

Abstract
Sal-like 4 (SALL4) is a transcription factor that enhances proliferation and migration in breast cancer cells. SALL4 expression therefore has the potential to promote cancer malignancy. However, the regulatory mechanisms involved in SALL4 protein expression have not been thoroughly elucidated. In this study, we observed that treating MCF-7 and SUM159 breast cancer cell lines with a proteasome inhibitor increases SALL4 protein levels, suggesting that SALL4 is degraded by the ubiquitin–proteasome system. Using immunoprecipitation to uncover SALL4-binding proteins, we identified an E3 ubiquitin-protein ligase, tripartite motif–containing 21 (TRIM21). Using an EGFP reporter probe of the major SALL4 isoform SALL4B, we observed that shRNA-mediated knockdown of TRIM21 increases cellular SALL4B levels. Immunostaining experiments revealed that TRIM21 localizes to the nucleus, and a K64R substitution in the nuclear localization motif in SALL4B increased SALL4B levels in the cytoplasm. These results suggested that TRIM21 is involved in nuclear SALL4 degradation. To identify the amino acid residue that is targeted by TRIM21, we fragmented the SALL4B sequence, fused it to EGFP, and identified Lys-190 in SALL4B as TRIM21's target residue. Amino acid sequence alignments of SALL family members indicated that the region around SALL4 Lys-190 is conserved in both SALL1 and SALL3. Because SALL1 and SALL4 have similar functions, we constructed a SALL1-EGFP probe and found that the TRIM21 knockdown increases SALL1 levels, indicating that TRIM21 degrades both SALL1 and SALL4. Our findings extend our understanding of SALL4 and SALL1 regulation and may contribute to the development of SALL4-targeting therapies.
Keywords: E3 ubiquitin ligase, breast cancer, molecular biology, protein degradation, transcription factor, Ro52, SALL family, SALL4, TRIM21
Introduction
The expression of certain transcription factors induces oncogenic and malignant transformations by altering the gene expression pattern. Therefore, studies on the regulatory mechanisms of oncogenic and malignant transcription factors may contribute to the understanding of cancer progression and the development of cancer therapy.
Sal-like 4 (SALL4)2 is a transcription factor. In various types of cancers, SALL4 expression correlates to poor prognosis (1–3). In the cancer genome atlas data, high SALL4 expression was observed in malignant basal-like breast cancer (4, 5). SALL4 binds to genomic DNA with its zinc finger motifs (6) and recruits histone deacetylase complex (7). Several studies have reported the regulation of SALL4 expression. β-Catenin/lymphoid enhancer factor 1 and β-catenin/T-cell factor 4E complexes directly up-regulate SALL4 transcription (8). Signal transducer and activator of transcription 3 up-regulates SALL4 expression by binding to and activating the SALL4 promoter in breast cancer cells (9). SALL4 protein suppresses SALL4 transcription (10). MicroRNAs regulate gene expression post-transcriptionally. MicroRNA-107 suppresses SALL4 expression and reduces glioma cell proliferation (11). In breast cancer, SALL4 mRNA is targeted by microRNA-33b (12). SUMOylation on the lysine residue is one type of post-translational modification. SALL4 has four SUMOylation sites, and SUMOylation promotes SALL4 activity (13). In addition, nuclear receptor–binding protein 1 is involved in the regulation of SALL4 protein levels (14). However, the regulatory mechanism for SALL4 expression has not been fully understood, especially in post-translational regulation.
Therefore, we aimed to reveal the mechanism of post-translational regulation of SALL4 using breast cancer cell lines because SALL4 is known as a malignant transcription factor in breast cancer cells (5, 15) and is involved in breast cancer progression (16). In breast cancer cells, SALL4 up-regulates the expression of BMI-1 (15) similar to its function in leukemic cells (17). BMI-1 suppresses the expression of INK4 locus genes, which encode cell cycle inhibitors (18). In addition, SALL4 up-regulates the expression of integrin genes ITGA6 and ITGB1 for cell migration in basal-like breast cancer cells (5). In this study, we observed an increase in SALL4 protein levels in the cells treated with a proteasome inhibitor. We identified tripartite motif–containing 21 (TRIM21; also known as Ro52) as a degradation factor for SALL4. TRIM21's target residue was identified. This study demonstrates TRIM21-mediated SALL4 post-translational regulation, which may facilitate the understanding of SALL4 regulation and the development of SALL4-targeted therapies.
Results
TRIM21 is involved in SALL4 degradation
In breast cancer cell lines, a low SALL4 protein level suggests the possibility that SALL4 is regulated post-translationally (Fig. 1A), although the protein level is regulated by several mechanisms. Because the ubiquitin–proteasome system degrades particular proteins, we treated breast cancer cell lines MCF-7 and SUM159 with the proteasome inhibitor MG-132 and analyzed the SALL4 protein level. We observed an increase in the level of SALL4B, an isoform of SALL4, in the cells treated with MG-132 (Fig. 1A). This finding suggests that SALL4 is degraded by the ubiquitin–proteasome system.
Figure 1.

TRIM21 is involved in SALL4 degradation. A, immunoblot image of SALL4 protein is shown. MCF-7 and SUM159 cells were treated with MG-132 (n = 3). The arrow indicates SALL4B bands. B, SALL4B-EGFP probe is depicted. C, relative mRNA levels of TRIM21 are shown. shRNA-mediated TRIM21 knockdown was performed in MCF-7 cells. D, immunoblot image of SALL4B-EGFP probe is shown (n = 4 TRIM21 knockdown constructs). The arrow indicates the position of SALL4B-EGFP. shScr was used as a control. E, immunoblot for EGFP is shown (n = 3). shTRIM21-5 was used. F, relative mRNA levels of TRIM21 and SALL4 are shown. TRIM21 knockdown with shTRIM21-5 was performed in SUM159 cells. n.s., not significant; **, p < 0.01 compared with shScr control (Student's t test). Error bars indicate standard deviations (n = 3).
