

Abstract
Background
Complete mitochondrial (mt) genomes have been sequenced for thousands of animals and represent a molecule of choice for many evolutionary studies. Nevertheless, some animal groups have remained under-sampled. Ctenophora (comb jellies) is one such example, with only two complete mt sequences determined hitherto for this phylum, which encompasses ca. 150–200 described species. This lack of data derives from the extremely fast mt evolutionary rate in this lineage, complicating primer design and DNA amplification. Indeed, in the two ctenophore mt genomes sequenced to date, i.e. those of Mnemiopsis leidyi (order Lobata) and Pleurobrachia bachei (order Cydippida), both rRNA and protein coding genes exhibit an extraordinary size reduction and have highly derived sequences. Additionally, all tRNAs, and the atp6 and atp8 genes are absent. In order to determine whether these characteristics are shared by other ctenophores, we obtained the complete mt genomes of three benthic ctenophores belonging to the so far unsampled order of Platyctenida: Coeloplana loyai, Coeloplana yulianicorum and Vallicula multiformis.
Results
The mt genomes of benthic ctenophores reveal the same peculiarities found in Mnemiopsis and Pleurobrachia, demonstrating that the fast evolutionary rate is a general trait of the ctenophore mt genomes. Our results also indicate that this high evolutionary rate not only affects the nucleotide substitution but also gene rearrangements. Indeed, gene order was highly rearranged among representatives of the different taxonomic orders in which it was close to random, but also quite variable within Platyctenida, in which the genera Coeloplana and Vallicula share only four conserved synteny blocks. However, the two congeneric Coeloplana species display exactly the same gene order. Because of the extreme evolutionary rate, our phylogenetic analyses were unable to resolve the phylogenetic position of ctenophores within metazoans or the relationships among the different Ctenophora orders. Comparative sequence-analyses allowed us to correct the annotation of the Pleurobrachia mt genome, confirming the absence of tRNAs, the presence of both rRNA genes, and the existence of a reassignment of codon TGA from tryptophan to serine for this species.
Conclusions
Since Platyctenida is an early diverging lineage among Ctenophora, our findings suggest that the mt traits described above are ancestral characteristics of this phylum.
Electronic supplementary material
The online version of this article (10.1186/s12862-018-1186-1) contains supplementary material, which is available to authorized users.
Keywords: Coeloplana, Vallicula, mtDNA, Gene loss, Extreme evolutionary rate
Background
The typical metazoan mitochondrial (mt) genome is a small (~ 20 kb), circular DNA molecule containing 37 genes – 13 protein coding genes (the cytochrome oxidase 1–3 [cox1–3]; the ATPase subunit 6 and 8 [atp6/8]; the cytochrome b [cob]; the NADH dehydrogenase subunit 1–6 [nad1–6]), 22 transfer RNA (tRNAs) genes and 2 ribosomal RNA (rRNA) genes (the small subunit ribosomal RNA [rns] and the large subunit ribosomal RNA [rnl]) [1, 2]. This compact molecule possesses several characteristics that have made it a perfect sequencing target. First, mt genomes are present in high copy numbers [3], since eukaryotic cells usually contain several mitochondria and each mitochondrion possesses numerous copies of the mt genome. Second, mt genomes are extremely compact – most are intron-less and non-coding regions are extremely small [3]. Consequently, it is usually easier to amplify and sequence mt genes than nuclear ones. Finally, the same set of orthologous single-copy genes is conserved in most animal taxa, thus facilitating sequence comparisons and, above all, allowing the reconstruction of gene trees that correspond to species trees [4].
The above-noted features have made the mt genome a molecule of choice for many fields of genomics and molecular evolution. For example, complete mt genome sequences have been used to resolve the phylogenetic relationships of many animal groups [5], to study the transfer of organelle genes to the nucleus [6] and to study the evolution of gene rearrangement [7]. Additionally, the mt cox1 gene is used as a universal “DNA barcode” [8] for animals and, consequently, for a given species, a complete mt genome is often sequenced from one individual as a prerequisite of population studies [9].
One direct consequence of the high interest in mt genomes is the large and increasing number of complete mt genomes available in public databases (i.e., over 7300 complete mt genomes of animals are available at the National Center for Biotechnology Information [NCBI] - https://www.ncbi.nlm.nih.gov/genome/browse/) [3], and numerous databases and web servers are dedicated to their annotations and comparisons [10, 11]. However, some animal phyla, such as Ctenophora, are under-represented in these databases [12].
Ctenophora (comb jellies) comprise a rather small phylum of marine animals that encompasses about 150–200 species [13]. Most ctenophores are planktonic predators with a gelatinous body similar to jellyfish (Cnidaria, Medusozoa), for which they are often mistaken. Unlike jellyfish and other cnidarians, ctenophores lack stinging cells and are characterized by the presence of comb rows composed of cilia, which are used for swimming, and tentacles, which possess a special cell type, the colloblasts, used for grasping and feeding. Ctenophores are traditionally divided into two classes, Tentaculata and Nuda, based on the presence or absence of tentacles throughout their lives, respectively [13]. However, molecular analyses suggest that Tentaculata, which include eight orders, is paraphyletic, and that the single Nuda order – Beroida – is nested within Tentaculata [14–17]. Among ctenophores, Platyctenida is the only order that includes benthic species. Most members of the Platyctenida do not possess comb rows in the adult stage and externally resemble flat worms, except for the presence of two long retractile tentacles. Interestingly, molecular studies suggest that Platyctenida is not the first ctenophore lineage to diverge, and that the adaptation to a benthic environment is a derived characteristic [14–17].
To date, only two complete mt genomes have been sequenced in Ctenophora: the genome of Mnemiopsis leidyi (order Lobata) [12] and that of Pleurobrachia bachei (order Cydippida) [18]. Both these genomes strongly diverge from the “typical” mt genome described above (see Lavrov and Pett [1]). Indeed, they are of very small size (10–11 kilobases [kb]); lack the atp8, atp6 and most/all tRNA genes; demonstrate a length reduction; and a high sequence divergence in both protein coding and rRNA genes. These mt genomes thus reveal an extremely high evolutionary rate. It is unclear, however, whether all these features are shared by all ctenophores or only by the few analysed lineages to date.
