

Abstract
Thrombotic cardiovascular disease, including acute myocardial infarction, ischemic stroke, and venous thromboembolic disease, is the leading cause of morbidity and mortality worldwide. While reperfusion therapy with thrombolytic agents reduces mortality from acute myocardial infarction and disability from stroke, thrombolysis is generally less effective than mechanical reperfusion and is associated with fatal intracerebral hemorrhage in up to 2–5% of patients. To address these limitations, we propose the tobacco mosaic virus (TMV)-based platform technology for targeted delivery of thrombolytic therapies. TMV is a plant virus-based nanoparticle with a high aspect ratio shape measuring 300 × 18 nm. These soft matter nanorods have favorable flow and margination properties allowing the targeting of the diseased vessel wall. We have previously shown that TMV homes to thrombi in a photochemical mouse model of arterial thrombosis. Here we report the synthesis of TMV conjugates loaded with streptokinase (STK). Various TMV-STK formulations were produced through bioconjugation of STK to TMV via intervening PEG linkers. TMV-STK was characterized using SDS–PAGE and Western blot, transmission electron microscopy, cryo-electron microscopy, and cryo-electron tomography. We investigated the thrombolytic activity of TMV-STK in vitro using static phantom clots, and in a physiologically relevant hydrodynamic model of shear-induced thrombosis. Our findings demonstrate that conjugation of STK to the TMV surface does not compromise the activity of STK. Moreover, the nanoparticle conjugate significantly enhances thrombolysis under flow conditions, which can likely be attributed to TMV’s shape-mediated flow properties resulting in enhanced thrombus accumulation and dissolution. Together, these data suggest TMV to be a promising platform for the delivery of thrombolytics to enhance clot localization and potentially minimize bleeding risk.
Keywords: tobacco mosaic virus (TMV), streptokinase (STK), plasminogen activator, drug delivery, cardiovascular disease, thrombosis
Graphical abstract
INTRODUCTION
Thrombosis is a pathological process caused by undesired blood coagulation leading to obstruction of blood flow and ischemia.1 Thrombotic cardiovascular diseases, which include acute myocardial infarction (AMI), ischemic stroke, and venous thromboembolic disease (VTE; including deep vein thrombosis, DVT; and pulmonary embolism, PE), are the leading cause of death in developed countries.2 Total U.S. healthcare expenditures in 2009 for coronary heart disease and stroke were a staggering $165.4 billion and $68.9 billion, respectively, with pharmacologic therapies estimated to exceed $20 billion worldwide.2 VTE is the third most common cause of death from cardiovascular disease after myocardial infarction and stroke.3 VTE is associated with a high case fatality rate with 10–30% of patients dying within one month of diagnosis.4 Estimates suggest that 60,000–100,000 people in the U.S. die of DVT/PE, which is the most common cause of death after elective surgery and pregnancy as well as the leading cause of preventable hospital deaths.4
Antithrombotic therapies for the prevention and treatment of thrombosis include antiplatelet agents (e.g., aspirin3 and P2Y12 ADP receptor5 and glycoprotein IIb/IIa receptor6 antagonists), anticoagulants (e.g., warfarin,7 rivaroxaban,3 apixaban,3 edoxaban,8 and dabigatran3), and thrombolytic agents (e.g., streptokinase,9–11 alteplase,10–12 and tenecteplase11,12). While reperfusion therapy with thrombolytic agents reduces mortality from acute myocardial infarction and disability from stroke, thrombolysis is less effective than mechanical reperfusion13 (i.e., thrombus aspiration and balloon angioplasty/stenting) for both AMI and stroke and is associated with fatal intracerebral hemorrhage in up to 2–5% of patients.14 Efficiency of reperfusion with thrombolytic agents can be enhanced by increasing fibrin binding (tissue-type plasminogen activator, t-PA, compared to streptokinase)15,16 and by introducing mutations that endow t-PA with resistance to its endogenous inhibitor, plasminogen activator inhibitor-1 (tenecteplase compared to t-PA).11 Although thrombolytic agents may be administered more rapidly and do not require specialized interventional capabilities for mechanical reperfusion, thrombolysis is utilized in a minority of AMI and ischemic stroke cases due to limited therapeutic “time windows” defined in clinical trials and to high rates of nonfatal and fatal hemorrhagic complications.17,18
Nanoparticle technologies hold promise to target therapies to the site of disease or injury. Multiple approaches have been developed to enhance efficacy and prevent side effects of thrombolytic therapies. For example, PEGylated drug conjugates,19–21 thrombolytic-containing liposomes22,23 and polymer-based nanoparticles,24–27 etc. were developed with the aim of enhancing the pharmacokinetic profiles of thrombolytics. It had been expected that prolonged circulation would translate into enhanced delivery and efficacy. However, long circulating thrombolytics can elevate the risk of hemorrhage. Additionally, “stealthing” often decreases all molecular interactions, including interactions with thrombi. To overcome these issues, multiple targeting strategies (e.g., toward endothelium, or targeting conformational changes taking place after conversion of fibrinogen to fibrin) are being developed.1,28 Potential disadvantages are poor specificity and/or decrease of thrombus penetration, and indeed data indicate an inverse correlation between target affinity and thrombus penetration.28,29 Finally, most nanoparticle-based therapies that are being developed rely on spherical (often synthetic) carriers, which might not have optimal flow and margination properties.30–33
In our approach we utilize tobacco mosaic virus (TMV), a plant virus-based nanoparticle (VNP) currently undergoing development for applications in biotechnology and medicine.34 TMV is a soft matter nanotube measuring 300 × 18 nm with a 4 nm wide interior channel. Proteinaceous nanoparticles formed by TMV (and other VNPs) are biocompatible, biodegradable, and nonpathogenic in humans.35,36 Based on its elongated geometry, TMV “tumbles” in the blood flow and marginates toward the vascular walls, allowing for enhanced interactions with the diseased vessel.31,33,37 Building on these properties, we have previously demonstrated molecular imaging of atherosclerotic plaques in mice using TMV loaded with contrast agents and targeted to areas of inflammation.38 Moreover, we have shown that, based on their high aspect ratio shape, TMV nanoparticles marginate toward and accumulate at sites of thrombosis.30
Together these properties make TMV a candidate carrier for the delivery of thrombolytics. Toward this goal, we have synthesized and studied conjugates of TMV and streptokinase (STK), a thrombolytic plasminogen (PG) activator derived from Streptococcus bacteria. In blood, STK binds to free PG, forming a proteolytic complex capable of hydrolyzing the Arg560/Val561 bond in neighboring PG proteins, resulting in the formation of plasmin,9 a physiological regulator of coagulation. It should be noted that alteplase, a recombinant PG activator, is currently the clinical standard in developed countries, due to its faster thrombolysis kinetics and lack of risks associated with immune response. However, STK is still widely used in developing nations,7,39 due to its significantly (approximately 10 times) lower cost, and comparable therapeutic efficacy.12 Another benefit of STK is its lower risk of bleeding after administration compared to alteplase, although STK still has a relatively high risk of bleeding. Thus, STK could be a drug of preference for reperfusion therapy in high hemorrhage risk patients, e.g., in elderly or obese people or patients with hypertension.10 Here, we present a study in which we investigate the thrombolytic activity of TMV-STK compared to free STK using a combination of an ex situ phantom clot dissolution assay and an in situ perfusion chamber assay, a physiologically relevant shear-induced model of thrombosis.
