

Abstract
Statins mediate vascular protection and reduce the prevalence of cardiovascular diseases. Recent work indicates that statins have anticonvulsive effects in the brain; however, little is known about the precise mechanism for its protective effect in kainic acid (KA)-induced seizures. Here, we investigated the protective effects of atorvastatin pretreatment on KA-induced neuroinflammation and hippocampal cell death. Mice were treated via intragastric administration of atorvastatin for 7 days, injected with KA, and then sacrificed after 24 h. We observed that atorvastatin pretreatment reduced KA-induced seizure activity, hippocampal cell death, and neuroinflammation. Atorvastatin pretreatment also inhibited KA-induced lipocalin-2 expression in the hippocampus and attenuated KA-induced hippocampal cyclooxygenase-2 expression and glial activation. Moreover, AKT phosphorylation in KA-treated hippocampus was inhibited by atorvastatin pretreatment. These findings suggest that atorvastatin pretreatment may protect hippocampal neurons during seizures by controlling lipocalin-2-associated neuroinflammation.
Keywords: Atorvastatin, Hippocampal cell death, Kainic acid, Lipocalin-2, Seizures
INTRODUCTION
Kainic acid (KA), a glutamate derivative, is an epileptogenic agent known to induce neuronal cell death [1]. The hippocampus is vulnerable to KA-induced damage, which leads to neuronal cell death and seizures [2]. KA injection activates both astrocytes and microglia related to cell apoptosis and increases inflammatory cytokines in damaged hippocampal neuronal cells [3]. KA-induced blood-brain barrier (BBB) leakage is associated with ionic imbalance and nuclear factor-κB (NF-κB) activation [4,5]. Thus, inhibition of neuroinflammation and neurovascular breakdown may be critical to protect against seizures-induced neuronal death.
Statins are recommended by American Heart Association as a standard element of stroke treatment due to their apparent neuroprotective role after stroke. In stroke patients, 35% with early onset seizures and 90% with late onset seizures developed epilepsy [6]. Several studies have provided convincing evidence that short-term statin treatment in acute stroke patients may have neuroprotective effects [7]. Atorvastatin inhibits 3-hydroxy-3-methylglutaryl coenzyme-A (HMG Co-A) reductase, which plays an important role in the production of cholesterol biosynthesis. Atorvastatin has vascular protective effects and many other pleiotropic effects such as anti-inflammatory and neuroprotective effects regardless of changes in cholesterol levels [8]. Previous studies reported that pretreatment with atorvastatin or other statins may have beneficial effects, including infarct volume reduction, improvement of neurological deficits in mouse models of cerebral ischemia [9], and inhibition of neuroinflammation and hippocampal cell death in KA-induced seizures [10,11]. Little is known about the precise mechanism for the protective effects of atorvastatin in KA-induced neuronal cell death.
Lipocalin-2 (LCN2) was originally shown to sequester iron and thereby induce anti-microbial defense responses [12]. LCN2 is important for cellular apoptosis in vitro [13]. Lipopolysaccharide (LPS) treatment upregulates LCN2 mRNA expression by activating the Toll-like receptor 4 (TLR4) signaling pathway [14]. LCN2 acts as an adipokine inducer in the brain [15], and studies in animal brain injury models, such as LPS-induced neuroinflammation, encephalomyelitis, and cerebral ischemia, report that LCN2 contributes to neuronal cell death [16]. In particular, reactive astrogliosis is closely associated with LCN2 expression [17]. No studies on the neuroprotective effects of atorvastatin (AS) on hippocampal LCN2 expression in KA-induced neuronal cell death have been reported. Thus, our study aims to investigate the neuroprotective effect of AS pretreatment on KA-induced neuronal cell death. We explored the anti-inflammatory effects of AS LCN2 expression and neuroinflammation in a KA-induced mouse seizure model.
METHODS
Animals
Male ICR (25-30 g) mice were purchased from KOATECH Co. (Pyeongtaek, South Korea) and housed under a 12/12-h light/dark schedule. Mice were provided with water and standard chow ad libitum for 1 week prior to any experimental procedures. All animal experiments were approved by the Institute of Animal Care and Use Committee of Gyeongsang National University (GNU-140922-M0046).
Drug treatment and seizure induction
Experiment 1
Mice were treated with an intraperitoneal injection of 30 mg/kg KA (Abcam, Cambridge, MA, USA). Control mice (n=5) were injected with an equivalent volume of 0.9% normal saline. Mice in the experimental group (n=6 mice per group) were sacrificed 2, 6, or 24 h after systemic KA injection.