Here, we report the sequencing and analysis of the complete mt genomes of three benthic ctenophore species belonging to the order Platyctenida and the family Coeloplanidae: Coeloplana loyai, Coeloplana yulianicorum and Vallicula multiformis. The analyses of these new species have enabled us to amend the annotation of the Pleurobrachia mt genome and to confirm a modification of the genetic code in the latter.
Results
General features of the Coeloplanidae mt genomes
The circular mt genomes of C. loyai, C. yulianicorum and V. multiformis are 11,574, 11,551, and 9961 base pairs (bp) long, respectively. The three sequenced species contain the same set of genes: 11 protein coding genes (all those present in typical animal mt genomes, except for atp6 and atp8) and 2 rRNAs (rnl and rns). In addition, from 1 to 4 unassigned ORFs (Unidentified Reading Frames, named URFs 1–4), ranging from 102 to 687 bp, were identified in each species (Fig. 1). All annotated URFs were predicted to encompass from 1 to 5 putative transmembrane domains. All these genes are encoded on the same strand (Fig. 1).
Fig. 1.

Organization of the Coeloplanidae mt genomes. a C. loyai. b C. yulianicorum. c V. multiformis. All genes are transcribed clockwise. Large and small ribosomal RNA subunits are marked in red. The protein coding genes are indicated by different colours: in purple - the cytochrome c oxidase genes (Complex VI), in blue - NADH dehydrogenase genes (Complex I), in yellow - the cytochrome b (complex III). Green areas are URFs (unidentified reading frames). Finally, noncoding regions are in white. A photo of each organism is provided in the centre of the corresponding mt genome
No tRNAs were identified. Indeed, all putative tRNAs detected by the three tRNA annotation software programs (see Methods) were located within ORFs and possessed a structure that did not fit the canonical mt tRNA structure [19]. They were thus considered to be false positive hits.
While gene order is conserved between the mt sequences of the two Coeloplana species, only four synteny blocks are conserved between the two genera Coeloplana and Vallicula (cox1-cox2-nad4, nad4L-nad1, cob-cox3-nad3, and rnl-rns) (Fig. 2). The gene order is thus conserved at the congeneric level but already quite variable at the intra-family level. Considering the other ctenophore taxonomic orders, only one synteny block is conserved (cox3-nd3) between Platyctenida and the Lobata M. leidyi, and no synteny blocks are shared between the Cydippida P. bachei (re-annotated, see below) and the other ctenophore species. The results of the breakpoint analyses support these observations (see Table 1, where values of the normalized breakpoint distances - BDn - close to one indicate extreme rearrangements close to random). Indeed, the BDn computed among Ctenophora orders range between 0.69 and 0.92.
Fig. 2.
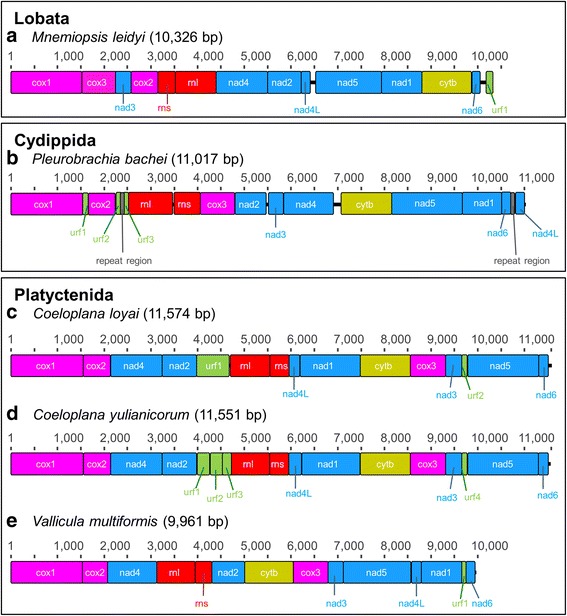
Comparison of mt gene order between Ctenophora orders. a M. leidyi. b P. bachei. c C. loyai. d C. yulianicorum. e V. multiformis. The genome of Pleurobrachia bachei has been re-annotated. Each orthologous protein coding or rRNA gene is indicated by a different colour. Black connecting lines between genes indicate noncoding regions. URFs are indicated in green. Remarkably, no syntenic regions were shared by all ctenophore species
Table 1.
Breakpoint distances computed for each pair of ctenophore mt genomes
| C. loyai | C. yulianicorum | V. multiformis | M. leidyi | P. bachei | |
|---|---|---|---|---|---|
| Coeloplana loyai | – | 0 | 5 | 9 | 11 |
| Coeloplana yulianicorum | 0 | – | 5 | 9 | 11 |
| Vallicula multiformis | 0.38 | 0.38 | – | 11 | 12 |
| Mnemiopsis leidyi | 0.69 | 0.69 | 0.85 | – | 12 |
| Pleurobrachia bachei | 0.85 | 0.85 | 0.77 | 0.92 | – |
Breakpoint distances [38] and normalized breakpoint distances are given above and below the diagonal respectively
Concerning base composition, the mt genomes of C. loyai, C. yulianicorum and V. multiformis are extremely AT rich, with AT contents of 84, 82.9 and 84.4%, respectively. The most frequent nucleotide in those genomes is T, which represents more than 50% of the nucleotide in all three species (60.2–60.6%).
No repeated region longer than 50 bp was found in these mt genomes. The longest repeated elements (31–21 bp) found in each species were always located in low complexity regions and composed mainly of poly-T repeats.
Re-annotation of the Pleurobrachia bachei mt genome
Analysis of the alignments of the mitochondrial protein genes led us to discover several errors in the original annotation of the Pleurobrachia mt genome (accession JN392469 [18]) (see Additional file 1). For example, we found that the nad4L gene had been erroneously annotated as nad3. The true nad3 was not identified by Kohn et al. [18] but included as part of the nad4 gene. Indeed, in Pleurobrachia, but not in other ctenophores, the nad3 gene shares the same translation frame as the downstream nad4 gene and possesses an incomplete stop codon. Similarly, the rns gene was originally annotated as rnl and the two tRNAs identified by Kohn et al. [18] were found in the region that we now recognize as the rnl. In other words, the two tRNAs annotated by Kohn et al. [18] are now considered as being only tRNA-like structures (Additional file 1). Our amended annotation thus confirms the absence of tRNA genes and the presence of rns and rnl in the P. bachei mt genome. Moreover, we definitively identified the nad2 and nad6 genes in two regions not corresponding to those suggested by Kohn et al. [18] (Additional file 1).