MATERIALS AND METHODS
Virus Propagation and Purification
Viruses were propagated by mechanical inoculation using 5–10 μg of virus per leaf. TMV-Lys mutants were propagated in Nicotiana benthamiana. The isolation of VNPs using established procedures yielded approximately 1–10 mg of virus per gram of infected leaf material.40
TMV-PEG8/28-STK Synthesis
Streptokinase (STK, MBS142456; MyBioSource) was conjugated to the external surface of TMV-Lys using different length PEG spacers (8-mer and 28-mer PEG), for comparison of activity, using a three-step reaction: (1) The NHS ester-to-lysine binding between STK and NHS-PEG4-SAc (26099; Thermo Fisher) was performed by mixing STK (3 mg/mL final concentration) and NHS-PEG4-SAc (4-mer PEG linker) in 1:1 stoichiometric ratio. The reaction was carried out in 0.01 M phosphate buffer + 0.125 M saline (PBS; pH 7.4) containing 10% (v/v) dimethyl sulfoxide (DMSO); the reaction was allowed to proceed overnight at room temperature (RT). To deprotect the –SH group, deacetylation solution (0.5 M hydroxylamine, 25 mM EDTA in PBS, pH 7.2–7.5) was added to give a final concentration of 10% (v/v) deprotection solution to reaction buffer. (2) The NHS ester-to-lysine binding between TMV-Lys and either 4-mer or 24-mer NHS-PEG4/24-mal linker (22104; Thermo Fisher) was performed by mixing TMV-Lys (2 mg/mL final concentration) and NHS-PEG4/24-mal using a 10-fold excess of NHS-PEG4/24-mal per TMV-Lys coat protein. The reaction was carried out in 0.01 M potassium phosphate buffer pH 7.4 containing 10% (v/v) DMSO; the reaction was allowed to proceed for 2 h at RT. The TMV-PEG4/24-mal particles were purified using PD MiniTrap G-25 desalting columns (28-9180-08; GE) and combined with product (1) in step 3. (3) The maleimide-to-thiol coupling between TMV-PEG4/24-mal and STK-PEG4-SH was carried out by mixing 2 equiv of STK per TMV-Lys coat protein overnight at RT; then the reaction was quenched for 1 h at RT by addition of excess glycine/L-cysteine. The TMV-PEG8/28-STK particles were then purified by ultracentrifugation at 112 × g (TLA-55 rotor, Beckman Coulter) for 1 h over a 40% (w/v) sucrose cushion and washed twice with PBS with subsequent purification steps using ultracentrifugation over 40% sucrose cushion (see above).
TMV Conjugation with PEG
TMV-PEG4 particles were obtained by conjugation of NHS-PEG4-mal (22114; Thermo Fisher) to the external surface of TMV-Lys. The reaction was performed by mixing TMV-Lys (2 mg/mL final concentration) and NHS-PEG4-mal at 10 equiv per TMV-Lys coat protein in 0.01 M potassium phosphate buffer pH 7.4 containing 10% (v/v) DMSO; the reaction was allowed to proceed overnight at RT. Then the reaction was quenched by addition of excess glycine/L-cysteine, and quenching was allowed to proceed for 1 h at RT. The products were then purified using PD MiniTrap G-25 desalting columns (28-9180-08; GE).
UV–Vis Spectroscopy
The concentrations of TMV were determined by the Beer–Lambert law using an extinction coefficient of TMV εTMV(260 nm) = 3 mg−1 mL cm−1 and a molecular weight of TMV MWTMV = 39.4 MDa.
SDS–PAGE and Western Blot (WB)
STK/PEG-labeled and unlabeled TMV samples (20 μg) were denatured by boiling at 100 °C for 7 min in gel loading buffer (62.5 mM Tris-HCl pH 6.8, 2% (w/v) SDS, 10% (v/v) glycerol, 0.01% (w/v) bromophenol blue, 10% (v/v) 2-mercaptoethanol). Denatured protein samples were then separated on 4–12% NuPAGE polyacrylamide gels in 1× MOPS running buffer (Invitrogen) at 200 V for 40 min. The gels were stained with Coomassie Brilliant Blue and visualized using an AlphaImager imaging system (Biosciences).
For WB, samples separated by SDS–PAGE were transferred from the gel onto nitrocellulose (88018, Thermo Scientific) under a voltage of 30 V for 1 h. The membranes were then incubated at RT for 1 h in blocking solution using 5% (w/v) skimmed milk in TBST (150 mM NaCl, 10 mM Tris HCl, 0.1% (v/v) Tween-20, pH 7.5). Then, blots were incubated overnight at 4 °C with (a) 1 μg/mL rabbit polyclonal antibody against streptokinase (NB100-62936; Novus Biologicals) or (b) 1 μg/mL rabbit anti-TMV antibody (custom-made; Pacific Immunology) in blocking solution and subsequently washed 3× for 5 min in TBST. After washing, membranes were incubated with 2 μg/mL of alkaline phosphatase goat anti-rabbit antibody in blocking solution for 1 h at RT and washed 3× for 15 min in TBST and 1× for 5 min in Milli-Q water. Specific antibody binding was visualized using Novex AP Chromogenic Substrate (BCIP/NBT) (WP20001; Invitrogen).
Negative Staining TEM
Particles were adsorbed to carbon-coated copper grids (01754-F, TED PELLA) at a concentration of 0.1 mg/mL (2 μL per grid), rinsed with deionized water, and negatively stained with 2% (w/v) uranyl acetate for 5 min before analysis with a Tecnai TF30 ST TEM at 300 kV.
Cryo-EM Grid Preparation, Imaging, and Tomography
A small amount (∼4.5 μL) of TMV-STK at a concentration of 0.1 mg/mL was mixed with 0.5 μL of fiducial nanogold (10 nm, Aurion). The suspension was applied to Quantifoil holey carbon EM grids (R2/2, 200 mesh; EMS) glow-discharged for 20 s at 25 mA. Grids were then blotted and plunge-frozen into liquid ethane by using a manual plunger. Frozen vitrified grids were transferred into liquid nitrogen for storage and imaging. Imaging was performed on a JEOL 2200FS transmission electron microscope (200 kV, FEG, in-column energy filter). Cryo-electron micrographs were collected using a DE20 direct electron detector (Direct Electron, LP, USA) with a defocus range of 4–5 μm and with 2 s exposure per movie, equating to a total electron dose of <100 e−/A2. Individual movie frames were then motion-corrected using custom EMAN241 scripts to produce final micrographs. Tilt series were collected on the JEOL 2200FS microscope using a Tietz TVIPS 4k × 4k CMOS camera with SerialEM.42 The tilt range was −60° to +60° with a defocus of −5.5 μm. The tilt-series data was processed with the IMOD software package,43 and the resulting tomogram was displayed with UCSF Chimera.44
Fibrin Clot Thrombolysis Assay
1 mg/mL of non-PG depleted fibrinogen (Enzyme Research Laboratories) was polymerized with 1 IU/mL of thrombin in the presence of 3.5 mM CaCl2 in PBS to form 2 mL fibrin clots in D = 35 mm Petri dishes. After 3 h, 1 μL of different amounts of STK and TMV-PEG8/28-STK were added in triplicates to the center and top of the in vitro formed thrombus followed by incubation for a total of 20 h; then photographs were taken to document the lysis zone.