In the ubiquitin–proteasome system, E3 ubiquitin-protein ligase binds to a target protein for ubiquitination. To identify the proteins binding to SALL4, we performed immunoprecipitation with anti-SALL4 antibody in SUM159 and MDA-MB-231 cells followed by MS. In the results, we identified an E3 ubiquitin-protein ligase, TRIM21 (Table 1). This suggests that TRIM21 targets SALL4.
Table 1.
Mass spectrometry results of SALL4-binding proteins
Only proteins identified in both cell lines are shown.
| Protein name | Coverage |
Number of peptides (confidence ≥95%) |
||
|---|---|---|---|---|
| SUM159 cells | MDA-MB-231 cells | SUM159 cells | MDA-MB-231 cells | |
| % | ||||
| E3 ubiquitin-protein ligase TRIM21 | 18.3 | 16.6 | 7 | 6 |
| Nuclease-sensitive element–binding protein 1 | 17.9 | 11.7 | 4 | 3 |
| Protein disulfide-isomerase | 15.0 | 1.6 | 8 | 1 |
| T-complex protein 1 subunit η | 11.2 | 2.9 | 5 | 2 |
| T-complex protein 1 subunit θ | 10.4 | 13.0 | 5 | 7 |
| Arfaptin-1 | 6.4 | 9.4 | 1 | 2 |
To monitor the SALL4 protein level, we constructed the SALL4B-EGFP construct (Fig. 1B). This construct was transcribed from a CMV promoter, and the mRNA does not have the SALL4 3′-UTR, indicating that the protein level of SALL4B-EGFP reflects the post-translational regulation of SALL4. To determine whether TRIM21 is involved in SALL4 degradation, we performed TRIM21 knockdown in breast cancer cell lines in which the SALL4B-EGFP construct was introduced and analyzed the SALL4B-EGFP level. We observed an increase in the SALL4B-EGFP level in TRIM21 knockdown cells (Fig. 1, C and D). TRIM21 knockdown did not reduce the EGFP protein level in the cells expressing only EGFP (Fig. 1E) and did not change the SALL4 mRNA level (Fig. 1F). These results indicate that TRIM21 is involved in the post-translational regulation of SALL4.
TRIM21 reduces nuclear SALL4 level
SALL4 is a transcription factor that localizes in the nucleus (19). To investigate where TRIM21 localizes, we performed immunohistochemistry with anti-TRIM21 antibody. We observed nuclear localization of TRIM21 in breast tumor tissues, including benign, ductal carcinoma in situ, and invasive ductal carcinoma (Fig. 2A). Although the number of samples was small, invasive ductal carcinoma may tend to have negative/low TRIM21 expression (four of six samples). Moreover, we immunostained breast cancer cell lines and conducted confocal microscopy. We observed TRIM21 signals in the nuclei (Fig. 2B). The results of line profiling showed overlap of TRIM21 signal and nuclear staining (Fig. 2B). These observations indicate that TRIM21 mainly localizes in the nucleus in breast cancer cells.
Figure 2.

TRIM21 regulates nuclear SALL4 level. A, immunohistochemical staining for TRIM21 was performed. Images of benign (n = 3), ductal carcinoma in situ (DCIS) (n = 1), and invasive ductal carcinoma (IDC) (n = 6) are shown. The inset is the magnified image of each boxed area. Arrowheads indicate TRIM21 signals. For simplicity, not all signals are indicated with arrowheads. Scale bar, 100 μm. B, confocal images of TRIM21 staining are shown. Hoechst was used for nuclear staining. Signal intensities of the line indicated by arrows are graphed. Scale bar, 10 μm. C, K64R mutation is depicted. The immunoblot image shows SALL4B-EGFP level (arrow) (n = 3). shTRIM21-5 was used. D, confocal images show change in subcellular localization of SALL4B-EGFP probe. Arrowheads indicate cytoplasmic SALL4-K64R signals. Scale bar, 10 μm.
In SALL4's nuclear localization signal, Lys-64 is necessary for localization in the nucleus, and K64R mutation increases the cytoplasmic level of SALL4 (19). To determine whether SALL4 is degraded by TRIM21 in the nucleus, we introduced the K64R mutation to the SALL4B-EGFP probe (Fig. 2C). We observed SALL4B-EGFP signal in the cytoplasm (Fig. 2D). In the immunoblotting results, we observed an increase in SALL4B-EGFP signal in cells expressing SALL4B K64R-EGFP (Fig. 2C), suggesting that SALL4 is degraded in the nucleus. TRIM21 knockdown slightly increased GFP immunoreaction (Fig. 2C) because SALL4B K64R localizes in both the cytoplasm and nucleus (19), and TRIM21 may be able to reduce the nuclear SALL4B K64R level. Cytoplasmic SALL4B K64R may escape from TRIM21-mediated degradation. These results suggest that TRIM21 reduces the SALL4 level in the nucleus.