The protein gene alignments also indicated that the 5′ region of the cox1 gene was truncated in P. bachei, since it lacks 25 amino acid positions highly conserved in the remaining ctenophores (Additional file 2). The region upstream to the cox1 gene includes several poly-T, and thus, we suspected the introduction of a frame shift due to erroneous sequencing of the number of T in these homopolymers by Kohn et al. [18]. To verify this, we downloaded Illumina reads used for assembling the P. bachei nuclear genome (run SRR1174875 [14] of the Sequence Read Archive of NCBI - https://trace.ncbi.nlm.nih.gov/Traces/sra/) and mapped them against the mt sequence of P. bachei (accession JN392469). This alignment confirmed that a T is missing eight bp upstream to the start of the cox1 gene as defined by Kohn et al. [18]. The inclusion of this missing T extended the reading frame and enabled identification of the missing 5′ region of the P. bachei cox1 (Additional file 2).
One and two URFs (length: 108–111 bp) were found upstream and downstream of cox2 respectively, while a 84 bp long sequence was found to make up a perfect repeat about 8.2 kb apart from each other.
Finally, investigation of codon usage in conserved gene regions using GenDecoder v.1.6 [20] confirmed that in P. bachei the codon TGA, which codes for tryptophan in other ctenophores, is reassigned to serine as indicated by Pett and Lavrov [21]. An example of conserved serine positions coded by TGA codons in P. bachei is given in Additional file 2.
An updated annotation of the P. bachei genome is provided in Additional file 3.
Protein-coding genes
Codon usage of the three benthic ctenophores C. loyai, C. yulianicorum and V. multiformis is provided in Additional file 4. Unlike for P. bachei, we did not detect any change in the mt genetic code of these species. In agreement with the observation that these sequences are AT-rich, the most abundant codons were TTT (Phe; 26.35, 26.29 and 26.54%, respectively), ATT (Ile; 8.72, 8.27, and 7.37%, respectively) and TTA (Leu; 9.74, 7.98 and 10.13%, respectively). Correspondingly, C- or G- rich codons demonstrated lower frequencies.
Concerning start codons, four start codons are used in Coeloplanidae (ATA, ATG, ATT, and TTA), with ATT and ATG being the favoured ones. In C. loyai and C. yulianicorum ATG is the most used (0.31 and 0.28% respectively), while ATT is the most abundant in V. multiformis (0.2%). In contrast, among canonical mt genes, TTA is only identified as a start codon of the nad1 gene of C. yulianicorum and of the nad2 gene of P. bachei. It is also the start codon of two of the URFs of C. yulianicorum. Because this codon is poorly used, it is thus possible that these URFs are in fact false positive. Concerning stop codons, both TAA and TAG are used in Ctenophora, with TAG being used only once in both Coeloplana species and in Mnemiopsis. Incomplete stop codons were only found in the nad2 and nad4L of V. multiformis, the nad3 of P. bachei.
rRNA genes
The rRNA genes were identified in C. loyai, C. yulianicorum and in the re-annotated P. bachei using a secondary structure model (see Methods) based on M. leidyi rRNA sequences. This approach, however, failed to detect the rns gene in V. multiformis (i.e., no hits were obtained with an E-value < 0.1). The rns of V. multiformis was consequently detected using standard Blastn searches using the rns sequence of Coeloplana as query. The lengths of the rRNA genes in C. loyai, C. yulianicorum, V. multiformis and P. bachei were: 413, 408, 355 and 563 nt for the rns, respectively; 843, 847, 825 and 943 nt for the rnl. These values are similar to the rRNA lengths observed in M. leidyi (i.e., 368 and 878 nt for the rns and rnl respectively). Alignment of the rRNA sequences revealed that there are very few shared positions between the five ctenophore sequences, indicating an extremely fast evolutionary rate of the rRNA genes (Additional files 5 and 6). Accordingly, only a few of the helices observed in the rRNAs of M. leidyi [12] were conserved among ctenophores. Specifically, the rnl helices 73, 74, 89, 90, 92 and 93 (Additional file 5) and the rns helices 18, 28 and 44 (Additional file 6) were the only conserved helices among the five species. Indeed, in the region of helices 29, 30 and 43, the rns of P. bachei contains novel insertions that are not shared by the other ctenophores.
Non-coding regions
Table 2 provides statistics on non-coding regions (NC) of the five ctenophores mt genomes. It should be noted that all ctenophore mt genomes (even Mnemiopsis) present URFs longer than 100 bp: according to the results of Blastn/Blastp against the NCBI nucleotide/protein databases, these URFs have no similarities to known peptides, so they could also be non-coding regions with accidental ORFs.
Table 2.
Statistics on non-coding sequences (NC) in Ctenophora mitochondrial genomes
| Genome length (bp) | NC length (bp) | N° NC > 20 bp | Longest NC | ||||||
|---|---|---|---|---|---|---|---|---|---|
| with URFs | without URFs | with URFs | without URFs | with URFs | without URFs | ||||
| bp | location | bp | location | ||||||
| Coeloplana loyai | 11,574 | 930 | 132 | 4 | 2 | 687 | nad2-rnl | 59 | nad6-cox1 |
| Coeloplana yulianicorum | 11,551 | 908 | 90 | 5 | 1 | 318 | nad2-rnl | 41 | nad6-cox1 |
| Vallicula multiformis | 9961 | 119 | 25 | 1 | 0 | 94 | nad1-nad6 | 13 | nad6-cox1 |
| Mnemiopsis leidyi | 10,326 | 406 | 265 | 3 | 2 | 276 | nad6-cox1 | 132 | nad6-urf1 |
| Pleurobrachia bachei | 11,016 | 755 | 429 | 8 | 5 | 276 | cox2-rn | 161 | nad4-cytb |
When URFs are considered to be non-coding, C. loyai and C. yulianicorum possess the largest proportion of non-coding regions: 8 and 7.9%, respectively. In both these species, the longest non-coding regions are located between nad2 and rnl and are 687 and 318 bp long, respectively. However, when URFs are treated as coding genes, P. bachei and M. leidyi possess the largest proportion of non-coding regions: 3.9 and 2.6% bp, respectively.