Perfusion Chamber Assay
A perfusion chamber (Portola Pharmaceuticals) was used to evaluate the effect of drug and nanoparticle formulation on thrombus formation time. Factor Xa inhibitor anticoagulated blood was labeled with 5 μg/mL Rhodamine 6G (Sigma-Aldrich) for 10 min. STK and TMV formulations were added at final concentrations of 250 and 1000 IU of STK/mL (assuming no loss of activity for TMV-conjugated STK); then blood was perfused through the human type III collagen-coated rectangular capillaries at an arterial shear rate of 600 s−1. Thrombus formation under flow was then visualized in real time and average fluorescence of platelet aggregation over time quantified. The correlation between the mass and activity of STK is expressed by the following equation: 1 mg of STK = 80000 IU (based on STK specifications provided by the manufacturer).
RESULTS AND DISCUSSION
Mutant TMV particles containing a solvent-exposed and chemically addressable lysine side chain at the C-terminus of the coat protein (i.e., Lys-158) were utilized in this study and are referred to as TMV-Lys. TMV-Lys was propagated in and purified from N. benthamiana plants using previously established protocols.40,45
Two types of TMV-based particles with STK conjugated were prepared using different length PEG spacers, an 8-mer or 28-mer PEG linker to yield TMV-PEG8-STK and TMV-PEG28-STK, respectively. The generic abbreviations TMV-STK or TMV-PEG8/28-STK will be used throughout the manuscript. TMV-PEG8/28-STK particles were prepared using a 3-step reaction (Figure 1): (a) conjugation of 4-mer or 24-mer NHS ester-PEG4/24-maleimide linker to TMV-Lys (reaction between NHS ester and NH2 group of TMV’s lysine side chain); (b) conjugation of 4-mer NHS ester-PEG4-SAc (acetyl-protected thiol) linker to STK (reaction between NHS ester and NH2 group of STK’s lysine side chains); (c) deprotection of thiol groups and conjugation of product (a) with product (b) (reaction between the maleimide installed on TMV and the thiol group introduced to STK).
Figure 1.
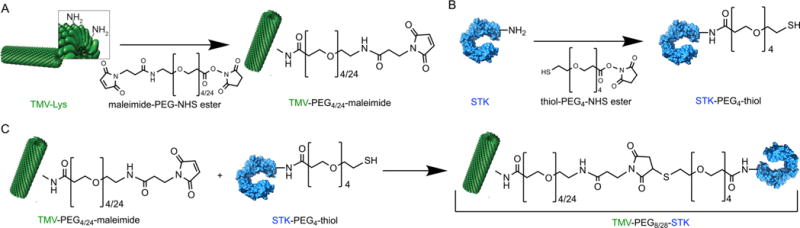
Schematic representation of TMV-PEG8/28-STK particle synthesis. (A) Functionalization of TMV-Lys particles with maleimide groups. (B) Labeling STK molecules with thiol groups. (C) Bioconjugation of STK-PEG4-SH ligands to TMV-PEG4/24-maleimide particle to form final TMV-PEG8/28-STK particles. TMV’s structural data46 (ID: 2TMV) and STK’s structural data47 (ID: 1BML) obtained from the Protein Data Bank (PDB) were used to represent these components using UCSF Chimera software.44 Note that the TMV fragment and STK structures are not shown on the same scale.
Following the 3-step reaction and purification, TMV-STK particles were characterized to confirm the structural integrity of the formulation and to determine the degree of STK conjugation. Denaturing polyacrylamide gel electrophoresis (SDS–PAGE) and Western blot (WB) analysis confirmed successful conjugation of STK to TMV. The conjugate of the TMV coat protein (TMVcp)-PEG8/28-STK was apparent as multiple bands with molecular weights (MW) equal to or higher than 64 kDa, which is the theoretical MW of the TMVcp-STK conjugate. The TMVcp has a MW of ∼17 kDa and STK of ∼47 kDa; see Figure 2A. Since each TMVcp contains precisely one lysine residue, the presence of multiple “conjugate” bands is possibly a result of a single STK molecule being conjugated to one or more neighboring TMVcps. This is possible because STK contains multiple solvent exposed lysine side chains that could be modified with more than one linker for conjugation, resulting in the formation of multimeric interactions. However, it is also possible that the modified proteins interact and get entangled through noncovalent interactions, and/or are incompletely denatured, which has been observed for other chemically modified VNP formulations.48,49 The presence of both TMVcp and STK components in the ≥64 kDa conjugate bands was confirmed by Western blotting using TMV- and STK-specific antibodies (Figure 2B,C). To quantify the number of STK molecules per TMV, densitometric analysis was performed on SDS–PAGE gels from two batches of synthesized TMV-PEG8/28-STK particles. The densitometry analysis revealed the presence of ∼767 (±210) STK molecules on average per TMV for TMV-PEG8-STK; and ∼111 (±39) STK molecules on average per TMV for TMV-PEG28-STK (Tables S1 and S2). The batch-to-batch variation could be explained by variability of gel staining and the need for multiple bioconjugation steps, which could be mitigated in the future through either genetic introduction of azide/alkyne-modified non-natural amino acids or the use of orthogonal click chemistries.
Figure 2.
