significantly increased the macrophage/monocyte population in the aged cerebellum. The aged cerebellum exhibited significantly increased macrophage/monocyte population compared with the young cerebellum after acute O3 exposure. G, CD45 and Ly6C expression in the aged cerebellum; the inflammatory monocyte population was denoted as CD11b+F4/80+CD45highLy6C+. H, Effects of O3 exposure on the inflammatory monocyte population in the aged cerebellum. I, Acute O3 exposure significantly increased the inflammatory monocyte population in the aged cerebellum (n = 3 per group, *p < .05; **p < .01; ***p < .001; ****p < .0001 by 2-way analysis of variance (ANOVA), all values represent mean ± SEM).
Table 1.
Flow Cytometry Markers for Immune Cells of Interest
| Cell type | CD45 | CD11b | F4/80 | MHCII | 1A8 | Ly6C | Nomenclature |
|---|---|---|---|---|---|---|---|
| Neutrophils | + | + | + | +/− | + | − | CD45+1A8+CD11b+ |
| Microglia | + | + | + | + | − | − | CD45low1A8−CD11b+F4/80+ |
| Activated microglia | + | High | High | High | − | − | CD45lowCD11b+F4/80highMHCIIhigh |
| Macrophages/monocytes | High | + | + | + | − | − | 1A8−CD11b+F4/80+CD45high |
| Infiltrating monocytes | + | + | + | − | − | + | CD11b+F4/80+CD45+Ly6C+ |
The macrophage/monocyte subpopulation was further analyzed for Ly6C expression, a marker for circulating inflammatory monocytes in the blood. Figures 2G and 2H demonstrate a 44.3% increase in the population of inflammatory monocytes (CD11b+CD45highLy6C+) in the aged cerebellum after acute O3 exposure. Two-way ANOVA statistical analysis of the inflammatory monocyte population demonstrates a main effect of O3 exposure (p < .05) but no main effect of age and no interaction. A significant difference (p < .05) in infiltrating inflammatory monocytes was found between aged FA and aged O3 exposure groups (Figure 2I). Significant monocyte infiltration in response to acute O3 exposure was not observed for total brain tissue (total brain without cerebellum and brain stem, referred to as “brain”); data are not shown due to lack of statistical significance. However, region specificity must be considered when determining the impacts of inhaled toxicants on peripheral immune infiltration in the brain. As such, it is likely that regional heterogeneity impacted gross flow cytometric analysis.
The Cerebellar Microglia Population is Sensitive to Acute O3 Exposure in Aged Animals
We identified the live leukocyte subpopulation with the following expression pattern as microglia: 1A8−CD11b+F4/80+CD45low (see Figure 1 for gating strategy). MFIs were determined for CD11b, F4/80, and MHCII expression on microglia in either the cerebellum or ROB (brain) where n = 3 mice per group. Based on this expression pattern, we observed an increase in CD11b MFI, indicative of increased microglial “activation” as a function of both age and in response to O3 exposure (Figure 3A). Two-way ANOVA analysis demonstrates a significant main effect of age (p < .01) and O3 exposure (p < .0001) with no interaction. Multiple comparison analyses with Bonferroni correction revealed a significant difference in the young versus aged cerebellum microglia population (p < .05), a significant effect of O3 in the young cerebellum (p < .001), and a significant effect of O3 in the aged cerebellum (p < .05) as seen in Figure 3A.
Figure 3.

Acute O3 exposure increases primed microglia in the aged cerebellum. The microglia population was further analyzed for CD11b, F4/80, and MHCII expression levels, as measured indicative of their “activation” phenotype, or reactive gliosis. A, Bar graph illustrating the CD11b mean fluorescence intensity (MFI) of microglia in the cerebellum. Age significantly increases this population and acute O3 exposure increases this population in both young and aged cerebellar tissue. B, Bar graph illustrating the F4/80 MFI of the microglia population indicative of reactive gliosis or “priming” of microglia in the cerebellum. Age significantly increases this population, and acute O3 exposure increases this population only in the aged cerebellum. F4/80 expression in the aged cerebellum after acute O3 exposure is significantly higher than in the young cerebellum after exposure. C, Bar graph illustrating the MHCII MFI of the microglia population indicative of reactive gliosis or “priming” of microglia in the cerebellum. Age significantly increases this population, and acute O3 exposure increases this population only in the aged cerebellum. MHCII expression in the aged cerebellum after acute O3 exposure is significantly higher than in the young cerebellum after exposure (n = 3 per group, *p < .05; **p < .01; ***p < .001; ****p < .0001 by 2-way ANOVA; all values represent mean ± SEM).
The current state of research focusing on neuroinflammation has reconceived the notion of “activated” microglia; M1 and M2 phenotypes are now considered archaic nomenclature for assigning functionality to these cells (Burda and Sofroniew, 2014; Ransohoff, 2016a). However, microglia cells that express high F4/80 or MHCII are “reactive” in that they exhibit larger soma, shortened arborizations, and enhanced phagocytic action, characteristics that are antithetical to their normal positive physiological function (Greter et al., 2015). Thus, to determine if microglia cell present in the brain are “reactive” or exhibit a phagocytic phenotype that underlies characteristic neuroinflammation, we assessed expression of F4/80 and MHCII. It is well understood that the presence of these membrane-bound proteins on microglia is associated with an inflammatory niche within the brain (Greter et al., 2015). Two-way ANOVA analysis of F4/80 expression demonstrates a significant effect of both age (p < .05) and acute O3 exposure (p < .001) with no significant interaction. F4/80 expression on CD11b+ microglia cells increases with age (p < .01). Acute O3 exposure only increases F4/80 expression in the aged cerebellum (p < .05), whereas aged animals have increased reactive microglia with more F4/80 compared with young animals after acute O3 exposure (p < .001) (Figure 3B). The same pattern was observed for MHCII expression on CD11b+ microglia cells: 2-way ANOVA analysis demonstrates a significant effect of age (p < .01) and O3 exposure (p < .001) with no interaction. MHCII expression increases with age (p < .05) and after acute O3 expression in the aged cerebellum (p < .05) (Figure 3B). Aged animals have increased reactive microglia with more MHCII compared with young animals after acute O3 exposure (p < .01).
The Microglia Population in Aged Animals is More Sensitive to Acute O3 Exposure in the Brain
Although we did not observe an effect of acute O3 exposure on infiltrating immune cells in the brain (cerebellum and brainstem removed, data not shown), we did observe an effect on the microglia. Two-way ANOVA analysis indicated a significant effect of acute O3 exposure (p < .0001), a significant effect of age (p < .01), and a significant interaction (p < .05), on CD11b expression (Figure 4A). This suggests that acute O3 exposure increases reactive microgliosis in the brain dependent on age, with more severe effects occurring in the aged brain. Posthoc analysis with Bonferonni correction confirms an increase in microglia reactivity in the young brain (p < .001) and in the aged brain (p < .0001) after acute O3 exposure. Aged animals have a greater reactive microgliosis population in the brain compared with young animals after acute O3 exposure (p < .01). Data are shown in Figure 4A. As with the analysis for cerebellum tissue, we confirmed this apparent reactive gliosis of microglia by assessing F4/80 and MHCII expression. Two-way ANOVA analysis of F4/80 expression demonstrates a significant effect of both age (p < .0001) and O3 exposure (p < .01) with a significant interaction (p < .001). However, as observed in the cerebellum, posthoc analysis suggests that acute O3 exposure increases F4/80 expression only in the aged brain (p < .001), although aged brains have more F4/80 expression on microglia than young brains (p < .0001) without any exposure. Aged animals have increased reactive microglia with more F4/80 in the brain compared with young animals after acute O3 exposure (p < .0001). Data are shown in Figure 4B. Interestingly, 2-way ANOVA analysis demonstrates a significant effect of age (p < .0001) on MHCII expression on microglial cells in the brain, but no effect of acute O3 exposure at either time point, suggesting a ceiling effect of age on microglia MHCII expression (Figure 4C). However, aged animals have increased reactive microglia with more MHCII in the brain compared with young animals after acute O3 exposure (p < .0001).