Among ctenophores, V. multiformis has the smallest number of non-coding regions: the total non-coding length is only 119 or 25 bp long, depending on whether URFs are included or excluded from non-coding, respectively (Table 2). The V. multiformis mt genome is consequently both the smallest ctenophore and metazoan mt genome reported to date (9961 bp long).
A correlation analysis indicates that the NC length strongly affects the overall size of the mt genome. In particular, plotting the mt genome size against the calculated NC lengths, we found the mt genome size to be significantly correlated to the NC length only when including URFs (r = 0.98).
Extreme evolutionary rate of ctenophore mitochondrial genome
We performed phylogenetic analyses based on the protein sequences from the seven most conserved mt genes (cob, cox1–3, nad1, nad3 and nad5), including species representatives of the main Metazoa phyla (see Methods). As exemplified in the reconstructed Bayesian phylogenetic tree (Fig. 3), all ctenophore mt genomes reveal very long branches and are therefore extremely fast evolving. Not only were ctenophores found to be distant from all other phyla considered but also between themselves. As a case in point, the branch length distance, as estimated in the phylogenetic tree, between the lobate Mnemiospsis and the benthic platyctenid Vallicula is far greater than the distance between human and sea urchin.
Fig. 3.
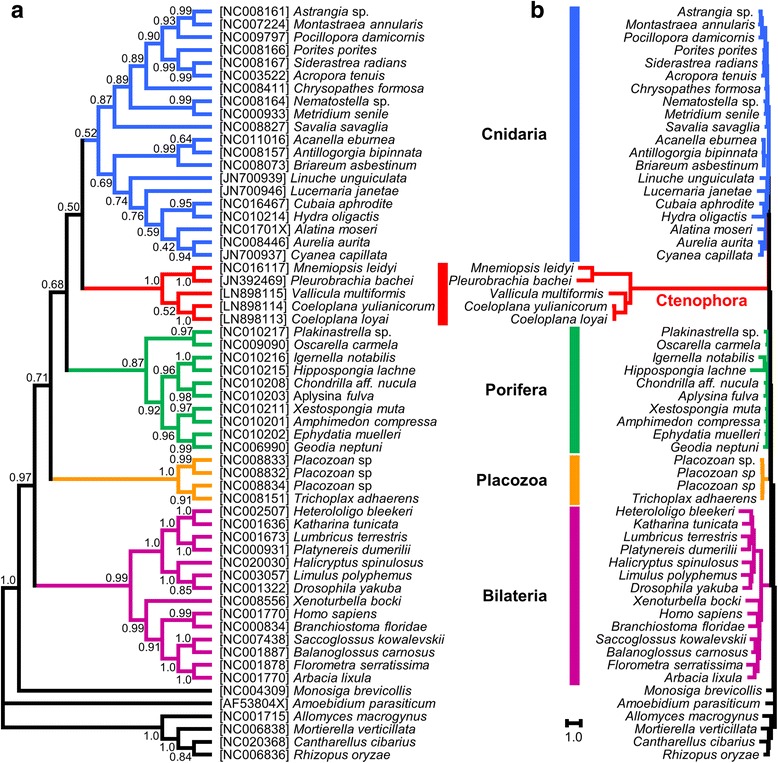
Phylogenetic position of Ctenophora within Metazoa using the most conserved mt genes. Bayesian tree reconstructed under the CAT+GTR + Γ mixture model from the seven most-conserved ctenophore genes (cob, cox1–3, nad1, nad3 and nad5). a Topology and posterior support values. b Tree with branch length. The long ctenophore branches, in blue, exemplify the fast evolutionary rate observed in this phylum compared to all other animal groups
Because of the extreme mt evolutionary rates, the phylogenetic position of the ctenophores among animals is not resolved by the analyses of mt sequences (the 0.50 posterior probability (PP) support value is unreliable). Concerning the relationships among the analysed ctenophore orders, the small taxon sampling prevents conclusive observations. However, it should be noted that members of the Coeloplanidae have shorter branches compared to the branch leading to the cluster Mnemiopsis plus Pleurobrachia (indicating a lower evolutionary rate). Moreover, the monophyly of Coeloplanidae was surprisingly recovered with low support (PP = 0.52) while it received high or maximal support in previous molecular studies based on 18S rRNA [15] and transcriptome analyses [14].
Discussion
The new mt sequences confirm that the key characteristics of the Mnemiopsis and Pleurobrachia mt genomes are also shared by benthic ctenophore species of the order Platyctenida. These characteristics include: the absence of atp6, atp8 and all mt tRNA genes (Fig. 2); a strong reduction in the overall size and number of conserved helices in the mt rRNA genes (both rns and rnl); the presence of all genes on the same strand (Figs. 1 and 2); a base composition with a high AT%; and an extremely fast evolution rate (Fig. 3) [12, 18]. Most of these characteristics have led to a reduction in size of the ctenophore mt genome when compared to other animals. Indeed, it should be noted that the V. multiformis mt genome (9961 bp long) is the smallest animal mt genome sequenced to date.
Since Platyctenida are considered an earlier diverging lineage among Ctenophora than Mnemiopsis and Pleurobrachia [14, 16, 17], our findings suggest that the traits described above are ancestral in Ctenophora. However, the mt genome of representatives from the families Euplokamididae and Mertensiidae (order Cydippida), which were reported to diverge before Platyctenida [14, 16, 17], should be sequenced in order to confirm this contention.
The loss of mt-tRNA in ctenophores has been found to be correlated with gene losses in the nuclear genome. As a case-in-point, most of the mt aminoacyl tRNA synthetases (aaRS) – the enzymes that attach an aminoacid to their corresponding tRNA – are absent from the nuclear genomes of ctenophores [21]. There are two exceptions to the aaRS loss in Ctenophora: the mt-PheRS, which is assumed to be retained because of import constraints; and the mt-TrpRS, which is considered to be retained by genetic code constraint [1, 21, 22]. Specifically, the mt-TrpRS is assumed to be required for the aminoacylation of the mt-tRNATrp, which recognizes the TGA codon in the “Mold, Protozoan and Coelenterate” mitochondrial code. In P. bachei, however, where the TGA codon is reassigned to Ser, it has been shown that the mt-TrpRS has been lost [1, 21]. Our analyses did not indicate any mt genetic-code change in Coeloplanidae. In agreement with this result, Pett and Lavrov [21] have shown the presence of a mt-TrpRS in the transcriptome of V. multiformis. Surprisingly, this enzyme was not detected in the transcriptome of Coeloplana astericola. However, this may be the result of a lower transcriptome quality for this species. Unfortunately, the sequencing methods used in this work do not allow us to investigate this issue in C. loyai and C. yulianicorum.