TRIM21 targets Lys-190 in SALL4 amino acid sequence
Ubiquitin ligases ubiquitinate the lysine residue of a target protein. To identify which lysine residue is targeted by TRIM21, we fragmented the SALL4B amino acid (aa) sequence (Fig. 3A). Because SALL4 is degraded in the nucleus, the nuclear localization signal was not removed in all fragmented SALL4B-EGFP probes. We introduced these constructs into MCF-7 cells and performed TRIM21 knockdown. We observed that expression of the probes having the SALL4B(1–255) and SALL4B(1–70,256–450) sequences was lower than that of the others. We observed an increase in GFP immunoreaction by TRIM21 knockdown only in the cells expressing the probe having the SALL4B(1–255) sequence, whereas the protein levels of the other probes were not changed by TRIM21 knockdown (Fig. 3B). This finding indicates that there is a TRIM21 target residue in the region between aa 172 and 255 (Fig. 3B).
Figure 3.

TRIM21 targets Lys-190 in SALL4 amino acid sequence. A, structures of fragmented-SALL4B-EGFP probes are shown. NLS, nuclear localization signal. B, EGFP signals of MCF-7 cells expressing the probes are shown (n = 3). TRIM21 knockdown was performed with shTRIM21-5. The identified TRIM21 target region (aa 172–255) is depicted. C, the immunoblot image shows GFP immunoreactions of MCF-7 cells expressing SALL4B(1–255)-EGFP probes with Lys to Arg mutations (mut.) at residues 173, 175, and 190 (n = 3). D, TRIM21 knockdown was performed in MCF-7 cells expressing the SALL4B(1–255) K190R-EGFP probe (n = 3). E, the immunoblot image shows the signal of the polyubiquitinated SALL4B(1–255)-EGFP probe (n = 3). MCF-7 cells were treated with MG-132. Input samples were used for the total amount of each probe (α-GFP) and internal control (α-β-actin). F, the immunoblot image shows overexpression of the SALL4B-EGFP probe with or without K190R mutation (n = 3). Short- and long-exposure images of GFP immunoreaction are shown. The results of migration assays are shown. Arrowheads indicate migrated cells. Scale bar, 100 μm. The graph shows the migrated cell numbers. n.s., not significant; *, p < 0.05 compared with EGFP vector (vec.) control (Dunnett's t test). Error bars indicate standard deviations (n = 3). ShTRIM21-5 was used (B and D). The arrow indicates the signal of each SALL4B-EGFP probe (B–F).
In this region, there are three lysine residues, Lys-173, Lys-175, and Lys-190. We introduced a Lys to Arg mutation in each candidate residue in the SALL4B(1–255)-EGFP probe. We observed an increase in protein level only in the K190R mutation (Fig. 3C). The protein level of SALL4B K190R-EGFP was not increased by TRIM21 knockdown (Fig. 3D). These results indicate that Lys-190 is the target residue of TRIM21-mediated degradation.
To analyze the ubiquitination of the SALL4B-EGFP probe, we used the tandem ubiquitin-binding entity (TUBE) system. TUBE binds to a ubiquitin tetramer (20), and purification with agarose-TUBE resins enriches polyubiquitinated proteins. With this resin, we purified samples from cells expressing SALL4B(1–255)-EGFP probe with or without the K190R mutation. No detectable EGFP band was observed in the purified sample of EGFP vector control (Fig. 3E). The estimated molecular masses of SALL4B(1–255)-EGFP probe and ubiquitin tetramer are ∼54.5 and 34.4 kDa, respectively. Therefore, the estimated molecular mass of SALL4B-EGFP probe with four ubiquitins is 88.9 kDa. In the purified samples, immunostaining with anti-GFP antibody showed bands between the 72.8 and 96.1 kDa markers, and the intensity of the band of the K190R mutation was weaker than that of the control (Fig. 3E), indicating that Lys-190 is one of the ubiquitination sites on the SALL4 aa sequence.
SALL4 enhances cell migration and is expressed in basal-like breast cancer cells that have high migratory ability (5). To analyze the effect of SALL4 overexpression in a low-migratory luminal breast cancer cell line, MCF-7, we introduced the K190R mutation in full-length SALL4B-EGFP probe and overexpressed it in MCF-7 cells. As expected, we observed high expression of SALL4B-EGFP probe in the K190R mutation group (Fig. 3F). In cell migration assays, SALL4B K190R-EGFP–expressing MCF-7 cells showed an increase in cell migration (Fig. 3F). These results indicate that Lys-190 is a residue involved in the regulation of SALL4 protein level, and loss of SALL4 protein regulation enhances cell migration in breast cancer cells.
TRIM21 down-regulates the SALL1 protein level
We aligned aa sequences of the SALL family and observed that the sequence containing Lys-190 is conserved in SALL1, SALL3, and SALL4 (Fig. 4A and Data S1). We therefore hypothesized that TRIM21 is involved in the degradation of other SALL proteins. The symptoms of patients with SALL1 and SALL4 mutations are similar (21, 22); their mutations cause autosomal-dominant developmental disorders. In mouse neural tube closure, Sall1 and Sall4 have redundancy (23). To determine whether TRIM21 degrades SALL1, we constructed SALL1-EGFP fusion and performed TRIM21 knockdown. The results indicate that the SALL1-EGFP level was increased by TRIM21 knockdown (Fig. 4B). Moreover, we performed TRIM21 knockdown in SUM159 cells and analyzed the SALL4 and SALL1 levels. We observed an increase in endogenous levels of SALL4 and SALL1, indicating that TRIM21 is involved in SALL4 and SALL1 degradation.
Figure 4.