Figure 4.

Acute O3 exposure exacerbates the effect of age on primed microglia. A, Bar graph illustrating the CD11b MFI indicative of microglia in the brain (sans cerebellum and brain stem). Age does not significantly increase this population; acute O3 exposure increases microglia in both the young and aged brain. The microglia population in the aged brain after acute O3 exposure is significantly higher than in the young brain after exposure. B, Bar graph illustrating the F4/80 MFI of the microglia population indicative of reactive gliosis or “priming” of microglia in the brain. Age significantly increases this population, and acute O3 exposure increases this population only in the aged brain. F4/80 expression in the aged brain after acute O3 exposure is significantly higher than in the young brain after exposure. C, MHCII MFI of the microglia population indicative of reactive gliosis or “priming” of microglia in the cerebellum. Age significantly increased this population. Acute O3 exposure does not change this population in the young or aged brain, yet microglia in the aged brain exhibit significantly greater MHCII expression after O3 exposure than microglia in young brains after exposure (n = 3 per group, *p < .05; **p < .01; ***p < .001; ****p < .0001 by 2-way ANOVA; all values represent mean ± SEM).
Microscopy Imaging Analyses of the BBB in Response to Acute O3 Exposure
These flow cytometry data suggest a discrepancy in response to acute O3 exposure based on region. Age alone did not impact the infiltrating immune cells in the brain, but aged animals had an exacerbated response to acute O3 exposure, particularly with respect to reactive microglia. Thus, we assessed the permeability of the BBB and the potentially reactive microglia using immunohistochemistry to determine regional differences. Aged male C57BL/6 mice were injected with a FSCN tracer 20-h postacute O3 exposure (or post-FA exposure). Immunohistochemical analysis of BBB permeability was assessed as the total fluorescence of FSCN that was able to penetrate the vasculature and leak into the brain. Figure 5A shows a representative image of the BBB in the hypothalamus taken at ×63 in an aged animal. The vasculature (red, PECAM) and FSCN dye (green) overlap in the merged image denoting leakage of the dye through the barrier. Figure 5B shows a representative image of the BBB in the hypothalamus taken at ×63 in an aged animal, demonstrating the potential overlap between FSCN dye penetrating the barrier and astrocyte localization around the vasculature (merged image, white). This merged image is provided in greater detail in Figure 5C. Figure 5D demonstrates the impact on acute O3 exposure in the aged hypothalamus, particularly when compared with the aged brain (FA) in Figure 5C. These images suggest potential uptake or overlap of the FSCN dye with astrocytes after acute O3 exposure; further evidence of this is demonstrated in Figures 5E and 5F in the dentate gyrus of the hippocampus. These images suggested the necessity of assessing FSCN alone: indeed, Figures 6A and 6B demonstrate better visualization of FSCN in the aged cortex with and without acute O3 exposure, again suggesting a substantial impact of O3 exposure. Further, attempting to separate the astrocyte and microglia contribution to BBB integrity was difficult, as seen in Figure 6C, particularly after acute O3 exposure (Figure 6D). Thus, we determined that separate quantitative analyses of FSCN and microglia fluorescence were necessary.
Figure 5.

Microscopy reveals overlapping blood–brain barrier (BBB) impairment and astrocyte activation at the neurovascular unit following acute O3 exposure. A, Representative image of the BBB in the hypothalamus of an aged, O3-exposed brain taken at ×63; PECAM (red) indicates vasculature, FSCN (green) indicates FSCN dye penetration into the brain parenchyma, DAPI (blue) indicates nuclear staining, and the Merge (white) shows overlap indicative of FSCN leakage from the vessels (overlap of PECAM and FSCN). B, Representative image of the BBB and astrocyte activation in the hypothalamus of an aged, FA-exposed animal taken with a ×63 objective; GFAP (purple) indicates astrocytes that are co-localized with the FSCN dye (FSCN, green) and vasculature (PECAM, red) in the Merge image (white). C, The Merge image from (B) in greater detail (×100). D, Representative Merge image of the aged hypothalamus after acute O3 exposure. E, Representative Merge image of the aged dentate gyrus at ×100. F, Representative Merge image of the aged dentate gyrus after acute O3 exposure at ×100. Potential uptake or overlap of the FSCN and GFAP (white) is demonstrated by (C)–(F).
Figure 6.

BBB permeability and reactive gliosis must be analyzed separately in the aged brain, particularly after acute O3 exposure. A, Representative Merge image of FSCN penetration in the aged cortex, where DAPI indicates nuclear staining. B, Representative Merge image of FSCN penetration in the aged cortex after acute O3 exposure. C, Representative Merge image of aged cortex labeling microglia (Iba-1), FSCN, and astrocytes (GFAP). D, After acute O3 exposure. Delineating the specific astrocyte and microglia contribution to BBB integrity was difficult, particularly after acute O3 exposure. Thus, separate quantitative analyses of FSCN dye penetration and microglia fluorescence was performed. All images were taken with a ×20 objective on a Zeiss LSM800 Airyscan.
The BBB is Highly Permeable in Aged Animals After Acute O3 Exposure in a Region-Specific Manner
Flow cytometry assessment suggested regional differences in susceptibility to acute O3 exposure; to further delve into this potential discrepancy, we quantified BBB permeability in the following regions: brainstem, cerebellum, dentate gyrus, frontal cortex, hippocampus (CA1), and hypothalamus. Each of these regions is either (1) very sensitive to insult or (2) has differential permeability of the BBB compared with other regions. For example, the BBB in the hypothalamus is highly permeable due to the nature of hypothalamic functionality in controlling central and peripheral hormonal signaling (Haddad-Tóvolli et al., 2017). Regional differences in vasculature density, and thus BBB permeability, occur in the hippocampus, thus the necessity for comparing the CA1 region to the dentate gyrus (Wilhelm et al., 2016). Neurodegenerative disorders adversely impact microvessel density, particularly in limbic and cortical regions, whereas contradicting neuroinflammatory and BBB permeability responses to pathogens and toxins occur in the cerebellum and cortex in mouse models (Fabis et al., 2008; Janota et al., 2015; Phares et al., 2006). Based on these findings, we have assessed BBB permeability and reactive gliosis in these particular regions of interest.