Previous studies have emphasized the fast mt evolutionary rates of ctenophores when compared to other animal phyla [12, 18]. The long branches of ctenophores in the metazoan tree (Fig. 3) and the high BDn found in inter-order comparisons (Table 1) indicate that not only mt sequence substitutions but also mt gene rearrangements are saturated. Specifically, our increased taxon sampling has revealed that the mt gene order is extremely rearranged among different taxonomic orders of ctenophores, quite variable at the intra-family level and conserved at the intra-genus level, at least in the analysed family Coeloplanidae. Concerning substitution rate, the fast mt evolutionary rate is not limited to a comparison between ctenophores and other animals but also among ctenophore orders. Indeed, the long branch of the Mnemiopsis and Pleurobrachia cluster compared to the Platyctenida branches (Fig. 3) suggests that the mt genome of ctenophores or its separate genes might present a good marker to investigate ctenophore relationships mainly at lower taxonomic levels, such as at the intra-family or species level (see Alamaru et al. [23]).
The high divergence of ctenophore mt sequences has also complicated their annotation. Our new sequences enabled comparative gene annotations that revealed errors in the original annotation of the Pleurobrachia mt genome [18] in gene boundary delimitation or in gene definition, as well as in the identification of missing genes (Additional file 1). However, sequence comparisons did not enable us to identify the atp8 gene. The atp8 is a canonical animal mitochondrial gene often overlooked, since it is rather short and poorly conserved. For example, the atp8 gene was originally believed to be absent in the mt genomes of the tunicate Halocynthia roretzi [24], but was later identified there [25]. The presence of URFs in all ctenophore mt sequences suggests that the atp8 gene might still be encoded in their mt genome. Moreover, unlike the atp6 gene, the atp8 has not been identified in the nuclear genome for either Pleurobrachia [18] or Mnemiopsis [12]. However, the URFs identified in the ctenophore mt genomes neither demonstrate similarity among themselves nor to any known atp8. Additional ctenophore mt sequences are therefore needed in order to characterize the atp8 gene in this phylum.
The ctenophore rRNA genes were found to be about half the size of classical animal mt rRNAs and to share only a few of the helix structures identified in Mnemiopsis [12]. Surprisingly, the rns gene of Pleurobrachia was found to include insertions that were not shared by other ctenophore species. Since the order Cydippida appears well nested within Ctenophora in phylogenetic trees based on nuclear genes [14, 17], it is possible that a secondary size reduction has been occurring in members of this group, while the tremendous size reduction of the rRNA is an ancestral trait.
Conclusions
In conclusion, the new Platyctenida mt sequences obtained in this work has enabled us to assess for the first time the mt evolutionary rates among and within ctenophore orders. Given their small and compact genomes, Ctenophora could constitute good models in the search to understand the mechanisms leading to variation in mt gene order.
Methods
DNA extractions and mt genome sequencing
The benthic ctenophore specimens used in this study (C. loyai sample 2013/9; C. yulianicorum sample 2002/6; V. multiformis sample 2007/1) were collected in the Gulf of Aqaba (29°30′ N, 34°56′ E) in 2012–2013 under permit 2010/37891 from the Israel Nature and National Parks Authority [26].
Genomic DNA (gDNA) extractions were performed with the Qiagen Blood & Tissue kit (Qiagen, Venlo, Netherlands) following the manufacturer’s instructions and using a single individual for each species. To reduce the sequencing cost, we followed the approach of Rubinstein et al. [27] and obtained complete mt sequence from sequencing mixed gDNA samples of several species without barcode. Specifically, the gDNA of the three Coeloplanidae species was mixed with DNA of non-ctenophore species (tunicates) that had participated in other studies [28]. To avoid the assembly of chimera sequences during the assembly step, the two Coeloplana species were sequenced as part of a different mix. Library construction and sequencing was performed by the Technion Genome Center (Haifa, Israel). The sequencing of V. multiformis and C. yulianicorum was performed on an Illumina HiSeq 2000 sequencer with paired reads 100 bp long. The sequencing of C. loyai was performed separately on an Illumina MiSeq with paired reads 250 bp long.
The program Cutadapt v1.10 [29] was used to trim reads from both 5′ and 3′ adapters. Rubinstein et al. [27] have shown that genomic assemblers can fail to assemble complete mitochondrial contigs from mixed gDNA samples. Consequently, following Rubinstein et al. [27], reads were assembled with the transcriptome assembler SOAPdenovo-Trans (Release 1.03, 07-25-2013) [30]. Blastn searches were conducted to identify mitogenome contigs using the cox1 sequence of Coeloplana sp. SHL-2011 and Coeloplana bocki [31] as query. The mt genome of V. multiformis was assembled on a single mt contig. In contrast, the mt genomes of the two Coeloplana species were initially each assembled on several contigs (8 for C. yulianicorum and 3 for C. loyai). Therefore, for these two species tBlastx searches were performed using the mt genome of Mnemiopsis leidyi as query [12] with the aim of identifying additional mt contigs. The mt contigs thereby identified were then elongated by read mapping: setting a minimum overlap of 25 bp between a read and a mt contig, we were able to extend a mt contig by at least 75 bp at each round of read mapping, since the single reads are at least 100 bp long. Several rounds of read mapping allowed us to elongate the mt contigs by ~ 500 bp on each end, and Blastn searches were then conducted to identify the contigs whose ends matched.
Once the circular structure of each mt chromosome had been determined, a last paired-read mapping analysis was performed for each species with Geneious Pro (Version 6.1.7, Biomatters, Auckland, New Zealand). This analysis allowed us to confirm the circular organization of the mt genome of each species as well as to verify manually the quality of the assembly for each base of the sequence. The mean coverages of the assembled genomes were 530 (standard deviation [SD] 190; range: 67–901), 11 (SD 4; range: 2–27) and 81 (SD 26; range: 22–160) for C. loyai, C. yulianicorum and V. multiformis, respectively.