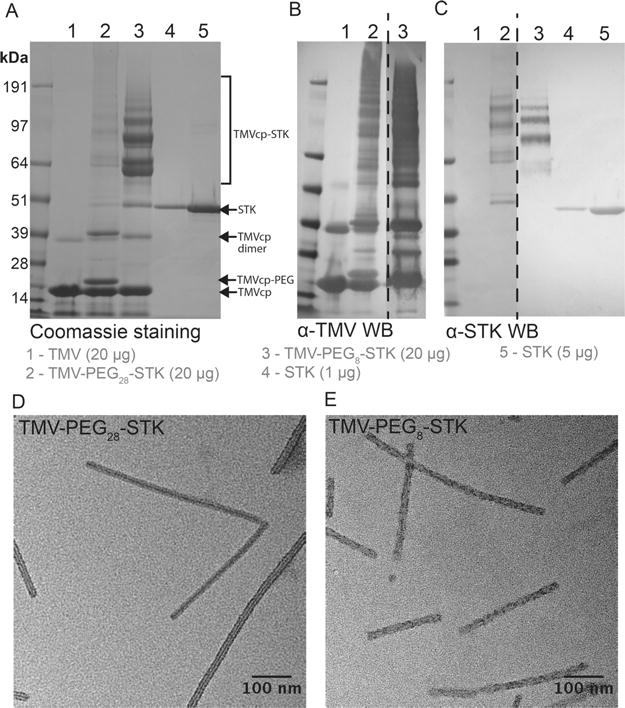
TMV-PEG8/28-STK characterization. (A–C) SDS–PAGE and Western Blot (WB) analysis of TMV-Lys particles before and after conjugation of STK. Free STK was used as reference. Bands corresponding to TMV capsid protein (TMVcp, MW = 17 kDa) and STK (MW = 47 kDa) are indicated with arrows. Successful TMV-STK conjugation is indicated by presence of multiple protein bands corresponding to TMVcp-PEG8/28-STK (multiple bands of apparent MW > 64 kDa; theoretical molecular weight of 1:1 SA:TMVcp monomer = 64 kDa) as shown by WB immune recognition. The low molecular weight bands (<14 kDa; panels A and B) correspond to TMV coat protein fragments, possibly resulting from degradation during sample preparation. (D, E) TEM images of TMV-PEG28-STK and TMV-PEG8-STK particles, respectively.
STK is a flexible molecule composed of three independently folded β-grasp domains (α, β, and γ) separated by two flexible coiled coils.47,50 Due to its flexibility, it is difficult to model the dimensions of an STK molecule. However, based on the crystallography data,47 (PDB database ID: 1BML) we approximated its dimensions as 9 × 6 × 2.5 nm and modeled it as an ellipsoid of corresponding Dx, Dy, and Dz diameters. The theoretical number of STK molecules needed for full coverage of TMV was estimated as either 400 or 1200, depending on whether the largest or smallest cross-sectional area of the ellipsoid was used. Provided that the 22 amine groups present on the STK surface are available for conjugation, in reality, the orientation of STK on TMV surface is most likely random (see Figure S1). We have recently shown random orientations of a conjugated protein on TMV using cryo-electron tomography combined with subtomogram averaging of a TMV–serum albumin conjugate synthesized using a similar bioconjugation strategy.51,52 As a consequence, the maximum theoretical number of STK molecules on TMV is difficult to determine and most likely falls within the range of 400–1200. It should also be noted that the flexible nature of solvated STK is not reflected in the crystallography data, thus our calculation should be treated as an approximation only.
The conjugation strategy using the 8-mer PEG linker yielded significantly higher STK-to-TMV coverage compared to conjugation with the 28-mer PEG linker. Two contributing factors may explain this variation in conjugation efficiency: (a) the longer PEG chains might result in lower PEG density on the TMV surface due to steric hindrance (Figure 1A), and therefore provide fewer attachment sites for STK; (b) lower accessibility of the maleimide groups in longer PEG chains (Figure 1A) for subsequent reaction with STK’s introduced thiols (Figure 1C), because the longer PEG chains likely adapt a “tangled” mushroom conformation at low surface grafting densities.48,53
Lastly, we confirmed the structural integrity of the TMV particles postconjugation using negative-stain transmission electron microscopy (TEM, Figure 2D,E). Additionally, cryo-electron microscopy (cryo-EM) revealed a dense coat of STK molecules on the TMV-PEG8-STK surface (Figure 3A). However, it was technically difficult to identify STK in cryo-EM images of TMV-PEG28-STK due to the scarcity of STK (see Figure S2B). The extent of the coverage of STK on the surface of TMV-PEG8-STK particles was further evaluated by cryo-electron tomography. The tomogram shows a uniform density of STK on the outer surface of TMV (Figure 3B,C and Movie S1).
Figure 3.

Cryo-EM imaging and cryo-electron tomography of TMV-PEG8-STK. (A) Cryo-electron micrograph of TMV-PEG8-STK collected with a nominal magnification of 50000×. STK is observed dotting the surface of the TMV particles. Several electron-dense nanogold fiducial markers are visible in the image. (B) Three-dimensional representation of TMV-PEG8-STK particles. (C) Single TMV-PEG8-STK rod (left) and with a superimposed cylinder (right) representing a bare TMV particle (blue, dimensions 300 × 18 nm). The scale bars represent 50 nm in panels A and B, and 25 nm in panel C.
Thrombolytic activity of TMV-PEG8/28-STK was evaluated and compared to that of free STK using in vitro formed fibrin clots. STK has no proteolytic activity of its own; its thrombolytic effect relies on formation of an active complex upon binding to PG and subsequent hydrolytic activation of blood PG to yield plasmin.9 Therefore, to prepare the phantom clots, non-PG depleted fibrinogen was mixed with 1 IU/mL of human thrombin in the presence of 3.5 mM CaCl2. In these conditions fibrinogen undergoes proteolytic cleavage by the serine protease thrombin to form insoluble fibrin that precipitates and forms a thrombus. Thrombolysis was evaluated by spotting 1 μL of STK or TMV-PEG8/28-STK formulations (ranging from 0.005 to 50 IU STK and equivalent mass of TMV-conjugated STK) onto the phantom clots; the lysis zone was documented 20 h post treatment at room temperature (see Materials and Methods). The 20 h time point was chosen to allow the development of large-scale lysis zones enabling distinction of small differences between the samples. It should be noted, however, that the thrombolytic effect of STK and TMV-PEG8/28-STK was already apparent 1–2 h post treatment (data not shown).
While we observed successful thrombolysis for sample groups, free STK showed higher activity compared to TMV-PEG8/28-STK when used in amounts greater than 0.05 IU (Figure 4A,B). The significant difference in dissolution area for 5–50 IU spots (Figure 4A,B) is likely a result of two effects: (a) Differences in the efficiency of complex formation between PG and free STK versus TMV-bound STK. Differences in the orientation and/or packing of STK on TMV surface could result in decreased binding of PG due to steric hindrance by TMV’s surface or neighboring STK molecules; Figure S1 demonstrates a model of a few such scenarios. (b) Differences in diffusion of free versus TMV-bound STK under the static conditions of the experiment. The MW of the TMV-PEG8-STK conjugate (MW TMV-PEG8-STK = 7.60 × 107 Da; calculated using MWTMV = 3.94 × 107 Da and total MW of 767 STK molecules of 3.60 × 107 Da; we estimated the same number of PEG linkers, i.e. 767 PEG8 with a total MW of 5.33 × 105 Da). As a result, the high MW complexes of TMV-PEG8/28-STK are expected to have limited diffusion through the fibrin matrix. Note that diffusion rates are inversely proportional to the MW of the molecule. To address this point, we investigated the diffusion behavior of TMV versus proteins through fibrin matrices. As expected, while proteins freely diffuse through the matrix, the much larger TMV formulation does not (Figure S3). As a consequence, the free STK is expected to penetrate and dissolve the phantom clot faster (from within) compared to TMV-STK. In other words, the thrombolytic activity of TMV-STK is constrained by the boundaries of the dissolution area.