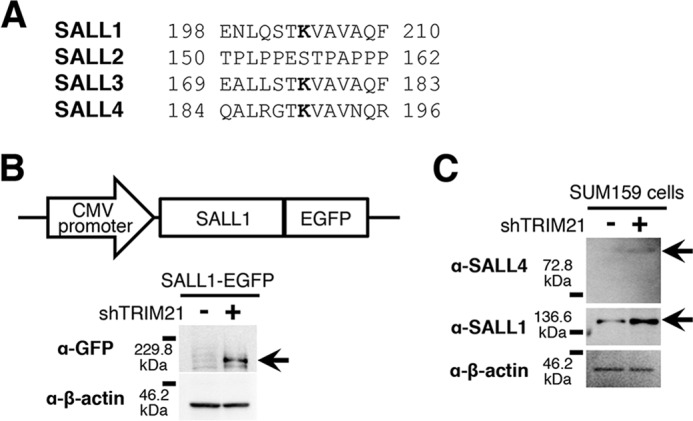
TRIM21 degrades SALL1. A, conservation of amino acid sequences around SALL4 Lys-190 is shown. Bold letters indicate conserved lysine residues. B, SALL1-EGFP probe was constructed and introduced into MCF-7 cells. The immunoblot shows the SALL1-EGFP signal (n = 3), which increased by TRIM21 knockdown with shTRIM21-5 (arrow). C, immunoblot images of SALL4 and SALL1 are shown (n = 3). TRIM21 knockdown was performed in SUM159 cells.
Discussion
SALL4 transcription is regulated by various factors (8, 9). SALL4 protein suppresses SALL4 transcription (10), and SALL4 mRNA is post-transcriptionally regulated (11, 12). This study revealed one of the mechanisms of SALL4 post-translational regulation in breast cancer cells (Fig. 5). We observed that SALL4 is degraded by the ubiquitin–proteasome system. We identified an E3 ubiquitin-protein ligase, TRIM21. TRIM21 knockdown increased the level of SALL4B-EGFP probe, indicating that SALL4 is degraded by TRIM21. In breast cancer cells, TRIM21 localizes in the nucleus. In the SALL4 aa sequence, we identified Lys-190 as the target of TRIM21. SALL4 is polyubiquitinated at Lys-190. Furthermore, we found conservation of aa sequences around Lys-190 in SALL1, SALL3, and SALL4 and revealed that TRIM21 degrades SALL1 as well as SALL4.
Figure 5.
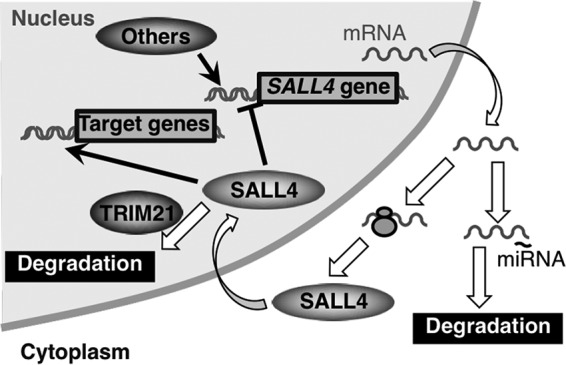
SALL4 expression is regulated post-translationally. In addition to transcriptional and post-transcriptional regulation, this study discovered one of the mechanisms of post-translational regulation of SALL4. TRIM21 degrades SALL4 protein in the nucleus. miRNA, microRNA.
SALL4 is a transcription factor that promotes proliferation, migration, and stemness (6). The factors for SALL4 transcriptional and post-transcriptional regulation have been identified (6, 8–12). It has been reported that SUMOylation controls SALL4 activity (13). Nuclear receptor–binding protein 1 is involved in SALL4 degradation (14). However, not all factors for SALL4 post-translational regulation have been identified to date. In this study, we showed that TRIM21 mediated the SALL4 degradation system in breast cancer cells. Because transcription factors change the expression of various genes, a high level of SALL4 causes cellular stress, and modulating SALL4 levels may benefit breast cancer cells. In the ubiquitin–proteasome system, an E3 ubiquitin-protein ligase binds to a target protein for ubiquitination, and a proteasome selectively degrades polyubiquitinated proteins. Although this study identified TRIM21, there might be unidentified E3 ubiquitin-protein ligases for SALL4 degradation.
The known function of TRIM21 is binding to internalized antibody-coated viruses for degradation (24). Additionally, TRIM21 acts as a E3 ubiquitin-protein ligase for certain proteins. TRIM21 targets Thr-187–phosphorylated p27 for cell cycle progression (25). In macrophages, TRIM21 activates interferon regulatory factor 8, which is a transcription factor for cytokine genes (26). TRIM21 degrades interferon regulatory factor 5 (27). In a loss of function study, TRIM21 showed reduced p62 polyubiquitination and increased antioxidant response (28). However, not all target proteins have been identified. This study discovered the novel TRIM21 target protein SALL4 and identified the TRIM21 target site in the SALL4 aa sequence. Although this study contributes to understanding TRIM21 function, we found that the phenotype of the TRIM21 knockdown differed from the phenotype of SALL4 overexpression,3 indicating that TRIM21 has other target proteins in breast cancer cells.