Acute O3 Exposure Increases BBB Permeability and the Reactive Microglia Population in Cortical and Limbic Brain Regions of Aged Animals
Representative images of FSCN penetration and microglia (Iba-1) in the aged cortex after FA (Figure 7A) and acute O3 exposure (Figure 7C) are shown in greyscale for better clarity. (Sections were imaged on a Zeiss AxioPlan2 Microscope with Nuance Multi-Spectral Camera.) Acute O3 exposure increased BBB permeability (p < .001) (Figs. 7A, 7C, and 7E) and microglia fluorescence (number and reactivity) (p < .01) in the frontal cortex of aged animals (Figs. 7B, 7D, and 7F). This same pattern of increased permeability and concurrent reactive gliosis after acute O3 exposure was found in the structures of the limbic system, which are particularly susceptible to neurodegeneration (Burgmans et al., 2011; Echávarri et al., 2011; Stoub et al., 2012). In the dentate gyrus of aged animals, acute O3 exposure increased both BBB permeability (Figure 8A, p < .05) and the reactive microglia population (Figure 8B, p < .01). The dentate gyrus harbors an adult stem cell niche that is particularly sensitive to extrinsic stimuli (whether positive or negative). Thus, we assessed the BBB permeability in another main hippocampal area, CA1. Acute O3 exposure increased the BBB permeability in the aged hippocampus (Figure 8C, p < .001) and increased microglia fluorescence (Figure 8D, p < .01). The hypothalamus has a specialized BBB, allowing for passage of hormone and nutrients for regulation and maintenance of endocrine responses and feeding behavior (Haddad-Tóvolli et al., 2017). We found increased BBB permeability after acute O3 exposure in the aged hypothalamus (Figure 8E, p < .001) and increased microglia fluorescence (Figure 8F, p < .01). Representative images of FSCN penetration and microglia fluorescence are provided in the Supplementary data for each of these structures.
Figure 7.

Acute O3 exposure increases the permeability of the BBB and induces reactive gliosis in cortex of aged animals. A, Representative image of BBB permeability as determined by FSCN penetration in the aged cortex. B, Representative image of microglia (Iba1) in the aged cortex. C, Acute O3 exposure increases FSCN fluorescence at the neurovascular unit and (D) Iba-1 fluorescence in the aged cortex. E, Bar graph depicting FSCN fluorescence quantification in animals exposed to FA (Aged FA) or acute O3 (Aged O3); acute O3 exposure increases fluorescence indicative of increased BBB permeability in the aged cortex (p < .001). F, Bar graph depicting Iba-1 fluorescence quantification in animals exposed to FA (Aged FA) or acute O3 (Aged O3); acute O3 exposure increases fluorescence indicative of reactive gliosis (neuroinflammation) in the aged cortex, (p < .01). Brain sections were imaged on a Zeiss AxioPlan2 Microscope with Nuance Multi-Spectral Camera, allowing for total fluorescence analysis (n = 6 per group, **p < .01; ***p < .001 by Student’s t test; all values represent mean ± SEM).
Figure 8.

Limbic structures of the aged brain are susceptible to increased BBB permeability and reactive gliosis after acute O3 exposure. A, In the aged dentate gyrus, acute O3 exposure increased BBB permeability, as determined by FSCN fluorescence, and (B) induces reactive gliosis, or microglia reactivity (neuroinflammation), as determined by Iba-1 fluorescence. C, In the aged hippocampus (CA1 area), acute O3 exposure increases BBB permeability and (D) induces reactive gliosis. E, In the aged hypothalamus, acute O3 exposure increases BBB permeability, and (F) induces reactive gliosis. Representative images of FSCN penetration and microglia fluorescence are provided the Supplementary data for each of these structures (n = 6 per group and 3 images per region, **p < .01; ***p < .001 by Student’s t test; all values represent mean ± SEM).
Acute O3 Exposure Does Not Alter BBB Permeability or Induce Reactive Microglia in the Brainstem or Cerebellum of Aged Animals
Acute O3 exposure did not significantly impact BBB permeability or microglia fluorescence in the brainstem or cerebellum in aged animals (data provided in the Supplementary Figs.). It should be noted that lobules IV and V of the cerebellum were assessed for fluorescence intensity. Potential reasons for the discrepancy in BBB permeability as determined by immunohistochemistry versus flow cytometry are discussed below.
Acute O3 Exposure Alters Microglia Number and Morphology in Cortical and Limbic Regions of the Aged Brain
Alterations in microglia morphology were quantitated using an activation index, determined by the ratio of soma size to territory occupied by an individual cell. Acute O3 exposure significantly increased the microglia activation index in the frontal cortex of aged animals (Figure 9A, p < .0001). These morphological changes were accompanied by an increase in total microglia number in the same region (Figure 9B, p < .05). Acute O3 exposure increased the microglia activation index in other limbic structures, including the dentate gyrus (Figure 9C, p < .01), the hippocampus (Figure 9E, p < .01), and the hypothalamus (Figure 9G, p < .0001). The number of microglia was also increased after acute O3 exposure in the dentate gyrus (Figure 9D, p < .001), the hippocampus (Figure 9F, p < .05), and the hypothalamus (Figure 9H, p < .01). Acute O3 exposure did not significantly alter the activation index nor the total number of microglia in the brainstem or cerebellum (Supplementary Figure 6).
Figure 9.