Figure 4.
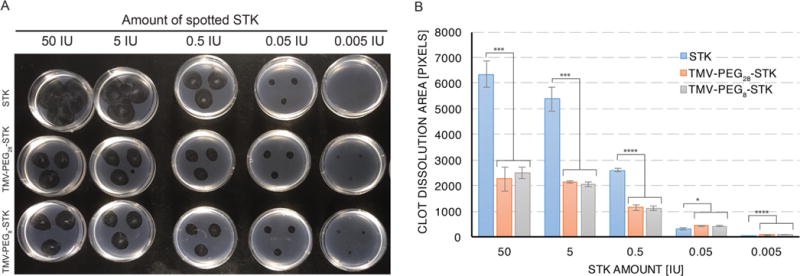
Static in vitro assay of thrombolytic activity: phantom clot dissolution. (A) Image of phantom clots 20 h after application of STK and TMV-PEG8/28-STK solutions. The dark colored circular areas result from dissolution of fibrin clot by STK. Samples were applied in triplicates. (B) Area-of-dissolution analysis of image (A). Data were analyzed using Student’s t test (2-tailed, unpaired); statistically significant differences are indicated by ****p ≤ 0.0001, ***p ≤ 0.001, *p ≤ 0.05. No statistically significant differences were found between TMV-PEG8-STK and TMV-PEG28-STK samples of corresponding concentrations.
Therefore, diffusion and efficiency in STK-PG complex formation may explain the enhanced efficacy of free STK versus TMV-STK at concentrations of 0.5 IU and higher. At lower concentrations, however, the TMV-STK complexes outperform free STK. We hypothesize that at low concentrations (≤0.05 IU) the disadvantageous drop of free STK concentration due to diffusion—below the critical value necessary to completely break down the clot—offsets the benefit of clot penetration, resulting in loss of efficacy compared to TMV-bound STK (Figure 4A,B). Indeed a diffusion-induced concentration drop was observed in the protein diffusion experiments (see Figure S3iii).
We did not observe significant differences in thrombolytic activity between TMV-PEG8-STK and TMV-PEG28-STK (when the amount of particles was normalized to provide the same STK content). Therefore, the data indicate that the length of the intervening PEG linker does not impact the activity of the formulation. In other words, the 8-mer PEG linker provides sufficient degrees of freedom for STK to bind and activate PG, and introduction of longer linkers does not provide any additional advantage.
To characterize the thrombolytic properties of developed TMV formulations in physiologically relevant conditions (i.e., under blood flow) we used an ex vivo model of shear-induced thrombosis. The experiments were performed in a perfusion chamber using blood labeled with Rhodamine 6G. The time dependent formation of a thrombus was monitored and quantified using fluorescent imaging of platelet aggregates in the presence of free STK and TMV-PEG8/28-STK (see Materials and Methods). TMV-PEG8-STK particles were chosen for this experiment based on their higher STK content (Figure 2). Particles were added to blood at a final STK concentration equivalent to 250 and 1000 IU of free STK/mL, after which blood was perfused through human type III collagen-coated rectangular capillaries at an arterial shear rate of 600/s to induce thrombosis.
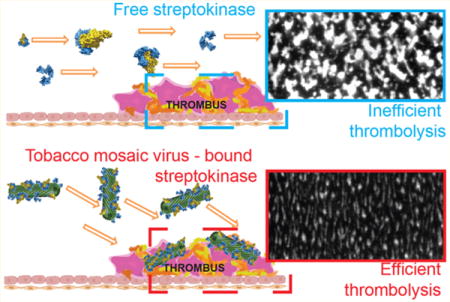
Around 10–30% reduction in thrombosis was observed in the presence of TMV-PEG8-STK particles at normalized concentration equivalent to 250 IU of free STK/mL. The reduction reached 60% at 1000 IU STK/mL concentration (Figure 5A–C). Surprisingly, no thrombolytic effect was observed in blood containing free STK. In the bloodstream, elongated particles tumble and rapidly marginate toward the walls of the blood vessel.31,33,54 Accordingly, TMV has been demonstrated to “passively” accumulate and penetrate cardiovascular thrombi (without the need of molecular recognition chemistry).30 We hypothesize that this is due to shape-mediated properties, such that TMV-PEG8-STK has enhanced interaction with thrombi under flow, leading to improved delivery of STK to the site of the thrombus (Figure 5E), localized conversion of PG to plasmin, and consequently enhanced efficacy. Some delay in thrombosis was also observed in the presence of high concentrations of TMV-PEG4 control particles. We attribute this to likely interference of TMV-PEG4 passively accumulating in thrombi with molecular recognition, recruitment, and adhesion of additional platelets in the thrombus. Free STK is a globular protein and thus is not expected to tumble or marginate efficiently to the disease site. In our experimental setup (and similarly in vivo), we expect free STK to flow in the center of capillary (or blood vessel in vivo) causing decreased interaction with the thrombus (Figure 5D), and delocalized generation of plasmin, leading to marginal efficacy during the limited time scale of the experiment.
Figure 5.
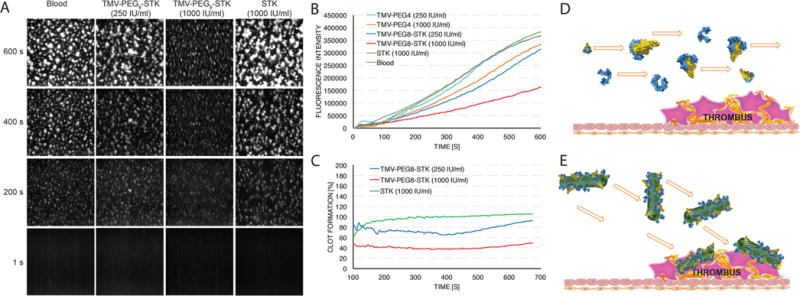
Dynamic ex vivo perfusion chamber assay of thrombolytic activity. (A) Images of fluorescent thrombus formation over time in the perfusion chamber capillary. White areas correspond to fluorescent platelet aggregates (thrombi). (B) Quantification of thrombus growth over time through measurement of fluorescence intensity. Slope of thrombus growth curves corresponds to rate of thrombus formation. Significant difference in thrombus formation rate between samples containing TMV-PEG8-STK and control TMV-PEG4 particles demonstrates the thrombolytic efficacy of TMV-PEG8-STK formulation. No thrombolysis has been observed for free STK. (C) Plot representing relative thrombus size normalized against control sample of pure blood, as a function of time. (D, E) Schematic representation of hydrodynamic properties of free STK and TMV-PEG8-STK in blood flow (schematics include monomeric STK and STK:PG active complexes). The flow direction of free STK is hypothesized to be parallel to the direction of blood flow, causing low interaction between STK and thrombi. The TMV-PEG8-STK particles marginate toward the sides of the blood vessel, causing enhanced interaction with and accumulation in thrombi.