Interestingly, the SALL4 aa sequence around the TRIM21 target lysine is conserved in SALL1 and SALL3. There is a functional similarity between SALL1 and SALL4 (21–23). SALL1 and SALL4 mutations cause the developmental disorders Townes-Brocks syndrome and Okihiro/Duane-radial ray syndrome, respectively (21, 22). Both patients have similar symptoms, such as anomalies of limb, kidney, and eye. In mouse embryos, Sall1+/−;Sall4+/gene trap and Sall1−/−;Sall4+/gene trap mutants have defective neural tube closure, whereas Sall1+/+;Sall4+/gene trap and SALL1−/−;SALL4+/+ mice showed no neural tube defects (23). This finding indicates that SALL1 and SALL4 have a functional redundancy. We showed that SALL1 was also degraded by TRIM21 in breast cancer cells. These results suggest that TRIM21 suppresses the compensation between SALL1 and SALL4, explaining why there is a system for SALL4 post-translational regulation despite the existence of transcriptional and post-transcriptional regulations.
In various types of cancers, the correlation between SALL4 expression and a poor prognosis has been reported, and SALL4-targeted therapy has been considered (1–3). A SALL4-targeting peptide reduced cell viability of acute myeloid leukemia cells (29) and tumor growth of xenografted hepatocellular carcinoma (1). This study revealed the TRIM21-mediated SALL4 degradation system. Because this system functions not only to reduce the SALL4 protein level but also to prevent compensation by SALL1, promotion of the TRIM21 function may reduce malignancy. Our findings reveal a novel mechanism of SALL4 regulation in breast cancer and may contribute to the development of future therapies.
Experimental procedures
Cell culture
Breast cancer cell lines MCF-7 and SUM159 were obtained from American Type Culture Collection (Manassas, VA) and Asterand (Detroit, MI), respectively. MCF-7 cells were maintained with RPMI 1640 medium containing 10% FBS, 1 nm β-estradiol (Sigma-Aldrich Chemie GmbH; E2758). SUM159 cells were maintained with Ham's F-12 nutrient mixture containing 5% FBS, 5 μg/ml insulin (I9278, Sigma-Aldrich Chemie GmbH), 1 μg/ml hydrocortisone (H0888, Sigma-Aldrich Chemie GmbH), and 10 mm HEPES (17557-94, Nacalai Tesque, Kyoto, Japan). To inhibit proteasome-mediated degradation, cells were treated with 10 μm MG-132 (AG-CP3-0011, Adipogen, Liestal, Switzerland) for 24 h. To investigate cell migration, Boyden chambers with 8.0-μm-pore membrane were used. Fifteen thousand cells were plated in the upper compartment with serum-free medium. Ten percent FBS-containing medium was added into the lower compartment, and cells were incubated for 24 h. After fixation, migrated cells were stained with crystal violet. Images were collected at room temperature with the all-in-one BZ-9000 microscope (Keyence, Osaka, Japan) equipped with a 10× plan apochromatic objective lens (NA, 0.45) and BZ-II Viewer software (Keyence).
Immunoprecipitation and MS
For immunoprecipitation with anti-SALL4 antibody (ab29112, lot number GR146101-1, Abcam, Cambridge, UK; validated by immunoblotting in SALL4 knockdown cells (15)), a Dynabeads Co-immunoprecipitation kit (DB14321, Veritas, Tokyo, Japan) was used. Normal rabbit IgG (148-09551, Wako, Osaka, Japan) was used as a control. Samples were obtained from cultures of SUM159 and MDA-MB-231 cells (American Type Culture Collection; maintained with RPMI 1640 medium containing 10% FBS). The precipitated samples were electrophoresed and silver-stained. Gels containing ∼30–60-kDa proteins were excised and digested with the In-Gel Tryptic Digestion kit (89871, Thermo Fisher Scientific, Waltham, MA). The digested samples were eluted from the gels, dried, and resuspended in 0.1% formic acid. The digests were separated using a nanoflow LC, Nano-LC-Ultra 2D-plus, equipped with cHiPLC Nanoflex (Eksigent Technologies, Dublin, CA) in a trap and elute mode. Separation was carried out with a binary gradient using solvent A (0.1% formic acid and water) and solvent B (0.1% formic acid and acetonitrile). The gradient program was 2–33.2% B in 50 min, 33.2–98% B in 2 min, 98% B for 5 min, 98–2% B in 0.1 min, and 2% B for 17.9 min at 300 nl/min. The analytical column was set to 40 °C. The eluted samples were directly introduced into the mass spectrometer, a TripleTOF 5600+ System coupled with a NanoSprayIII source and heated interface (SCIEX, Framingham, MA) and ionized in electrospray ionization–positive mode. Data sets were acquired with an information-dependent acquisition method. The acquired data sets were analyzed by ProteinPilot software version 4.5 beta (SCIEX) with the UniProtKB/Swiss-Prot database (May 2017, Homo sapiens) appended with the known common contaminant database (SCIEX). The quality of the database search was confirmed by a false discovery rate analysis in which the reversed amino acid sequences were used as the decoy. The reliabilities of the protein identification were evaluated by the numbers of distinctly identified peptides having at least 95% confidence.
Immunoblotting
The primary antibodies were anti-SALL4 antibody (ab29112, lot number GR146101-1, Abcam; 1:1000 dilution), anti-GFP antibody (ab290, lot number GR19413-1, Abcam; 1:1000 dilution; validated by immunoblotting in EGFP-overexpressing cells), anti-SALL1 antibody (AP17204a, lot number SA111125AA, Abgent, San Diego, CA; 1:1000 dilution; validated by immunoblotting in TRIM21 knockdown cells), and anti-β-actin antibody (ab6276, lot number 66278-2, Abcam; 1:5000 dilution). The secondary antibody for mouse primary antibody was goat anti-mouse IgG antibody conjugated to peroxidase (31340, lot number IA10516511, Pierce; 1:50,000 dilution). The Easy-Western-II detection system (BCL-EZS21, Beacle, Kyoto, Japan) was used to detect SALL4 and GFP signals. Polyubiquitinated proteins were precipitated with agarose-TUBE2 resin (UM402, Nacalai Tesque) from 80–90% confluent cultures in a 10-cm dish treated with 10 μm MG-132 for 24 h.