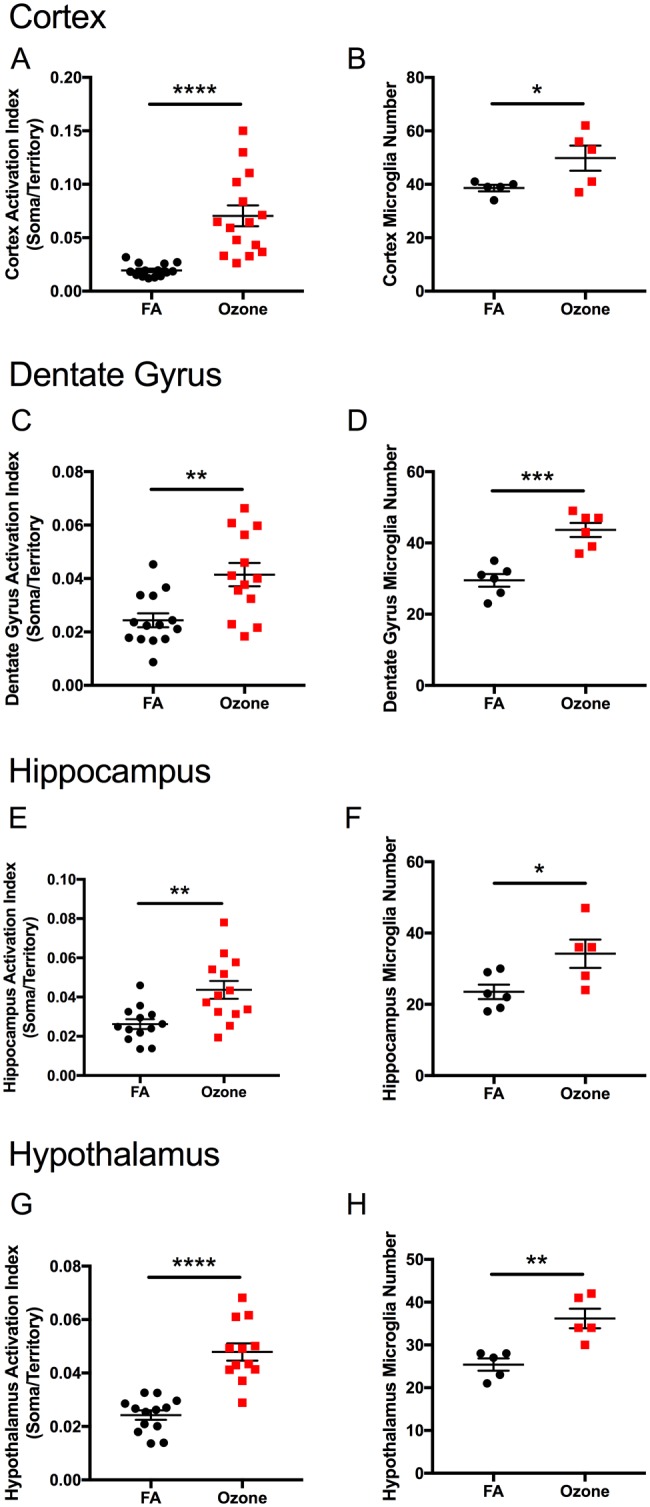
Acute O3 exposure alters microglia morphology and number in limbic structures of the aged brain. Acute O3 exposure significantly increases the activation index of microglia, as determined by the ratio of soma size to territory of processes in the aged (A) cortex, (C) dentate gyrus, (E) hippocampus, and (G) hypothalamus. Acute O3 exposure significantly increases reactive gliosis, including the number of microglia in the (B) cortex, (D) dentate gyrus, (F) hippocampus, and (H) hypothalamus (n = 11–15 individual cells per group and n = 5–6 images for total microglia quantification, *p < .05, **p < .01; ***p < .001, ****p < .0001 by Student’s t test; all values represent mean ± SEM).
Acute O3 Exposure Increased β-Amyloid Protein Expression in Cortical and Limbic Regions of the Aged Brain
We observed increased FSCN penetration of the BBB and increased microglia number and activation in the cortical and limbic regions of the brain (Figs 7–9). Both phenotypes underlie neuroinflammation, thought to be potentially causal in the onset of neurodegenerative disorders (Ransohoff, 2016 b). Thus, to determine the potential effect of acute O3 exposure on the development of neurodegeneration outcomes in these brain regions, we assessed β-amyloid protein expression in the aged brain. Acute O3 exposure increased β-amyloid protein expression in the brain of aged animals (Figure 10, p < .05).
Figure 10.
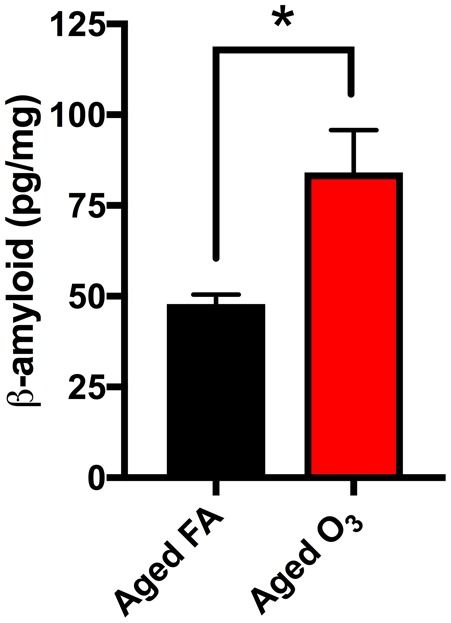
Acute O3 exposure increases β-amyloid protein expression in cortical and limbic regions of the aged brain. Increased β-amyloid protein accumulation in the brain was observed after acute O3 exposure (*p < .05 by Student’s t test; all values represent mean ± SEM).
DISCUSSION
In the present study, we hypothesized that the aged brain, due in part to increased BBB permeability, would exhibit vulnerability to neuroinflammatory outcomes of inhaled pollutants. To test the validity of this assertion, we optimized flow cytometry methodology for brain tissue, as previously described by Noor et al. (2017), to differentiate between populations of infiltrating leukocytes and resident microglia cells, including assessing the potential for reactive gliosis. O3 inhalation induced clear neuroinflammatory outcomes that were enhanced in mice of advanced age. Although O3 is certainly relevant as a ubiquitous, global air pollutant, its importance in the present study is also as a highly reactive gas that does not penetrate beyond the lung. As such, our results, demonstrating O3-induced BBB permeability and neuroinflammation in aged animals, provide important context for observed neuroinflammatory outcomes caused by pulmonary exposure to particulates, which have clear interactions in the lung, but may also translocate to the brain in limited quantities. Previous findings suggest that blood-borne factors arising from toxicant interactions in the lung may drive both BBB dysfunction and activation of microglia (Aragon et al., 2017; Mumaw et al., 2016), both of which may be influenced by aging-related changes in circulating factors (Villeda et al., 2011). Neuroinflammatory consequences following O3 exposure therefore strongly indicate that indirect pathways, stemming from pulmonary-derived circulating intermediate molecules, play a major role in driving neurological effects of inhaled pollutants.
Recent work demonstrates that exposure to many forms of air pollution, including particulate matter, induces hallmarks of neuroinflammation, notably increased proliferation and reactivity of microglia and astrocyte populations (Aragon et al., 2017; Levesque et al., 2011a, b; Mumaw et al., 2016; Oppenheim et al., 2013). This exposure-induced neuroinflammation has typically been reported in healthy, young research models. As neuroinflammation likely underlies the development of neurodegenerative disorders, understanding the contribution of environmental stressors, and their mechanisms of action, is of utmost importance (Jayaraj et al., 2017; Ransohoff, 2016 b). Despite the potential for increased BBB and neuroinflammation in older mice, recent studies of particulate matter impacts on neuronal atrophy in older animals has revealed a possible age-related ceiling effect (Woodward et al., 2017). Thus, it is perhaps an oversimplification to conclude that age confers neurological vulnerability to air pollutants, but evidence from the present study and others suggests a mechanistic pathway whereby circulating factors arising from the exposed lungs negatively impact BBB function and recruit neuro-supportive cells (astrocytes and microglia) to assist in protecting the brain. In doing so, the normal activities of microglia and astrocytes, such as aiding synaptic plasticity and removal of amyloid plaques, may be limited in the face of such environmental stressors. Although speculative, the recent findings of an age-ceiling effect in association with the present data of age-associated neuroinflammatory effects of O3, potentially suggest that young individuals may have diminished neuroinflammatory responses, but that their neuroplasticity and cognition is more sensitive to the modest BBB leakage indirectly induced by inhaled toxicants. Although we did not asses the perpetuation of O3-induced neuroinflammation, several studies report clear systemic physiological changes and some neuropathology in response to both acute and chronic exposure to O3 (Akhter et al., 2015; Gomez-Crisostomo et al., 2014; Thomson et al., 2013, 2016). Additionally, the potential compensatory molecular mechanisms combating O3-induced neuroinflammation in aged animals are the subject of further study.