The cox1 sequence of each mt genome was compared to the sequence obtained by Alamaru et al. [23] to confirm that the correct species had been amplified.
Mt. genome annotation
A preliminary annotation of the mitochondrial sequences was performed with the web server MITOS (revision 656) [10]. The MITOS annotation, however, contained several errors (e.g., absence of genes such as nad2 and nad3; miss-annotated genes such as atp6). Consequently, all ORFs detected in the Coeloplana and Vallicula mt genomes were aligned with the protein genes annotated in the Mnemiopsis and Pleurobrachia mt genomes. Start and stop codon positions were chosen based on two criteria: minimization of gene overlap and maximization of ctenophore sequence similarity [27]. URFs longer than 100 bp that encompassed putative transmembrane-domain were also annotated. Transmembrane helices were detected in protein sequences with the TMHMM Server v. 2.0 [32] using default settings.
rRNA genes were identified using covariance models as implemented in INFERNAL 1.1 [33]. Specifically, the programs Cmbuild and Cmcalibrate were used with default settings to construct covariance models based on the secondary structure predicted for the rnl and rns genes of M. leidyi [12]. Using these models (one for each rRNA), the program Cmsearch was run with the parameters: -g --notrunc --smxsize 3000 -E 10.000000. The RNA regions detected in each species were then aligned and the start and end of each rRNA gene were defined based on sequence similarity.
tRNA genes were searched with the programs tRNAscan [34], ARWEN [35] and MITOS (which implements MITFI) [10].
Base compositions were computed with Geneious (version R6.1). Potential modifications of the mold genetic code were investigated both manually and with the webserver GenDecoder v.1.6 [20] using default parameters.
Perfect repeats having a repeat unit longer than 20 bp were searched with the REPFIND webserver (http://zlab.bu.edu/repfind/form.html) [36] under default settings with and without the option "Filter low complexity sequence".
Genome rearrangement
Pairwise breakpoint distances (BD) were computed between each pair of ctenophore mt genomes using the web server CREx [37]. Normalized breakpoint distances (BDn) were obtained by dividing the breakpoint distance by 13 (i.e., the number of genes shared between the ctenophores mt genomes) [38].
Phylogenetic analysis
Mitochondrial protein coding sequences for representatives of the animal diversity, together with fungi and choanoflagellate outgroups, were downloaded from the NCBI organelle genome resource (https://www.ncbi.nlm.nih.gov/genome/organelle/). Our dataset comprises 20 Cnidaria species, 10 Porifera, 4 Placozoa and 14 Bilateria, plus 6 outgroups.
Translated sequences of cob, cox1–3, nad1, nad3 and nad5 (the most conserved ctenophore genes) were aligned at the amino acid level using MAFFT (version 7) [39] under the L-INS-i refinement strategy. The web server Guidance 2 [40] was used to remove positions with a low confidence score (i.e., below 0.93), as well as positions present in less than 25% of the species. The sequence alignments are provided in Additional file 7.
Phylogenetic inference was conducted using PhyloBayes v3.3b. We used the CAT + GTR + Γ mixture model for the Bayesian analysis. Bayesian analyses were conducted with three chains run for 90,000 cycles each and sampled every 10 cycles, with the first 2000 trees discarded as burn-in. The maximum and average differences, observed at the end of the run were 0.09 and 0.0053, respectively. Similarly, we verified that the effsize and rel_diff of all parameters were higher than 50 and lower or equal to 0.3, respectively, which indicates a correct chain convergence.
Additional files
Comparison between annotation of the mt genome of Pleurobrachia bachei (accession JN392469) [18] and the re-annotation performed in the current work. (PPT 224 kb)
Alignment of the 5′ region of the cox1 gene among Ctenophora. Figure, illustrating the difference in the 5′ region of the cox1 gene between Kohn et al. [18] annotation of the Pleurobrachia genome and the amended annotation performed in the current work. (DOCX 15 kb)
Reannotation of the mt genome of Pleurobrachia bachei. Flatfile in Genbank format. (TXT 26 kb)
Codon usage of Ctenophora species. (XLSX 29 kb)
Alignment of the rnl sequences of ctenophores. (DOCX 937 kb)
Alignment of the rns sequences of ctenophores. (DOCX 584 kb)
Sequence alignment file used in the phylogenetic analyses. DNA sequence alignment, in Nexus format, used to reconstruct the phylogenetic tree present in Fig. 3. (NEX 193 kb)
Acknowledgements
We thank the Steinhardt Museum of Natural History and National Research Center, as well as Tal Katz-Ezov and the Technion Genome Center for their technical help. We would like to express our gratitude to the editor and three reviewers for their comments, Naomi Paz for editing the text and Eran Brokovich for editing the photos. Finally, we would like to address special thanks and appreciation to Prof. Yossi Loya for his support and encouragement throughout the years.
Funding
The sampling of the biological material was funded by the Israel Taxonomy Initiative to AA, DH and YL. The sequencing and data analyses were supported by the Israel Science Foundation (ISF) grant No. 161/15 to DH. Finally, CG research was suported by the Molecular Biodiversity Laboratory of the Italian node of Lifewatch (CNR-Italy).
Availability of data and materials
The mtDNA sequences of C. loyai, C. yulianicorum and V. multiformis have been deposited at the EMBL-EBI European Nucleotide Archive under accession numbers LN898113, LN898114 and LN898115 respectively. The data matrix generated and analysed during this study is included in the Additional files (Additional file 7).
Abbreviations
- aaRS
Aminoacyl tRNA synthetase
- atp6/8
ATPase subunit 6/8 gene
- BD
Pairwise breakpoint distance
- BDn
Normalized breakpoint distance
- bp
Base pair
- cob
Cytochrome b gene
- cox1–3
Cytochrome oxidase 1–3 gene
- gDNA
Genomic DNA
- kb
Kilobase
- mt
Mitochondrial
- nad1–6
NADH dehydrogenase subunit 1–6 gene
- NC
Non-coding region
- NCBI
National Center for Biotechnology Information
- PP
Posterior probability
- rnl
Large subunit ribosomal RNA
- rns
Small subunit ribosomal RNA
- rRNAs
Ribosomal ribonucleic acid
- SD
Standard deviation
- tRNA
Transfer RNA acid
- URF
Unidentified reading frames
Authors’ contributions
AA and DH conceived the study. DH supervised and supported the research. AA collected the samples and performed the DNA extractions. DH assembled the mt sequences from the reads. DH and HA conducted the phylogenetic analysis, drew the figures and drafted the manuscript. CG made substantial contributions to the analysis of the data. All authors assisted in writing and revising the manuscript and approved the final version of the text.