CONCLUSIONS
In conclusion, we have designed a novel platform for delivery of thrombolytics utilizing a high aspect ratio plant virus-based nanoparticle: the tobacco mosaic virus. Our platform allows for control of STK payload achieving densities as high as ∼1 mg of STK per mg of TMV (Figure 2). The ability to control the density of PEG and STK on the TMV scaffold could allow for tailored interactions between TMV and the thrombus to modulate and maximize therapeutic efficacy. We confirmed the structural integrity of the TMV-STK formulations with TEM, cryo-EM, and cryo-electron tomography, and their thrombolytic activity with an in vitro phantom clot dissolution assay (Figures 2–4).
The TMV-STK formulation shows a clear advantage over free STK in a physiologically relevant ex vivo shear-induced model of thrombosis (Figure 5). The higher margination rate of the high aspect ratio TMV-STK toward the vessel wall31,33,37 allows for enhanced interaction with cardiovascular thrombi.30 This “passive” thrombus homing confers enhanced efficacy of TMV-STK versus free STK under flow. In other words, despite likely loss of TMV-conjugated STK “biological activity” (defined as ratio between number of biologically active STK molecules and total number of STK) due to conjugation procedure and steric hindrance by TMV surface, the “effective activity” (seen as reduction in thrombosis in ex vivo perfusion chamber assay) is increased, due to enhanced targeting to thrombi. We expect that TMV-mediated delivery of other thrombolytics, such as tPA or tenecteplase with enhanced thrombus specificity, could result in even more efficient thrombolysis.
Delivery of thrombolytics using TMV or other similar high aspect ratio carriers in reperfusion therapy could become an advantageous option for treatment of acute stroke and thromboembolism. Localized delivery and a decrease in the dose necessary to achieve therapeutic effect are expected to alleviate bleeding risks associated with administration of thrombolytics. We have previously reported that TMV-based nanoparticles have rapid blood clearance (within tens of minutes),36 an advantage for application as thrombolytic therapy as the clearance of nonutilized, i.e., non-thrombus-bound, excess drug would further decrease systemic bleeding risks. Future studies will be necessary to evaluate the in vivo performance of TMV-based thrombolytics.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institutes of Health (NIH): NHLBI R21 HL121130 and R01 HL137674 (to N.F.S.). We thank Prof. Christina Wege and her team from the University of Stuttgart, Germany, for the TMV-Lys mutant. We thank CWRU Farm for help with the scaled-up production and growth of N. benthamiana plants. We thank Jing Pu and Stephen Wang for the assistance during the TMV purification. We thank Sudheer Molugu for assistance with cryo-electron tomography data collection and Neetu Gulati for helpful tips for using the Chimera package. Selected molecular graphics and analyses were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41-GM103311). We acknowledge the Case Western Reserve High Performance Computing Cluster for processing of the DE micrographs.
ABBREVIATIONS USED
- VTE
venous thromboembolic disease
- AMI
acute myocardial infarction
- DVT
deep vein thrombosis
- PE
pulmonary embolism
- VNP
viral nanoparticle
- TMV
tobacco mosaic virus
- TMV-Lys
lysine mutant of tobacco mosaic virus
- TMVcp
tobacco mosaic virus capsid protein
- STK
streptokinase
- PG
plasminogen
- PEGn
n-mer of poly ethylene glycol
- SDS–PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- WB
Western blot
- DMSO
dimethyl sulfoxide
- MRI
magnetic resonance imaging
- RT
room temperature
- Cryo-EM
cryo-electron microscopy
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.7b00559.
Model of STK interactions with PG and TMV, cryo-EM images, diffusion of molecules through fibrin matrix of in vitro phantom clots, SDS–PAGE densitometry data, and labeling of TMV and HSA with Cy5 fluorophores (PDF) Movie of TMV-PEG8-STK tomogram (AVI)
ORCID
Andrzej S. Pitek: 0000-0002-4021-835X
Nicole F. Steinmetz: 0000-0002-0130-0481
Author Contributions
A.S.P. produced and characterized the particles and performed phantom clot assays; A.S.P., Y.W., and H.G. performed the perfusion chamber experiments. S.G. and P.L.S. performed the cryo-electron microscopy and tomography. N.F.S. conceived and developed the study. A.S.P. and N.F.S. wrote the paper. All authors have read and edited the paper and given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Carnemolla R, Muzykantov VR. Vascular Targeting of Antithrombotic Agents. IUBMB Life. 2011;63(8):632–639. doi: 10.1002/iub.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, Ford E, Furie K, Go A, Greenlund K, Haase N, Hailpern S, Ho M, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott M, Meigs J, Mozaffarian D, Nichol G, O’Donnell C, Roger V, Rosamond W, Sacco R, Sorlie P, Stafford R, Steinberger J, Thom T, Wasserthiel-Smoller S, Wong N, Wylie-Rosett J, Hong Y. Heart Disease and Stroke Statistics–2009 Update: a Report From the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119(3):e21–e181. doi: 10.1161/CIRCULATIONAHA.108.191261. [DOI] [PubMed] [Google Scholar]
- 3.Goldhaber SZ, Bounameaux H. Pulmonary Embolism and Deep Vein Thrombosis. Lancet. 2012;379(9828):1835–1846. doi: 10.1016/S0140-6736(11)61904-1. [DOI] [PubMed] [Google Scholar]
- 4.Beckman MG, Hooper WC, Critchley SE, Ortel TL. Venous Thromboembolism. Am J Prev Med. 2010;38(4):S495–S501. doi: 10.1016/j.amepre.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 5.Andre P, Delaney SM, LaRocca T, Vincent D, DeGuzman F, Jurek M, Koller B, Phillips DR, Conley PB. P2Y12 Regulates Platelet Adhesion/Activation, Thrombus Growth, and Thrombus Stability in Injured Arteries. J Clin Invest. 2003;112(3):398–406. doi: 10.1172/JCI17864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.The GUSTO IV-ACS Investigators. Effect of Glycoprotein IIb/IIIa Receptor Blocker Abciximab on Outcome in Patients with Acute Coronary Syndromes Without Early Coronary Revascularisation: the GUSTO IV-ACS Randomised Trial. Lancet. 2001;357(9272):1915–1924. doi: 10.1016/s0140-6736(00)05060-1. [DOI] [PubMed] [Google Scholar]