Our flow cytometry analysis suggests that age alone does not increase the population of infiltrating peripheral immune cells in the brain. However, acute O3 exposure increased the populations of neutrophils, macrophages and/or monocytes, and inflammatory monocytes in the aged cerebellum presumably via permissive infiltration through an already vulnerable BBB. These populations were not significantly increased in the young cerebellum after acute O3 exposure, suggesting a discrepancy in response to an inhaled pollutant. We did not detect substantial effects of age or O3 exposure on the infiltration of immune cells for brain homogenate tissue (sans cerebellum and brain stem), although we may have been underpowered to detect smaller influences of aging. Selective permeability exacerbated by aging is dependent on region (Mooradian, 1988; Wilson et al., 2008); thus, we were not able to discern these regional differences using flow cytometry analysis on brain homogenate tissue. Potentially, the effects we observed would be more pronounced provided a larger pool of subjects.
Microglia cells, as part of innate immunity, respond to tissue insult, particularly BBB damage, with inflammatory actions that surpass their normal phagocytic behavior in supporting structure and function of the CNS (Streit et al., 2004). Under inflammatory conditions, microglia lose their “ramified” morphology, adopt an amoeboid shape, and increase their expression of Iba-1 (Greter et al., 2015). We assessed total Iba-1 fluorescence as a proxy for reactive gliosis, or “activated” microglia, indicative of neuroinflammation. Microglia fluorescence directly correlated with BBB permeability, as determined by FSCN penetration through the vasculature, in all brain regions. Microglia morphological assessment was quantitated as an activation index, derived from the ratio of the soma size to the territory covered by the microglia based on the perimeter from the most extensive processes. Acute O3 exposure increased all measures of reactive gliosis, including microglia fluorescence, proliferation, and the activation index (altered morphology) in cortical and limbic regions. Thus, acute O3 exposure concurrently increased BBB permeability and neuroinflammation in the cortex, dentate gyrus, hippocampus (CA1), and hypothalamus, but not in the cerebellum or brain stem of aged animals.
To further determine if microglia have a “reactive gliosis” phenotype simply due to age, we assessed the population of microglia and its expression of F4/80 and MHCII. As elevated CD45+ expression is present in both activated microglia and infiltrating monocytes, further gating on F4/80 expression was performed for positive identification of microglia (Figure 1). For total brain homogenate, the microglia population was not significantly increased by age; however, the microglia population of the cerebellum was increased in aged animals compared with young ones. In both cases, of the microglia present in the aged brain, a significant proportion of the population expressed more F4/80 and MHCII than microglia in the young brain, suggesting potential priming ahead of an insult (Hart et al., 2012; Norden and Godbout, 2013). Further, acute O3 exposure increased the population of microglia, in both young and aged brains, but significantly increased the F4/80 and MHCII expression only in the aged brain. Thus, acute O3 exposure not only increased the population of already primed microglia in the aged brain, but also further exacerbated their reactivity. Although acute O3 induced gliosis in the young brain, these microglia cells are likely not adopting a reactive, harmful phenotype, indicating potential resilience precluding damage by O3 exposure (Nikodemova et al., 2016). This resilience has been observed in other studies demonstrating that immune challenge induces greater neuroinflammatory outcomes in the aged versus young brain (Hart et al., 2012; Norden and Godbout, 2013). Given these data, the aged brain is potentially more vulnerable to insult and its resident immune cells, microglia, are primed such that insult drives a neuroinflammatory response, which is not observed in young animals.
The breakdown of BBB permeability may be an initiating mechanism of neurodegeneration eventually resulting in cognitive decline (Montagne et al., 2015). Additionally, infiltration of peripheral immune cells may induce neurodegenerative outcomes, including AD pathology (Prinz and Priller, 2017; Rezai-Zadeh et al., 2009). AD is a neurological disorder that results in severe memory deficits and is characterized by neuronal loss, increased accumulation of toxic isoforms of β-amyloid peptide and tau in brain parenchyma, and neurovascular dysfunction (Sagare et al., 2012). Acute exposure to O3 increased the Aβ1–42 peptide in aged animals, although we did not confirm the permanence of these effects. Some reports suggest that β-amyloid production is a protective response to foreign invaders, including viruses (Bourgade et al., 2016; Castellani et al., 2009). Potentially, the increase we observed was a compensatory response; however, increased accumulation of β-amyloid can induce neuroinflammatory responses including reactive gliosis of microglia. This reactivity can be both protective (in facilitating the elimination Aβ) or detrimental in the setting of a prolonged chronic neuroinflammatory state, contributing to neurodegenerative outcomes (Wang et al., 2015). Few reports show no change in Aβ accumulation after exposure to air pollution (Bhatt et al., 2015). Many studies, using either acute or chronic exposure to some type of inhaled pollutant, have reported an increase in toxic Aβ in the brain concurrent with memory impairment, though we did not assess functional or behavioral outcomes (Block and Calderón-Garcidueñas, 2009; Calderon-Garciduenas et al., 2016; Levesque et al., 2011a; Mumaw et al., 2016; Rivas-Arancibia et al., 2000). In aged animals, this increase, when coupled with primed microglia, likely induces a negative impact on the brain, promoting neuroinflammatory and neurodegenerative outcomes.
Current literature suggests that aging impairment of BBB permeability may vary substantially among individuals, potentially indicating a pathological mechanism that confers neurological vulnerability to environmental stressors such as air pollution (Gemechu and Bentivoglio, 2012). Enhanced BBB leakage may permit translocation of circulating molecules (proteins, lipids, metabolites) into the brain parenchyma, as well as permit greater recruitment of systemic immune cells. Further, a recent report demonstrated that normal aging disrupts tight junction proteins concurrent with BBB “leakage” but this does not result in increased leukocyte trafficking into the brain (Elahy et al., 2015). Thus, to quantify the discrepancies in brain region permeability, we assessed the translocation of a systemically-injected dye into specific brain regions in aged animals with and without acute O3 exposure. We found significant differences in BBB permeability, as measured by FSCN penetration in the brain, in cortical and limbic regions in aged animals after acute O3 exposure; however, as discussed above we did not find changes in permeability for these regions (collectively in homogenate brain tissue) using flow cytometry. Conversely, we found no significant differences in FSCN penetration in the brain in the cerebellum or brainstem, yet we measured increased leukocytes in the cerebellum using flow cytometry. These findings correlate with those of microglia populations. We found no significant changes in microglia fluorescence, proliferation, or morphology in the aged cerebellum after acute O3 exposure and yet, we did observe increases in the microglia population in the aged cerebellum compared with the young cerebellum. Potentially, this discrepancy in microscopy and cytometry data may be due to a ceiling effect: the cerebellar BBB has been shown to be more sensitive than the cortical BBB (Phares et al., 2006), thus visualizing a potentially increased fluorescence signal is not as sensitive as high-throughput single cell analysis. Changes in cerebellar-specific neuroinflammation, including cytokine production, can potentially be decoupled from neuropathology, suggesting another mechanism of action that requires further investigation (Hurley and Tizabi, 2013; Streit et al., 1998). Alternatively, regional differences in BBB permeability have been observed in other studies assessing viral clearance suggesting that low molecular weight markers (below 150 Da) are needed to observe cerebellar leakage after insult (Fabis et al., 2008). These data may be similar to a previous study showing regional BBB permeability without increased leukocyte trafficking (for cortical and limbic regions) or may underscore the importance of delineating brain regions when assessing peripheral immune infiltration via flow cytometry. Whether leukocyte extravasation occurs due to increased BBB permeability or due to circulating peripheral immune factors that aid in homing to the brain is unclear. What is apparent, from our studies and the literature, is that the aged brain is more sensitive to this environmental stressor than the young brain (Blau et al., 2012; Lee et al., 2012), resulting in concurrently increased BBB permeability and leukocyte trafficking.