Ethics approval and consent to participate
The studied organisms were collected in the Gulf of Eilat under permit 2010/37891 issued by the Israel Nature and National Park Protection Authority.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12862-018-1186-1) contains supplementary material, which is available to authorized users.
Contributor Information
Hanan Arafat, Email: hanan.arafat281@gmail.com.
Ada Alamaru, Email: alamarua@gmail.com.
Carmela Gissi, Email: carmela.gissi@uniba.it.
Dorothée Huchon, Email: huchond@post.tau.ac.il.
References
- 1.Lavrov DV, Pett W. Animal mitochondrial DNA as we do not know it: mt-genome organization and evolution in nonbilaterian lineages. Genome Biol Evol. 2016;8:2896–2913. doi: 10.1093/gbe/evw195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gissi C, Iannelli F, Pesole G. Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity. 2008;101:301–320. doi: 10.1038/hdy.2008.62. [DOI] [PubMed] [Google Scholar]
- 3.Smith DR. The past, present and future of mitochondrial genomics: have we sequenced enough mtDNAs? Brief Funct Genomics. 2016;15:47–54. doi: 10.1093/bfgp/elv027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y, Coleman-Derr D, Chen G, Gu YQ. OrthoVenn: a web server for genome wide comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 2015;43:78–84. doi: 10.1093/nar/gkv487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh TR, Tsagkogeorga G, Delsuc F, Blanquart S, Shenkar N, Loya Y, Douzery EJP, Huchon D. Tunicate mitogenomics and phylogenetics: peculiarities of the Herdmania momus mitochondrial genome and support for the new chordate phylogeny. BMC Genomics. 2009;10:534. doi: 10.1186/1471-2164-10-534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hazkani-Covo E, Zeller RM, Martin W. Molecular poltergeists: mitochondrial DNA copies (numts) in sequenced nuclear genomes. PLoS Genet. 2010;6:e1000834. doi: 10.1371/journal.pgen.1000834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gissi C, Pesole G, Mastrototaro F, Iannelli F, Guida V, Griggio F. Hypervariability of ascidian mitochondrial gene order: exposing the myth of deuterostome organelle genome stability. Mol Biol Evol. 2009;27:211–215. doi: 10.1093/molbev/msp234. [DOI] [PubMed] [Google Scholar]
- 8.Hebert PD, Cywinska A, Ball SL. Biological identifications through DNA barcodes. Proc R Soc Lond B Biol Sci. 2003;270:313–321. doi: 10.1098/rspb.2002.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanabe AS, Toju H. Two new computational methods for universal DNA barcoding: a benchmark using barcode sequences of bacteria, archaea, animals, fungi, and land plants. PLoS One. 2013;8:e76910. doi: 10.1371/journal.pone.0076910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bernt M, Donath A, Jühling F, Externbrink F, Florentz C, Fritzsch G, Pütz J, Middendorf M, Stadler PF. MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol. 2013;69:313–319. doi: 10.1016/j.ympev.2012.08.023. [DOI] [PubMed] [Google Scholar]
- 11.De Meo PDO, D’Antonio M, Griggio F, Lupi R, Borsani M, Pavesi G, Castrignanò T, Pesole G, Gissi C. MitoZoa 2.0: a database resource and search tools for comparative and evolutionary analyses of mitochondrial genomes in Metazoa. Nucleic Acids Res. 2012;40:D1168–72. [DOI] [PMC free article] [PubMed]
- 12.Pett W, Ryan JF, Pang K, Mullikin JC, Martindale MQ, Baxevanis AD, Lavrov DV. Extreme mitochondrial evolution in the ctenophore Mnemiopsis leidyi: insight from mtDNA and the nuclear genome. Mitochondrial DNA. 2011;22:130–142. doi: 10.3109/19401736.2011.624611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mills CE. Phylum Ctenophora: list of all valid species names. [Internet Document]. Available from: http://faculty.washington.edu/cemills/Ctenolist.html. Last Update Date: 30 Mar 2017. 1998-Present.