- 7.Sikri N, Bardia A. A History of Streptokinase Use in Acute Myocardial Infarction. Vol. 34. Texas Heart Institute; 2007. pp. 318–327. [PMC free article] [PubMed] [Google Scholar]
- 8.Nyberg J, Karlsson KE, Jönsson S, Yin O, Miller R, Karlsson MO, Simonsson U. Edoxaban Exposure-Response Analysis and Clinical Utility Index Assessment in Patients with Symptomatic Deep-Vein Thrombosis or Pulmonary Embolism. CPT: Pharmacometrics Syst Pharmacol. 2016;5(4):222–232. doi: 10.1002/psp4.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kunamneni A, Abdelghani TTA, Ellaiah P. Streptokinase–the Drug of Choice for Thrombolytic Therapy. J Thromb Thrombolysis. 2007;23(1):9–23. doi: 10.1007/s11239-006-9011-x. [DOI] [PubMed] [Google Scholar]
- 10.Hunt D. Alteplase (R-TPA) vs Streptokinase. Aust N Z J Med. 1998;28(4):514–517. doi: 10.1111/j.1445-5994.1998.tb02102.x. [DOI] [PubMed] [Google Scholar]
- 11.Bivard A, Lin L, Parsonsb MW. Review of Stroke Thrombolytics. Journal of Stroke. 2013;15(2):90–98. doi: 10.5853/jos.2013.15.2.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dundar Y, Hill R, Dickson R, Walley T. Comparative Efficacy of Thrombolytics in Acute Myocardial Infarction: a Systematic Review. QJM. 2003;96(2):103–113. doi: 10.1093/qjmed/hcg016. [DOI] [PubMed] [Google Scholar]
- 13.Widimsky P, Coram R, Abou-Chebl A. Reperfusion Therapy of Acute Ischaemic Stroke and Acute Myocardial Infarction: Similarities and Differences. Eur Heart J. 2014;35(3):147–155. doi: 10.1093/eurheartj/eht409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller DJ, Simpson JR, Silver B. Safety of Thrombolysis in Acute Ischemic Stroke: a Review of Complications, Risk Factors, and Newer Technologies. Neurohospitalist. 2011;1(3):138–147. doi: 10.1177/1941875211408731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The GUSTO Angiographic Investigators. The Effects of Tissue Plasminogen Activator, Streptokinase, or Both on Coronary-Artery Patency, Ventricular Function, and Survival After Acute Myocardial Infarction. N Engl J Med. 1993;329(22):1615–1622. doi: 10.1056/NEJM199311253292204. [DOI] [PubMed] [Google Scholar]
- 16.The GUSTO Investigators. An International Randomized Trial Comparing Four Thrombolytic Strategies for Acute Myocardial Infarction. the GUSTO Investigators. N Engl J Med. 1993;329(10):673–682. doi: 10.1056/NEJM199309023291001. [DOI] [PubMed] [Google Scholar]
- 17.Latchaw RE, Alberts MJ, Lev MH, Connors JJ, Harbaugh RE, Higashida RT, Hobson R, Kidwell CS, Koroshetz WJ, Mathews V, Villablanca P, Warach S, Walters B. American Heart Association Council on Cardiovascular Radiology and Intervention, Stroke Council, and the Interdisciplinary Council on Peripheral Vascular Disease. Recommendations for Imaging of Acute Ischemic Stroke: a Scientific Statement From the American Heart Association. Stroke. 2009;40(11):3646–3678. doi: 10.1161/STROKEAHA.108.192616. [DOI] [PubMed] [Google Scholar]
- 18.Boersma E, Maas AC, Deckers JW, Simoons ML. Early Thrombolytic Treatment in Acute Myocardial Infarction: Reappraisal of the Golden Hour. Lancet. 1996;348(9030):771–775. doi: 10.1016/S0140-6736(96)02514-7. [DOI] [PubMed] [Google Scholar]
- 19.Berger H, Pizzo SV. Preparation of Polyethylene Glycol-Tissue Plasminogen Activator Adducts That Retain Functional Activity: Characteristics and Behavior in Three Animal Species. Blood. 1988;71(6):1641–1647. [PubMed] [Google Scholar]
- 20.Rajagopalan S, Gonias SL, Pizzo SV. A Nonantigenic Covalent Streptokinase-Polyethylene Glycol Complex with Plasminogen Activator Function. J Clin Invest. 1985;75(2):413–419. doi: 10.1172/JCI111715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakuragawa N, Shimizu K, Kondo K, Kondo S, Niwa M. Studies on the Effect of PEG-Modified Urokinase on Coagulation-Fibrinolysis Using Beagles. Thromb Res. 1986;41(5):627–635. doi: 10.1016/0049-3848(86)90359-2. [DOI] [PubMed] [Google Scholar]
- 22.Heeremans JL, Prevost R, Bekkers ME, Los P, Emeis JJ, Kluft C, Crommelin DJ. Thrombolytic Treatment with Tissue-Type Plasminogen Activator (T-PA) Containing Liposomes in Rabbits: a Comparison with Free T-PA. Thromb Haemost. 1995;73(3):488–494. [PubMed] [Google Scholar]
- 23.Kim IS, Choi HG, Choi HS, Kim BK, Kim CK. Prolonged Systemic Delivery of Streptokinase Using Liposome. Arch Pharmacal Res. 1998;21(3):248–252. doi: 10.1007/BF02975283. [DOI] [PubMed] [Google Scholar]
- 24.Leach JK, Patterson E, O’Rear EA. Improving Thrombolysis with Encapsulated Plasminogen Activators and Clinical Relevance to Myocardial Infarction and Stroke. Clin Hemorheol Microcirc. 2004;30(3–4):225–228. [PubMed] [Google Scholar]
- 25.Fernandes EGR, de Queiroz AAA, Abraham GA, San Román J. Antithrombogenic Properties of Bioconjugate Streptokinase-Polyglycerol Dendrimers. J Mater Sci: Mater Med. 2006;17(2):105–111. doi: 10.1007/s10856-006-6813-5. [DOI] [PubMed] [Google Scholar]
- 26.Wang X, Inapagolla R, Kannan S, Lieh-Lai M, Kannan RM. Synthesis, Characterization, and in Vitro Activity of Dendrimer–Streptokinase Conjugates. Bioconjugate Chem. 2007;18(3):791–799. doi: 10.1021/bc060322d. [DOI] [PubMed] [Google Scholar]
- 27.Wang SS, Chou NK, Chung TW. The T-PA-Encapsulated PLGA Nanoparticles Shelled with CS or CS-GRGD Alter Both Permeation Through and Dissolving Patterns of Blood Clots Compared with T-PA Solution: an in Vitro Thrombolysis Study. J Biomed Mater Res, Part A. 2009;91A(3):753–761. doi: 10.1002/jbm.a.32234. [DOI] [PubMed] [Google Scholar]
- 28.Greineder CF, Howard MD, Carnemolla R, Cines DB, Muzykantov VR. Advanced Drug Delivery Systems for Antithrombotic Agents. Blood. 2013;122(9):1565–1575. doi: 10.1182/blood-2013-03-453498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakharov DV, Rijken DC. Superficial Accumulation of Plasminogen During Plasma Clot Lysis. Circulation. 1995;92(7):1883–1890. doi: 10.1161/01.cir.92.7.1883. [DOI] [PubMed] [Google Scholar]
