
Figure 7.
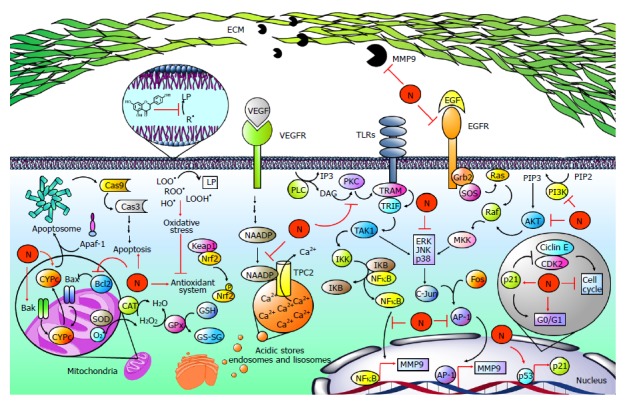
Naringenin in cancer development. Hepatocellular carcinoma is strongly associated with elevated levels of free radicals such as lipid hydroperoxides (LOOH•), peroxyl radicals (ROO•), and hydroxyl radicals (OH•), leading to the development of lipid peroxidation (LP), oxidative stress and finally to an imbalance of the endogenous antioxidant system. Naringenin (N) inhibits oxidative stress by its intrinsic antioxidant properties and by improving the endogenous antioxidant system. Notably, oxidative stress has been linked to the hepatocarcinogenic process because it is implicated in the activation of mitogen activated protein kinases (MAPKs), nuclear factor-kappa B (NF-κB), or phosphatidylinositol-3-kinase (PI3K/AKT) pathways, increasing the production and activity of metalloprotease 9 (MMP9), which is involved in migration and invasion processes. When toll-like receptors (TLRs) are activated, TRAMP is recruited to activate TRIF; in turn, it promotes transforming growth factor beta-activated kinase 1 (TAK1) activation, which phosphorylates IκB kinase (IKK). Then, IKK promotes NF-κB release via inhibitor κB (IKB) phosphorylation. On the other hand, phospholipase C (PLC) catalyzes phospholipid hydrolysis, generating inositol triphosphate (IP3) and diacylglycerol (DAG); the latter is an activator of protein kinase C (PKC), which can induce the NF-κB pathway in a TRAMP-dependent manner. Then, NF-κB induces the expression of MMP9. Epidermal growth factor (EGF) is highly involved in carcinogenic pathways; it binds to epidermal growth factor receptor (EGFR) promoting Grb2, SOS, Ras, Raf and mitogen-activated protein kinase kinase (MKK) activation, which participates in extracellular signal-regulated protein kinase (ERK), c-Jun N-terminal kinase (JNK) and p38 (MAPK) phosphorylation and activation. Then, MAPKs promote activator protein 1 (AP-1) activation by c-Jun and Fos dimerization. After that, AP-1 induces the expression of MMP9. Alternatively, MAPKs are activated via PI3K/AKT. PI3K produces phosphatidylinositol (3, 4, 5)-trisphosphate (PIP3) from phosphatidylinositol 4,5-bisphosphate (PIP2); PIP3 activates AKT, which promotes MAPK activation in a Ras-dependent pathway. It has been reported that naringenin inhibits MM9 expression and secretion through diminution of p38, JNK, ERK, IKB, and PI3K/AKT phosphorylation as well as NF-κB and AP-1-DNA binding. In addition, naringenin inhibits PKC cytoplasm-to-membrane translocation. Notably, naringenin induces p53 accumulation, leading to p21 expression. Then, p21 inhibits cyclin E/cyclin-dependent kinase 2 (CDK2) complex, which participates in proliferation. p53 accumulation results in naringenin-induced G0/G1 phase arrests. An important mechanism for the elimination of cancer cells is apoptosis. Naringenin induces apoptosis by increased cytochrome c (CYPc) release, as well as BCL2-associated X protein (Bax), BCL2-antagonist/killer 1 (Bak) and Caspase 3 (Cas3) elevation. Additionally, naringenin inhibited B-cell CLL/lymphoma 2 (Bcl-2) an antiapoptotic protein. Two-pore channels (TPCs) are members of the voltage-gated ion channel superfamily localized in acidic calcium (Ca2+) stores and have been implicated in angiogenic processes. Vascular endothelial growth factor (VEGF) and its receptor vascular endothelial growth factor receptor (VEGFR) promotes TPC activation via nicotinic acid adenine dinucleotide phosphate (NAADP); then, Ca2+ is transported to the cytoplasm through TPCs, activating angiogenic signals. Naringenin inhibits VEGF angiogenesis induction blocking NAADP activation and NAADP/TPC association.