CONCLUSION
Aging exacerbates the neuroinflammatory impact of O3 exposure, potentially due to impaired BBB functionality. We propose that increased permeability of the BBB due to a pulmonary insult activates already primed microglia in the aged brain. This activation along with concurrent infiltration of peripheral leukocytes may collectively contribute to the accumulation of β-amyloid protein, which likely further intensifies neuroinflammatory outcomes and initiates or perpetuates neurodegenerative phenotypes. Long-term neurological consequences of such effects of O3 remain unknown, but given the rapidly aging global urban population exposed to O3 and related pollutants, further investigation to understand the mechanism of toxicity and underpinnings of vulnerability are warranted.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
Supplementary Material
ACKNOWLEDGMENTS
Images in this paper were generated in the University of New Mexico & Cancer Center Fluorescence Microscopy Shared Resource, funded as detailed on: http://hsc.unm.edu/crtc/microscopy/acknowledgement.shtml.
FUNDING
This work was funded by National Institutes of Health, NIEHS (ES0014639 to M.J.C) and NIAAA (AA023051 to E.D.M and AA022534 to K.K.C.).
REFERENCES
- Ailshire J. A., Crimmins E. M. (2014). Fine particulate matter air pollution and cognitive function among older US adults. Am. J. Epidemiol. 180, 359–366.http://dx.doi.org/10.1093/aje/kwu155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhter H., Ballinger C., Liu N., Van Groen T., Postlethwait E. M., Liu R. M. (2015). Cyclic ozone exposure induces gender-dependent neuropathology and memory decline in an animal model of Alzheimer’s disease. Toxicol. Sci. 147, 222–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragon M. J., Topper L., Tyler C. R., Sanchez B., Zychowski K., Young T., Herbert G., Hall P., Erdely A., Eye T. et al. , (2017). Serum-borne bioactivity caused by pulmonary multiwalled carbon nanotubes induces neuroinflammation via blood-brain barrier impairment. Proc. Natl. Acad. Sci. U. S. A. 114, E1968–E1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt D. P., Puig K. L., Gorr M. W., Wold L. E., Combs C. K., Block M. L. (2015). A pilot study to assess effects of long-term inhalation of airborne particulate matter on early Alzheimer-like changes in the mouse brain. PLoS One 10, e0127102–e0127120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blau C. W., Cowley T. R., O'Sullivan J., Grehan B., Browne T. C., Kelly L., Birch A., Murphy N., Kelly A. M., Kerskens C. M. et al. , (2012). The age-related deficit in LTP is associated with changes in perfusion and blood-brain barrier permeability. Neurobiol. Aging 33, 1005.e23. [DOI] [PubMed] [Google Scholar]
- Block M. L., Calderón-Garcidueñas L. (2009). Air pollution: Mechanisms of neuroinflammation and CNS disease. Trends Neurosci. 32, 506–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgade K., Le Page A., Bocti C., Witkowski J. M., Dupuis G., Frost E. H., Fülöp T., Otth C. (2016). Protective effect of amyloid-β peptides against herpes simplex virus-1 infection in a neuronal cell culture model. J. Alzheimer’s Dis. 50, 1227–1241. [DOI] [PubMed] [Google Scholar]
- Burda J. E., Sofroniew M. V. (2014). Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81, 229–248.http://dx.doi.org/10.1016/j.neuron.2013.12.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgmans S., van Boxtel M. P. J., van den Berg K. E. M., Gronenschild E. H. B. M., Jacobs H. I. L., Jolles J., Uylings H. B. M. (2011). The posterior parahippocampal gyrus is preferentially affected in age-related memory decline. Neurobiol. Aging 32, 1572–1578. [DOI] [PubMed] [Google Scholar]
- Calderon-Garciduenas L., de la Monte S. M. (2017). Apolipoprotein E4, gender, body mass index, inflammation, insulin resistance, and air pollution interactions: Recipe for Alzheimer’s disease development in Mexico City Young Females. J. Alzheimer’s Dis. 58, 613–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderon-Garciduenas L., Kavanaugh M., Block M., D’Angiulli A., Delgado-Chavez R., Torres-Jardon R. et al. , (2012). Neuroinflammation, hyperphosphorylated tau, diffuse amyloid plaques, and down-regulation of the cellular prion protein in air pollution exposed children and young adults. J. Alzheimer’s Dis. 28, 93–107. [DOI] [PubMed] [Google Scholar]
- Calderon-Garciduenas L., Leray E., Heydarpour P., Torres-Jardon R., Reis J. (2016). Air pollution, a rising environmental risk factor for cognition, neu and neurodegeneration: The clinical impact on children and beyond. Rev. Neurol. (Paris) 172, 69–80. [DOI] [PubMed] [Google Scholar]
- Calderon-Garciduenas L., Solt A. C., Henríquez-Roldán C., Torres-Jardón R., Nuse B., Herritt L., Villarreal-Calderón R., Osnaya N., Stone I., García R. et al. , (2008). Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood-brain barrier, ultrafine particulate deposition, and accumulation of amyloid beta-42 and alpha-synuclein in children and young. Toxicol. Pathol. 36, 289–310. [DOI] [PubMed] [Google Scholar]
- Castellani R. J., Lee H-g., Siedlak S. L., Nunomura A., Hayashi T., Nakamura M., Zhu X., Perry G., Smith M. A. (2009). Reexamining Alzheimer’s disease: Evidence for a protective role for amyloid-β protein precursor and amyloid-β. J. Alzheimer’s Dis. 18, 447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H. H., Hao H., Sarnat S. E. (2014). A statistical modeling framework for projecting future ambient ozone and its health impact due to climate change. Atmos. Environ. (1994) 89, 290–297.http://dx.doi.org/10.1016/j.atmosenv.2014.02.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channell M. M., Paffett M. L., Devlin R. B., Madden M. C., Campen M. J. (2012). Circulating factors induce coronary endothelial cell activation following exposure to inhaled diesel exhaust and nitrogen dioxide in humans: Evidence from a novel translational in vitro model. Toxicol. Sci. 127, 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echávarri C., Aalten P., Uylings H. B. M., Jacobs H. I. L., Visser P. J., Gronenschild E. H. B. M., Verhey F. R. J., Burgmans S. (2011). Atrophy in the parahippocampal gyrus as an early biomarker of Alzheimer’s disease. Brain Struct. Funct. 215, 265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elahy M., Jackaman C., Mamo J., Lam V., Dhaliwal S. S., Giles C., Nelson D., Takechi R. (2015). Blood-brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immun. Ageing 12, 2.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt B., Vajkoczy P., Weller R. O. (2017). The movers and shapers in immune privilege of the CNS. Nat. Immunol. 18, 123–131.http://dx.doi.org/10.1038/ni.3666 [DOI] [PubMed] [Google Scholar]