Construction of SALL4-EGFP fusions
EGFP sequence was amplified with primers 5′-CACCATGGTGAGCAAGGGCGAG-3′ and 5′-CTTTACTTGTACAGCTCGTCCATG-3′. The PCR product was cloned into pENTR vector (45-0218, Life Technologies). Full-length and fragmented SALL4B sequences were fused to the NcoI site of the EGFP gene with the In-Fusion System (639641, Takara, Kusatsu, Japan). SALL4B-EGFP fusion genes were subcloned into lentiviral vector pLenti 6.3 (V533-06, Life Technologies). Infected cells were selected with 10 μg/ml blasticidin (ant-bl-1, Invivogen, San Diego, CA) for 7 days.
Gene knockdown
For TRIM21 knockdown, double-stranded oligos having shRNA sequences were cloned into lentiviral vector pLKO.1 (8453, Addgene, Cambridge, MA). The target sequences for TRIM21 knockdown were: 1, 5′-TGAGAAGTTGGAAGTGGAAAT-3′; 3, 5′-TGGAAGTGGAAATTGCAATAA-3′; 4, 5′-GAGTTGGCTGAGAAGTTGGAA-3′; 5, 5′-CAATCCGTGGCTGATACTTTC-3′. For a control, shScr (5′-CCTAAGGTTAAGTCGCCCTCG-3′) was used. Infected cells were selected with 1 μg/ml puromycin (ant-pr-1, Invivogen) for 4 days.
mRNA quantification
To extract RNA, cells were treated with TRIzol reagent (15596026, Thermo Fisher Scientific). Complementary DNAs were synthesized using SuperScript III reverse transcriptase (18080044, Thermo Fisher Scientific) and (dT)18 primer. Primers for quantitative RT-PCR were: EF1A1-forward, 5′-AAATGACCCACCAATGGAAGCAGC-3′; EF1A1-reverse, 5′-TGAGCCGTGTGGCAATCCAATACA-3′; TRIM21-forward, 5′-TGCTGCAGGAGGTGATAAT-3′; TRIM21-reverse, 5′-TCCTGAGTTCTGGAGAGGTAATA-3′; SALL4-forward, 5′-TACCACCAAAGGCAACTTAAAG-3′; SALL4-reverse, 5′-GTTCTCGATGGCCAACTTCC-3′. Relative mRNA levels were calculated by the ΔΔCt method.
Immunostaining
A tissue microarray of breast tumors obtained with the patients' informed consent (ab178114, Abcam) was purchased and stained with the anti-TRIM21 antibody clone D101D (92043, lot number 1, Cell Signaling Technology, Danvers, MA; 1:50 dilution; validated by immunofluorescence in TRIM21 knockdown cells). Subsequently, the sample was treated with the VECTASTAIN Elite ABC-HRP kit for rabbit IgG (PK-6101, Vector Laboratories, Burlingame, CA). Signals were developed with the NovaRED substrate kit (SK-4800, Vector Laboratories). Hematoxylin was used for counterstaining. Dehydrated sample was mounted with Malinol (2009-1, Muto Pure Chemicals, Tokyo, Japan). Images were collected at room temperature with the all-in-one BZ-9000 microscope equipped with a 20× plan apochromatic objective lens (NA, 0.75) and BZ-II Viewer software. Investigations were performed according to the principles expressed in the Declaration of Helsinki.
Breast cancer cells were cultured on a chamber slide (SCS-008, Matsunami Glass, Osaka, Japan) for 2 days. Fixed cells were stained with anti-TRIM21 antibody clone D101D or anti-GFP antibody clone GF090R (04404-26, lot number M7M9359, Nacalai Tesque; 1:500 dilution; validated by immunofluorescence in EGFP-overexpressing cells). The secondary antibodies were anti-rabbit IgG antibody conjugated to Alexa Fluor 488 (A1108, lot number 913909, Life Technologies; 1:1000 dilution) and anti-rat IgG antibody conjugated to Alexa Fluor 488 (4416, Cell Signaling Technology; 1:1000 dilution). Nuclei were stained with Hoechst 33342 (346-07951, Dojindo, Kamimashiki, Japan: 1:1000 dilution). Stained cells were mounted with Fluoromount-G (0100-01, SouthernBiotech, Birmingham, AL). A confocal microscope platform, TCS SP8 (Leica, Tokyo, Japan), with a 100× plan apochromatic objective lens (NA, 1.40) was used for fluorescence imaging. Hoechst 33342 signal was excited by 405 nm light and detected with 410–470 nm light at room temperature. Alexa Fluor 488 signal was excited by 488 nm light and detected with 500–550 nm light at room temperature. Line profiling was performed with LAS AF software (Leica).
Amino acid sequence alignment
The DNA sequences of the coding regions of the SALL family genes were obtained from the mRNA sequences registered in GenBankTM. The accession numbers were: SALL1, NM_002968.2; SALL2, NM_005407.2; SALL3, NM_171999.3; SALL4B, NM_001318031.1. The coding regions were translated with ApE software. Alignment was performed with ClustalW software.