- 14.Moroz LL, Kocot KM, Citarella MR, Dosung S, Norekian TP, Povolotskaya IS, Grigorenko AP, Dailey C, Berezikov E, Buckley KM. The ctenophore genome and the evolutionary origins of neural systems. Nature. 2014;510:109–114. doi: 10.1038/nature13400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Podar M, Haddock SH, Sogin ML, Harbison GR. A molecular phylogenetic framework for the phylum Ctenophora using 18S rRNA genes. Mol Phylogenet Evol. 2001;21:218–230. doi: 10.1006/mpev.2001.1036. [DOI] [PubMed] [Google Scholar]
- 16.Simion P, Bekkouche N, Jager M, Quéinnec É, Manuel M. Exploring the potential of small RNA subunit and ITS sequences for resolving phylogenetic relationships within the phylum Ctenophora. Zoology. 2015;118:102–14. [DOI] [PubMed]
- 17.Simion P, Philippe H, Baurain D, Jager M, Richter DJ, Di Franco A, Roure B, Satoh N, Quéinnec É, Ereskovsky A. A large and consistent phylogenomic dataset supports sponges as the sister group to all other animals. Curr Biol. 2017;27:958–967. doi: 10.1016/j.cub.2017.02.031. [DOI] [PubMed] [Google Scholar]
- 18.Kohn AB, Citarella MR, Kocot KM, Bobkova YV, Halanych KM, Moroz LL. Rapid evolution of the compact and unusual mitochondrial genome in the ctenophore, Pleurobrachia bachei. Mol Phylogenet Evol. 2012;63:203–207. doi: 10.1016/j.ympev.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giegé R, Jühling F, Pütz J, Stadler P, Sauter C, Florentz C. Structure of transfer RNAs: similarity and variability. Wiley Interdiscip Rev RNA. 2012;3:37–61. [DOI] [PubMed]
- 20.Abascal F, Zardoya R, Posada D. GenDecoder: genetic code prediction for metazoan mitochondria. Nucleic Acids Res. 2006;34:W389–W393. doi: 10.1093/nar/gkl044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pett W, Lavrov DV. Cytonuclear interactions in the evolution of animal mitochondrial tRNA metabolism. Genome Biol Evol. 2015;7:2089–2101. doi: 10.1093/gbe/evv124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haen KM, Pett W, Lavrov DV. Parallel loss of nuclear-encoded mitochondrial aminoacyl-tRNA synthetases and mtDNA-encoded tRNAs in Cnidaria. Mol Biol Evol. 2010;27:2216–2219. doi: 10.1093/molbev/msq112. [DOI] [PubMed] [Google Scholar]
- 23.Alamaru A, Hoeksema BW, van der Meij SE, Huchon D. Molecular diversity of benthic ctenophores (Coeloplanidae) Sci Rep. 2017;7:6365. doi: 10.1038/s41598-017-06505-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yokobori S-I, Ueda T, Feldmaier-Fuchs G, Pääbo S, Ueshima R, Kondow A, Nishikawa K, Watanabe K. Complete DNA sequence of the mitochondrial genome of the ascidian Halocynthia roretzi (Chordata, Urochordata) Genetics. 1999;153:1851–1862. doi: 10.1093/genetics/153.4.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gissi C, Iannelli F, Pesole G. Complete mtDNA of Ciona intestinalis reveals extensive gene rearrangement and the presence of an atp8 and an extra trnM gene in ascidians. J Mol Evol. 2004;58:376–389. doi: 10.1007/s00239-003-2559-6. [DOI] [PubMed] [Google Scholar]
- 26.Alamaru A, Brokovich E, Loya Y. Four new species and three new records of benthic ctenophores (Family: Coeloplanidae) from the Red Sea. Mar Biodivers. 2016;46:261–279. doi: 10.1007/s12526-015-0362-4. [DOI] [Google Scholar]
- 27.Rubinstein ND, Feldstein T, Shenkar N, Botero-Castro F, Griggio F, Mastrototaro F, Delsuc F, Douzery EJ, Gissi C, Huchon D. Deep sequencing of mixed total DNA without barcodes allows efficient assembly of highly plastic ascidian mitochondrial genomes. Genome Biol Evol. 2013;5:1185–1199. doi: 10.1093/gbe/evt081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shenkar N, Koplovitz G, Dray L, Gissi C, Huchon D. Back to solitude: solving the phylogenetic position of the Diazonidae using molecular and developmental characters. Mol Phylogenet Evol. 2016;100:51–56. doi: 10.1016/j.ympev.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 29.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–2.
- 30.Xie Y, Wu G, Tang J, Luo R, Patterson J, Liu S, Huang W, He G, Gu S, Li S. SOAPdenovo-trans: de novo transcriptome assembly with short RNA-Seq reads. Bioinformatics. 2014;30:1660–1666. doi: 10.1093/bioinformatics/btu077. [DOI] [PubMed] [Google Scholar]
- 31.Song J, Hwang S, Lee S, Park J. New records of creeping ctenophores, genus Coeloplana (Tentaculata: Platyctenida: Coeloplanidae), from Korea. Anim Syst Evol Divers. 2011;27:47–52. doi: 10.5635/KJSZ.2011.27.1.047. [DOI] [Google Scholar]
- 32.Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 33.Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 2013;29:2933–2935. doi: 10.1093/bioinformatics/btt509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bernt M, Braband A, Middendorf M, Misof B, Rota-Stabelli O, Stadler PF. Bioinformatics methods for the comparative analysis of metazoan mitochondrial genome sequences. Mol Phylogenet Evol. 2013;69:320–327. doi: 10.1016/j.ympev.2012.09.019. [DOI] [PubMed] [Google Scholar]
- 36.Betley JN, Frith MC, Graber JH, Choo S, Deshler JO. A ubiquitous and conserved signal for RNA localization in chordates. Curr Biol. 2002;12:1756–1761. doi: 10.1016/S0960-9822(02)01220-4. [DOI] [PubMed] [Google Scholar]
- 37.Bernt M, Merkle D, Ramsch K, Fritzsch G, Perseke M, Bernhard D, Schlegel M, Stadler PF, Middendorf M. CREx: inferring genomic rearrangements based on common intervals. Bioinformatics. 2007;23:2957–2958. doi: 10.1093/bioinformatics/btm468. [DOI] [PubMed] [Google Scholar]
- 38.Blanchette M, Bourque G, Sankoff D. Breakpoint phylogenies. Genome Inform Ser Workshop. 1997;8:25–34. [PubMed] [Google Scholar]
- 39.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sela I, Ashkenazy H, Katoh K, Pupko T. GUIDANCE2: accurate detection of unreliable alignment regions accounting for the uncertainty of multiple parameters. Nucleic Acids Res. 2015;43:W7–W14. doi: 10.1093/nar/gkv318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comparison between annotation of the mt genome of Pleurobrachia bachei (accession JN392469) [18] and the re-annotation performed in the current work. (PPT 224 kb)
Alignment of the 5′ region of the cox1 gene among Ctenophora. Figure, illustrating the difference in the 5′ region of the cox1 gene between Kohn et al. [18] annotation of the Pleurobrachia genome and the amended annotation performed in the current work. (DOCX 15 kb)
Reannotation of the mt genome of Pleurobrachia bachei. Flatfile in Genbank format. (TXT 26 kb)
Codon usage of Ctenophora species. (XLSX 29 kb)
Alignment of the rnl sequences of ctenophores. (DOCX 937 kb)
Alignment of the rns sequences of ctenophores. (DOCX 584 kb)
Sequence alignment file used in the phylogenetic analyses. DNA sequence alignment, in Nexus format, used to reconstruct the phylogenetic tree present in Fig. 3. (NEX 193 kb)
Data Availability Statement
The mtDNA sequences of C. loyai, C. yulianicorum and V. multiformis have been deposited at the EMBL-EBI European Nucleotide Archive under accession numbers LN898113, LN898114 and LN898115 respectively. The data matrix generated and analysed during this study is included in the Additional files (Additional file 7).