- Fabis M. J., Phares T. W., Kean R. B., Koprowski H., Hooper D. C. (2008). Blood-brain barrier changes and cell invasion differ between therapeutic immune clearance of neurotrophic virus and CNS autoimmunity. Proc. Natl. Acad. Sci. U. S. A. 105, 15511–15516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly K., Ettehadieh D., Upadhyay S., Takenaka S., Adler T., Karg E., Krombach F., Kreyling W. G., Schulz H., Schmid O. et al. , (2017). Early pulmonary response is critical for extra-pulmonary carbon nanoparticle mediated effects: Comparison of inhalation versus intra-arterial infusion exposures in mice. Part. Fibre Toxicol. 14, 19..http://dx.doi.org/10.1186/s12989-017-0200-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemechu J. M., Bentivoglio M. (2012). T cell recruitment in the brain during normal aging. Front. Cell Neurosci. 6, 38.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Crisostomo N. P., Rodriguez Martinez E., Rivas-Arancibia S. (2014). Oxidative stress activates the transcription factors FoxO 1a and FoxO 3a in the hippocampus of rats exposed to low doses of ozone. Oxid. Med. Cell. Longev. 2014, 1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Mejiba S. E., Zhai Z., Akram H., Pye Q. N., Hensley K., Kurien B. T., Scofield R. H., Ramirez D. C. (2009). Inhalation of environmental stressors & chronic inflammation: Autoimmunity and neurodegeneration. Mutat. Res. 674, 62–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greter M., Lelios I., Croxford A. L. (2015). Microglia versus myeloid cell nomenclature during brain inflammation. Front. Immunol. 6, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad-Tóvolli R., Dragano N. R. V., Ramalho A. F. S., Velloso L. A. (2017). Development and function of the blood-brain barrier in the context of metabolic control. Front. Neurosci. 11, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart A. D., Wyttenbach A., Hugh Perry V., Teeling J. L. (2012). Age related changes in microglial phenotype vary between CNS regions: Grey versus white matter differences. Brain Behav. Immun. 26, 754–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley L. L., Tizabi Y. (2013). Neuroinflammation, neurodegeneration and depression. Neurotox. Res. 23, 131–144.http://dx.doi.org/10.1007/s12640-012-9348-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janota C. S., Brites D., Lemere C. A., Brito M. A. (2015). Glio-vascular changes during ageing in wild-type and in Alzheimer’s disease-like APP/PS1 mice. Brain Res. 1620, 153–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraj R. L., Rodriguez E. A., Wang Y., Block M. L. (2017). Outdoor ambient air pollution and neurodegenerative diseases: The neuroinflammation hypothesis. Curr. Environ. Heal. Rep. 4, 166–179. [DOI] [PubMed] [Google Scholar]
- Lee P., Kim J., Williams R., Sandhir R., Gregory E., Brooks W. M., Berman N. E. J. (2012). Effects of aging on blood brain barrier and matrix metalloproteases following controlled cortical impact in mice. Exp. Neurol. 234, 50–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque S., Surace M. J., McDonald J., Block M. L. (2011). Air pollution & the brain: Subchronic diesel exhaust exposure causes neuroinflammation and elevates early markers of neurodegenerative disease. J. Neuroinflammation 8, 105.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque S., Taetzsch T., Lull M. E., Kodavanti U., Stadler K., Wagner A., Johnson J. A., Duke L., Kodavanti P., Surace M. J. et al. , (2011). Diesel exhaust activates and primes microglia: Air pollution, neuroinflammation, and regulation of dopaminergic neurotoxicity. Environ. Health Perspect. 119, 1149–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucero J., Suwannasual U., Herbert L. M., McDonald J. D., Lund A. K. (2017). The role of the lectin-like oxLDL receptor (LOX-1) in traffic-generated air pollution exposure-mediated alteration of the brain microvasculature in Apolipoprotein (Apo) E knockout mice. Inhal. Toxicol. 1–16. doi: 10.1080/08958378.2017.1357774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher B. A., Ahmed I. A. M., Karloukovski V., MacLaren D. A., Foulds P. G., Allsop D., Mann D. M. A., Torres-Jardón R., Calderon-Garciduenas L. (2016). Magnetite pollution nanoparticles in the human brain. Proc. Natl. Acad. Sci. U. S. A. 113, 10797–10801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer R. R., Scabilloni J. F., Hubbs A. F., Wang L., Battelli L. A., McKinney W., Castranova V., Porter D. W. (2013). Extrapulmonary transport of MWCNT following inhalation exposure. Part. Fibre Toxicol. 10, 38.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. R., Raftis J. B., Langrish J. P., McLean S. G., Samutrtai P., Connell S. P., Wilson S., Vesey A. T., Fokkens P. H. B., Boere A. J. F. et al. , (2017). Inhaled nanoparticles accumulate at sites of vascular disease. ACS Nano 11, 4542–4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne A., Barnes S. R., Sweeney M. D., Halliday M. R., Sagare A. P., Zhao Z., Toga A. W., Jacobs R. E., Liu C. Y., Amezcua L. et al. , (2015). Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85, 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooradian A. D. (1988). Effect of aging on the blood-brain barrier. Neurobiol. Aging 9, 31–39.http://dx.doi.org/10.1016/S0197-4580(88)80013-7 [DOI] [PubMed] [Google Scholar]
- Mumaw C. L., Levesque S., McGraw C., Robertson S., Lucas S., Stafflinger J. E., Campen M. J., Hall P., Norenberg J. P., Anderson T. et al. , (2016). Microglial priming through the lung-brain axis: The role of air pollution-induced circulating factors. FASEB J. 30, 1880–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niccoli T., Partridge L. (2012). Ageing as a risk factor for disease. Curr. Biol. 22, R741..http://dx.doi.org/10.1016/j.cub.2012.07.024 [DOI] [PubMed] [Google Scholar]
- Nikodemova M., Small A. L., Kimyon R. S., Watters J. J. (2016). Age-dependent differences in microglial responses to systemic inflammation are evident as early as middle age. Physiol. Genomics 48, 336–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor S., Sanchez J. J., Vanderwall A. G., Sun M. S., Maxwell J. R., Davies S., Jantzie L. L., Petersen T. R., Savage D. D., Milligan E. D. et al. , (2017). Prenatal alcohol exposure potentiates chronic neuropathic pain, spinal glial and immune cell activation and alters sciatic nerve and DRG cytokine levels. Brain Behav. Immun. 61, 80–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norden D. M., Godbout J. P. (2013). Review: Microglia of the aged brain: Primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 39, 19–34.http://dx.doi.org/10.1111/j.1365-2990.2012.01306.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdorster G., Sharp Z., Atudorei V., Elder A., Gelein R., Kreyling W., Cox C. (2004). Translocation of inhaled ultrafine particles to the brain. Inhal. Toxicol. 16, 437–445. [DOI] [PubMed] [Google Scholar]