Statistical analysis
Relative mRNA levels were analyzed by Student's t test. Migrated cell numbers were analyzed by Dunnett's test. p < 0.05 was considered to be statistically significant.
Author contributions
J. I. conceptualization; J. I. data curation; J. I. formal analysis; J. I. and M. T. funding acquisition; J. I., W. L., S. I., S. T., and Y. M. investigation; J. I. visualization; J. I. and S. I. methodology; J. I. writing-original draft; J. I. and M. T. project administration; S. I. and M. T. writing-review and editing; Y. M. and F. S. supervision; F. S. resources.
Supplementary Material
Acknowledgments
We thank the Medical Research Support Center of Kyoto University for technical assistance. The manuscript was proofread by American Journal Experts.
This work was supported by Taiho Pharmaceutical Co., Ltd. and by Japan Society for the Promotion of Science Grant-in-aid for Young Scientists (B) 25870384. J. I. is an employee of Kyoto University's Sponsored Research Program funded by Taiho Pharmaceutical Co., Ltd. M. T. received research funding from Taiho Pharmaceutical Co., Ltd.
This article contains Data S1.
J. Itou, W. Li, S. Ito, S. Tanaka, Y. Matsumoto, F. Sato, and M. Toi, unpublished observation.
- SALL
- Sal-like
- TRIM21
- tripartite motif–containing 21
- EGFP
- enhanced GFP
- SUMO
- small ubiquitin-like modifier
- aa
- amino acid
- TUBE
- tandem ubiquitin-binding entity
- NA
- numerical aperture.
References
- 1. Yong K. J., Gao C., Lim J. S., Yan B., Yang H., Dimitrov T., Kawasaki A., Ong C. W., Wong K. F., Lee S., Ravikumar S., Srivastava S., Tian X., Poon R. T., Fan S. T., et al. (2013) Oncofetal gene SALL4 in aggressive hepatocellular carcinoma. N. Engl. J. Med. 368, 2266–2276 10.1056/NEJMoa1300297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yang M., Xie X., and Ding Y. (2016) SALL4 is a marker of poor prognosis in serous ovarian carcinoma promoting invasion and metastasis. Oncol. Rep. 35, 1796–1806 10.3892/or.2016.4545 [DOI] [PubMed] [Google Scholar]
- 3. Nicolè L., Sanavia T., Veronese N., Cappellesso R., Luchini C., Dabrilli P., and Fassina A. (2017) Oncofetal gene SALL4 and prognosis in cancer: a systematic review with meta-analysis. Oncotarget 8, 22968–22979 10.18632/oncotarget.14952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cancer Genome Atlas Network (2012) Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 10.1038/nature11412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Itou J., Tanaka S., Li W., Iida A., Sehara-Fujisawa A., Sato F., and Toi M. (2017) The Sal-like 4-integrin alpha6beta1 network promotes cell migration for metastasis via activation of focal adhesion dynamics in basal-like breast cancer cells. Biochim. Biophys. Acta 1864, 76–88 10.1016/j.bbamcr.2016.10.012 [DOI] [PubMed] [Google Scholar]
- 6. Tatetsu H., Kong N. R., Chong G., Amabile G., Tenen D. G., and Chai L. (2016) SALL4, the missing link between stem cells, development and cancer. Gene 584, 111–119 10.1016/j.gene.2016.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lu J., Jeong H., Kong N., Yang Y., Carroll J., Luo H. R., Silberstein L. E., and Yupoma Chai L. (2009) Stem cell factor SALL4 represses the transcriptions of PTEN and SALL1 through an epigenetic repressor complex. PLoS One 4, e5577 10.1371/journal.pone.0005577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Böhm J., Sustmann C., Wilhelm C., and Kohlhase J. (2006) SALL4 is directly activated by TCF/LEF in the canonical Wnt signaling pathway. Biochem. Biophys. Res. Commun. 348, 898–907 10.1016/j.bbrc.2006.07.124 [DOI] [PubMed] [Google Scholar]
- 9. Bard J. D., Gelebart P., Amin H. M., Young L. C., Ma Y., and Lai R. (2009) Signal transducer and activator of transcription 3 is a transcriptional factor regulating the gene expression of SALL4. FASEB J. 23, 1405–1414 10.1096/fj.08-117721 [DOI] [PubMed] [Google Scholar]
- 10. Yang J., Gao C., Chai L., and Ma Y. (2010) A novel SALL4/OCT4 transcriptional feedback network for pluripotency of embryonic stem cells. PLoS One 5, e10766 10.1371/journal.pone.0010766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. He J., Zhang W., Zhou Q., Zhao T., Song Y., Chai L., and Li Y. (2013) Low-expression of microRNA-107 inhibits cell apoptosis in glioma by upregulation of SALL4. Int. J. Biochem. Cell Biol. 45, 1962–1973 10.1016/j.biocel.2013.06.008 [DOI] [PubMed] [Google Scholar]