- 30.Wen AM, Wang Y, Jiang K, Hsu GC, Gao H, Lee KL, Yang AC, Yu X, Simon DI, Steinmetz NF. Shaping Bio-Inspired Nanotechnologies to Target Thrombosis for Dual Optical-Magnetic Resonance Imaging. J Mater Chem B. 2015;3(29):6037–6045. doi: 10.1039/C5TB00879D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee S-Y, Ferrari M, Decuzzi P. Shaping Nano-/Micro-Particles for Enhanced Vascular Interaction in Laminar Flows. Nanotechnology. 2009;20(49):495101. doi: 10.1088/0957-4484/20/49/495101. [DOI] [PubMed] [Google Scholar]
- 32.Caldorera-Moore M, Guimard N, Shi L, Roy K. Designer Nanoparticles: Incorporating Size, Shape and Triggered Release Into Nanoscale Drug Carriers. Expert Opin Drug Delivery. 2010;7(4):479–495. doi: 10.1517/17425240903579971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gentile F, Chiappini C, Fine D, Bhavane RC, Peluccio MS, Cheng MM-C, Liu X, Ferrari M, Decuzzi P. The Effect of Shape on the Margination Dynamics of Non-Neutrally Buoyant Particles in Two-Dimensional Shear Flows. J Biomech. 2008;41(10):2312–2318. doi: 10.1016/j.jbiomech.2008.03.021. [DOI] [PubMed] [Google Scholar]
- 34.Wen AM, Steinmetz NF. Design of Virus-Based Nanomaterials for Medicine, Biotechnology, and Energy. Chem Soc Rev. 2016;45(15):4074–4126. doi: 10.1039/c5cs00287g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koudelka KJ, Pitek AS, Manchester M, Steinmetz NF. Virus-Based Nanoparticles as Versatile Nanomachines. Annu Rev Virol. 2015;2(1):379–401. doi: 10.1146/annurev-virology-100114-055141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bruckman MA, Randolph LN, VanMeter A, Hern S, Shoffstall AJ, Taurog RE, Steinmetz NF. Biodistribution, Pharmacokinetics, and Blood Compatibility of Native and PEGylated Tobacco Mosaic Virus Nano-Rods and -Spheres in Mice. Virology. 2014;449:163–173. doi: 10.1016/j.virol.2013.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toy R, Hayden E, Shoup C, Baskaran H, Karathanasis E. The Effects of Particle Size, Density and Shape on Margination of Nanoparticles in Microcirculation. Nanotechnology. 2011;22(11):115101. doi: 10.1088/0957-4484/22/11/115101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bruckman MA, Jiang K, Simpson EJ, Randolph LN, Luyt LG, Yu X, Steinmetz NF. Dual-Modal Magnetic Resonance and Fluorescence Imaging of Atherosclerotic Plaques in Vivo Using VCAM-1 Targeted Tobacco Mosaic Virus. Nano Lett. 2014;14(3):1551–1558. doi: 10.1021/nl404816m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bryan J. The Rise and Fall of the Clot Buster: a Review on the History of Streptokinase. Pharm J. 2014 doi: 10.1211/PJ.2014.20065679. [DOI] [Google Scholar]
- 40.Bruckman MA, Steinmetz NF. Chemical Modification of the Inner and Outer Surfaces of Tobacco Mosaic Virus (TMV) Methods Mol Biol. 2014;1108:173–185. doi: 10.1007/978-1-62703-751-8_13. Chapter13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an Extensible Image Processing Suite for Electron Microscopy. J Struct Biol. 2007;157(1):38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 42.Mastronarde DN. Automated Electron Microscope Tomography Using Robust Prediction of Specimen Movements. J Struct Biol. 2005;152(1):36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 43.Kremer JR, Mastronarde DN, McIntosh JR. Computer Visualization of Three-Dimensional Image Data Using IMOD. J Struct Biol. 1996;116(1):71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 44.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a Visualization System for Exploratory Research and Analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 45.Geiger FC, Eber FJ, Eiben S, Mueller A, Jeske H, Spatz JP, Wege C. TMV Nanorods with Programmed Longitudinal Domains of Differently Addressable Coat Proteins. Nanoscale. 2013;5(9):3808–3816. doi: 10.1039/c3nr33724c. [DOI] [PubMed] [Google Scholar]
- 46.Namba K, Pattanayek R, Stubbs G. Visualization of Protein-Nucleic Acid Interactions in a Virus. Refined Structure of Intact Tobacco Mosaic Virus at 2.9 a Resolution by X-Ray Fiber Diffraction. J Mol Biol. 1989;208(2):307–325. doi: 10.1016/0022-2836(89)90391-4. [DOI] [PubMed] [Google Scholar]
- 47.Wang X, Lin X, Loy JA, Tang J, Zhang XC. Crystal Structure of the Catalytic Domain of Human Plasmin Complexed with Streptokinase. Science. 1998;281(5383):1662–1665. doi: 10.1126/science.281.5383.1662. [DOI] [PubMed] [Google Scholar]
- 48.Lee KL, Shukla S, Wu M, Ayat NR, El Sanadi CE, Wen AM, Edelbrock JF, Pokorski JK, Commandeur U, Dubyak GR, Steinmetz NF. Stealth Filaments: Polymer Chain Length and Conformation Affect the in Vivo Fate of PEGylated Potato Virus X. Acta Biomater. 2015;19:166–179. doi: 10.1016/j.actbio.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Destito G, Yeh R, Rae CS, Finn MG, Manchester M. Folic Acid-Mediated Targeting of Cowpea Mosaic Virus Particles to Tumor Cells. Chem Biol. 2007;14(10):1152–1162. doi: 10.1016/j.chembiol.2007.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nolan M, Bouldin SD, Bock PE. Full Time Course Kinetics of the Streptokinase-Plasminogen Activation Pathway. J Biol Chem. 2013;288(41):29482–29493. doi: 10.1074/jbc.M113.477935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pitek AS, Jameson SA, Veliz FA, Shukla S, Steinmetz NF. Serum Albumin “Camouflage” of Plant Virus Based Nanoparticles Prevents Their Antibody Recognition and Enhances Pharmacokinetics. Biomaterials. 2016;89:89–97. doi: 10.1016/j.biomaterials.2016.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gulati NM, Pitek AS, Steinmetz NF, Stewart PL. Cryo-Electron Tomography Investigation of Serum Albumin-Camouflaged Tobacco Mosaic Virus Nanoparticles. Nanoscale. 2017;9(10):3408–3415. doi: 10.1039/c6nr06948g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bludau H, Czapar AE, Pitek AS, Shukla S, Jordan R, Steinmetz NF. POxylation as an Alternative Stealth Coating for Biomedical Applications. Eur Polym J. 2017;88:679–688. doi: 10.1016/j.eurpolymj.2016.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Geng Y, Dalhaimer P, Cai S, Tsai R, Tewari M, Minko T, Discher DE. Shape Effects of Filaments Versus Spherical Particles in Flow and Drug Delivery. Nat Nanotechnol. 2007;2(4):249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.