- Oppenheim H. A., Lucero JAnn., Guyot A.-C., Herbert L. M., McDonald J. D., Mabondzo A., Lund A. K. (2013). Exposure to vehicle emissions results in altered blood brain barrier permeability and expression of matrix metalloproteinases and tight junction proteins in mice. Part. Fibre Toxicol. 10, 62.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A., Veronesi B., Calderón-Garcidueñas L., Gehr P., Chen L., Geiser M., Reed W., Rothen-Rutishauser B., Schürch S., Schulz H. et al. , (2006). Translocation and potential neurological effects of fine and ultrafine particles a critical update. Part. Fibre Toxicol. 3, 13.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phares T. W., Kean R. B., Mikheeva T., Hooper D. C. (2006). Regional differences in blood-brain barrier permeability changes and inflammation in the apathogenic clearance of virus from the central nervous system. J. Immunol. 176, 7666–7675. [DOI] [PubMed] [Google Scholar]
- Post E. S., Grambsch A., Weaver C., Morefield P., Huang J., Leung L.-Y., Nolte C. G., Adams P., Liang X.-Z., Zhu J.-H. et al. , (2012). Variation in estimated ozone-related health impacts of climate change due to modeling choices and assumptions. Environ. Health Perspect. 120, 1559–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postlethwait E. M., Langford S. D., Bidani A. (1994). Determinants of inhaled ozone absorption in isolated rat lungs. Toxicol. Appl. Pharmacol. 125, 77–89.http://dx.doi.org/10.1006/taap.1994.1051 [DOI] [PubMed] [Google Scholar]
- Power M. C., Adar S. D., Yanosky J. D., Weuve J. (2016). Exposure to air pollution as a potential contributor to cognitive function, cognitive decline, brain imaging, and dementia: A systematic review of epidemiologic research. Neurotoxicology 56, 235–253.http://dx.doi.org/10.1016/j.neuro.2016.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz M., Priller J. (2017). The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 20, 136–144.http://dx.doi.org/10.1038/nn.4475 [DOI] [PubMed] [Google Scholar]
- Ransohoff R. M. (2016). A polarizing question: Do M1 and M2 microglia exist?. Nat. Neurosci. 19, 987–991.http://dx.doi.org/10.1038/nn.4338 [DOI] [PubMed] [Google Scholar]
- Ransohoff R. M. (2016). How neuroinflammation contributes to neurodegeneration. Science 353, 777–783.http://dx.doi.org/10.1126/science.aag2590 [DOI] [PubMed] [Google Scholar]
- Rezai-Zadeh K., Gate D., Town T. (2009). CNS infiltration of peripheral immune cells: D-Day for neurodegenerative disease? J. Neuroimmune Pharmacol. 4, 462–475.http://dx.doi.org/10.1007/s11481-009-9166-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas-Arancibia S., Dorado-Martínez C., Borgonio-Pérez G., Hiriart-Urdanivia M., Verdugo-Díaz L., Durán-Vázquez A., Colin-Baranque L., Rosa Avila-Costa M., (2000). Effects of taurine on ozone-induced memory deficits and lipid peroxidation levels in brains of young, mature, and old rats. Environ. Res. 82, 7–17. [DOI] [PubMed] [Google Scholar]
- Robertson S., Colombo E. S., Lucas S. N., Hall P. R., Febbraio M., Paffett M. L., Campen M. J. (2013). CD36 mediates endothelial dysfunction downstream of circulating factors induced by O3 exposure. Toxicol. Sci. 134, 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagare A. P., Bell R. D., Zlokovic B. V. (2012). Neurovascular defects and faulty amyloid-β vascular clearance in Alzheimer’s disease. Adv. Alzheimer’s Dis. 3, 87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schisler J. C., Ronnebaum S. M., Madden M., Channell M., Campen M., Willis M. S. (2015). Endothelial inflammatory transcriptional responses to an altered plasma exposome following inhalation of diesel emissions. Inhal. Toxicol. 27, 272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoub T. R., Barnes C. A., Shah R. C., Stebbins G. T., Ferrari C., deToledo-Morrell L. (2012). Age-related changes in the mesial temporal lobe: The parahippocampal white matter region. Neurobiol. Aging 33, 1168–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowell J. D., Kim Y.-M., Gao Y., Fu J. S., Chang H. H., Liu Y. (2017). The impact of climate change and emissions control on future ozone levels: Implications for human health. Environ. Int. 108, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit W. J., Mrak R. E., Griffin W. S. T. (2004). Microglia and neuroinflammation: A pathological perspective. J. Neuroinflammation. 1, 14..http://dx.doi.org/10.1186/1742-2094-1-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit W. J., Semple-Rowland S. L., Hurley S. D., Miller R. C., Popovich P. G., Stokes B. T. (1998). Cytokine mRNA profiles in contused spinal cord and axotomized facial nucleus suggest a beneficial role for inflammation and gliosis. Exp. Neurol. 152, 74.. [DOI] [PubMed] [Google Scholar]
- Thomson E. M., Pal S., Guénette J., Wade M. G., Atlas E., Holloway A. C., Williams A., Vincent R. (2016). Ozone inhalation provokes glucocorticoid-dependent and -independent effects on inflammatory and metabolic pathways. Toxicol. Sci. 152, 17–28. [DOI] [PubMed] [Google Scholar]
- Thomson E. M., Vladisavljevic D., Mohottalage S., Kumarathasan P., Vincent R. (2013). Mapping acute systemic effects of inhaled particulate matter and ozone: Multiorgan gene expression and glucocorticoid activity. Toxicol. Sci. 135, 169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler C. R., Zychowski K. E., Sanchez B. N., Rivero V., Lucas S., Herbert G., Liu J., Irshad H., McDonald J. D., Bleske B. E. et al. , (2016). Surface area-dependence of gas-particle interactions influences pulmonary and neuroinflammatory outcomes. Part. Fibre Toxicol. 13, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villeda S. A., Luo J., Mosher K. I., Zou B., Britschgi M., Bieri G., Stan T. M., Fainberg N., Ding Z., Eggel A. et al. , (2011). The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477, 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.-Y., Tan M.-S., Yu J.-T., Tan L. (2015). Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 3, doi: 10.3978/j.issn.2305-5839.2015.03.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm I., Nyúl-Tóth Á., Suciu M., Hermenean A., Krizbai I. A. (2016). Heterogeneity of the blood-brain barrier. Tissue Barriers 4, e1143544.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A. C., Clemente L., Liu T., Bowen R. L., Meethal S. V., Atwood C. S. (2008). Reproductive hormones regulate the selective permeability of the blood-brain barrier. Biochim. Biophys. Acta - Mol. Basis Dis. 1782, 401–407. [DOI] [PubMed] [Google Scholar]
- Woodward N. C., Pakbin P., Saffari A., Shirmohammadi F., Haghani A., Sioutas C., Cacciottolo M., Morgan T. E., Finch C. E. (2017). Traffic-related air pollution impact on mouse brain accelerates myelin and neuritic aging changes with specificity for CA1 neurons. Neurobiol. Aging 53, 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.