- 12. Lin Y., Liu A. Y., Fan C., Zheng H., Li Y., Zhang C., Wu S., Yu D., Huang Z., Liu F., Luo Q., Yang C. J., and Ouyang G. (2015) MicroRNA-33b inhibits breast cancer metastasis by targeting HMGA2, SALL4 and Twist1. Sci. Rep. 5, 9995 10.1038/srep09995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang F., Yao Y., Jiang Y., Lu L., Ma Y., and Dai W. (2012) Sumoylation is important for stability, subcellular localization, and transcriptional activity of SALL4, an essential stem cell transcription factor. J. Biol. Chem. 287, 38600–38608 10.1074/jbc.M112.391441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wilson C. H., Crombie C., van der Weyden L., Poulogiannis G., Rust A. G., Pardo M., Gracia T., Yu L., Choudhary J., Poulin G. B., McIntyre R. E., Winton D. J., March H. N., Arends M. J., Fraser A. G., et al. (2012) Nuclear receptor binding protein 1 regulates intestinal progenitor cell homeostasis and tumour formation. EMBO J. 31, 2486–2497 10.1038/emboj.2012.91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Itou J., Matsumoto Y., Yoshikawa K., and Toi M. (2013) Sal-like 4 (SALL4) suppresses CDH1 expression and maintains cell dispersion in basal-like breast cancer. FEBS Lett. 587, 3115–3121 10.1016/j.febslet.2013.07.049 [DOI] [PubMed] [Google Scholar]
- 16. Dirican E., and Akkiprik M. (2016) Functional and clinical significance of SALL4 in breast cancer. Tumour Biol. 37, 11701–11709 10.1007/s13277-016-5150-7 [DOI] [PubMed] [Google Scholar]
- 17. Yang J., Chai L., Liu F., Fink L. M., Lin P., Silberstein L. E., Amin H. M., Ward D. C., and Ma Y. (2007) Bmi-1 is a target gene for SALL4 in hematopoietic and leukemic cells. Proc. Natl. Acad. Sci. U.S.A. 104, 10494–10499 10.1073/pnas.0704001104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang M. C., Li C. L., Cui J., Jiao M., Wu T., Jing L. I., and Nan K. J. (2015) BMI-1, a promising therapeutic target for human cancer. Oncol. Lett. 10, 583–588 10.3892/ol.2015.3361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu M., Yang F., Ren Z., Jiang Y., Ma Y., Chen Y., and Dai W. (2014) Identification of the nuclear localization signal of SALL4B, a stem cell transcription factor. Cell Cycle 13, 1456–1462 10.4161/cc.28418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hjerpe R., Aillet F., Lopitz-Otsoa F., Lang V., England P., and Rodriguez M. S. (2009) Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep. 10, 1250–1258 10.1038/embor.2009.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kohlhase J., Wischermann A., Reichenbach H., Froster U., and Engel W. (1998) Mutations in the SALL1 putative transcription factor gene cause Townes-Brocks syndrome. Nat. Genet. 18, 81–83 10.1038/ng0198-81 [DOI] [PubMed] [Google Scholar]
- 22. Kohlhase J., Heinrich M., Schubert L., Liebers M., Kispert A., Laccone F., Turnpenny P., Winter R. M., and Reardon W. (2002) Okihiro syndrome is caused by SALL4 mutations. Hum. Mol. Genet. 11, 2979–2987 10.1093/hmg/11.23.2979 [DOI] [PubMed] [Google Scholar]
- 23. Böhm J., Buck A., Borozdin W., Mannan A. U., Matysiak-Scholze U., Adham I., Schulz-Schaeffer W., Floss T., Wurst W., Kohlhase J., and Barrionuevo F. (2008) Sall1, sall2, and sall4 are required for neural tube closure in mice. Am. J. Pathol. 173, 1455–1463 10.2353/ajpath.2008.071039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mallery D. L., McEwan W. A., Bidgood S. R., Towers G. J., Johnson C. M., and James L. C. (2010) Antibodies mediate intracellular immunity through tripartite motif-containing 21 (TRIM21). Proc. Natl. Acad. Sci. U.S.A. 107, 19985–19990 10.1073/pnas.1014074107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sabile A., Meyer A. M., Wirbelauer C., Hess D., Kogel U., Scheffner M., and Krek W. (2006) Regulation of p27 degradation and S-phase progression by Ro52 RING finger protein. Mol. Cell. Biol. 26, 5994–6004 10.1128/MCB.01630-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kong H. J., Anderson D. E., Lee C. H., Jang M. K., Tamura T., Tailor P., Cho H. K., Cheong J., Xiong H., Morse H. C. 3rd, Ozato K. (2007) Cutting edge: autoantigen Ro52 is an interferon inducible E3 ligase that ubiquitinates IRF-8 and enhances cytokine expression in macrophages. J. Immunol. 179, 26–30 10.4049/jimmunol.179.1.26 [DOI] [PubMed] [Google Scholar]
- 27. Lazzari E., Korczeniewska J., Ní Gabhann J., Smith S., Barnes B. J., and Jefferies C. A. (2014) TRIpartite motif 21 (TRIM21) differentially regulates the stability of interferon regulatory factor 5 (IRF5) isoforms. PLoS One 9, e103609, 10.1371/journal.pone.0103609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pan J. A., Sun Y., Jiang Y. P., Bott A. J., Jaber N., Dou Z., Yang B., Chen J. S., Catanzaro J. M., Du C., Ding W. X., Diaz-Meco M. T., Moscat J., Ozato K., Lin R. Z., et al. (2016) TRIM21 ubiquitylates SQSTM1/p62 and suppresses protein sequestration to regulate redox homeostasis. Mol. Cell 61, 720–733 10.1016/j.molcel.2016.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gao C., Dimitrov T., Yong K. J., Tatetsu H., Jeong H. W., Luo H. R., Bradner J. E., Tenen D. G., and Chai L. (2013) Targeting transcription factor SALL4 in acute myeloid leukemia by interrupting its interaction with an epigenetic complex. Blood 121, 1413–1421 10.1182/blood-2012-04-424275 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.