Experiment 2
Animals were randomly divided into the following four groups: control (CTL, n=14), KA-induced seizure mice (KA, n=15), seizure mice pretreated with AS (Lipitor®, Daewoong Bio. Inc., South Korea) for 7 d (KA+AS, n=15), and mice treated only with AS (n=14). In order to evaluate the efficacy of AS in seizures mice, mice were treated orally with 10 mg/kg AS emulsified in saline once a day 7 before KA treatment. The dose of AS was based on previous studies in KA-treated seizure rats and has been shown to reduce seizure activity and hippocampal neuron death [10,18,19]. Then, mice were treated with an intraperitoneal injection of 30 mg/kg KA. Control mice were injected with an equivalent volume of normal saline. After KA or saline injection, the seizure activity was scored during the 2 h observation period. Seizure activity was evaluated using a six-point seizure scale, as described previously [20]. All mice were euthanized via decapitation 24 h after KA treatment.
Tissue preparation
For tissue preparation, mice (CTL or AS, n=5; KA or KA+AS, n=7) were deeply anesthetized with 5 mg/kg Zoletil (Virbac Laboratories, Carros, France), and subjected to transcardial perfusion of heparinized saline. Mice brains were fixed in 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS). After 6 h of post-fixation, the brains were immersed in 15% sucrose in 0.1 M PBS, and then in 30% sucrose in 0.1 M PBS at 4℃. Brains were embedded in OCT (Tissue-Teck), frozen, and then cut into 40-µm thick sections. Sections were performed cresyl violet.
TUNEL assay
TUNEL was performed on frozen tissue sections using an in situ cell death detection kit (Roche Molecular Biochemicals, Mannheim, Germany) according to the manufacturer's protocol. The sections were examined under a microscope (BX51-DSU, Olympus, Tokyo, Japan), and digital images were captured. For each treatment group, TUNEL-positive cells were manually counted in the CA3 region (50×50 µm) in three sections (n=5–7 mice/group). The cells were counted by observers who were blinded to the treatment conditions.
Immunofluorescence
Free-floating brain tissue sections were incubated with two antibodies: mouse anti-glial fibrillary acidic protein (GFAP, Santa Cruz Biotechnology, Dallas, TX, USA; 1:500) antibody; goat anti-LCN2 (Abcam, Cambridge, UK; 1:200) antibody; rabbit anti-Iba-1 (Wako Pure Chemical Industry Richmond, VA, USA; 1:200). Samples were washed with 0.1 M PBS and incubated with Alexa Fluor 488-conjugated donkey anti-mouse or 594-anti-goat secondary antibodies (Invitrogen Life Technologies, Carlsbad, CA, USA) for 1 h at room temperature. The sections imaging were carried out using a microscope and captured (BX51-DSU, Olympus), three consecutive sections per animal were analyzed and similar sections were used.
Immunohistochemistry
The brain sections were incubated with rabbit anti-cyclooxigenase-2 (COX-2, Cayman Chemical, Ann Arbor, MI, USA; 1:300). After 14 h incubation at 4℃, sections were washed and incubated for 1 h at RT with the biotinylated secondary antibody. Brain sections were processed with ABC solution (Vector Laboratories, CA, USA) and the DAB kit (Sigma, St. Louis, MO, USA). Sections were mounted on slides using standard protocols. The stained slides were examined and imaged under a microscope (Olympus).
Western blot analysis
Brains from mice that were anesthetized with Zoletil (CTL or AS, n=6; KA or KA+AS, n=7) were immediately removed from the skull. The hippocampus was dissected and frozen. Protein expression was analyzed as previously described [21]. Briefly, proteins were subjected to immunoblotting with anti-GFAP (Sigma, 1:1,000), anti-Iba1 (Wako, 1:1,000), anti-LCN2 (R&D Systems, Minneapolis, MN, USA; 1:1,000), anti-phospho AKT (Ser473, Cell Signaling Technology, Danvers, MA, USA; 1:1,000), anti-AKT (Cell Signaling, 1:1,000) and anti-COX-2 (Cayman Chemical, 1:1,000) antibodies. To determine relative protein amounts, β-actin (Sigma, 1:30,000) was used as an internal control. The PVDF membranes were visualized with an ECL substrate (Pierce, Rockford, IL, USA) and band intensities were measured with the Multi Gauge v 3.0 image analysis program (Fujifilm, Tokyo, Japan).
Statistical analysis
Differences between time-dependent groups or between the experimental groups (CTL, KA, KA+AS, and AS) were determined with one-way analysis of variance (ANOVA), followed by post-hoc analysis with a Student-Newman-Keuls test or a Student's t-test when only two groups were compared. Values are expressed as the mean±standard error of the mean (S.E.M.). A p-value <0.05 was considered statistically significant.
RESULTS
Effect of KA pretreatment on altered LCN2 expression in the mouse hippocampus
To determine whether KA treatment induces LCN2 expression in mice, we examined hippocampal LCN2 expression in mice 2, 6, and 24 h after systemic KA injection (Fig. 1). The expression levels of LCN2 in the hippocampus were significantly increased 6 h after KA treatment and were further increased 24 h after KA.
Fig. 1. Effect of KA treatment on hippocampal LCN2 expression in mice.
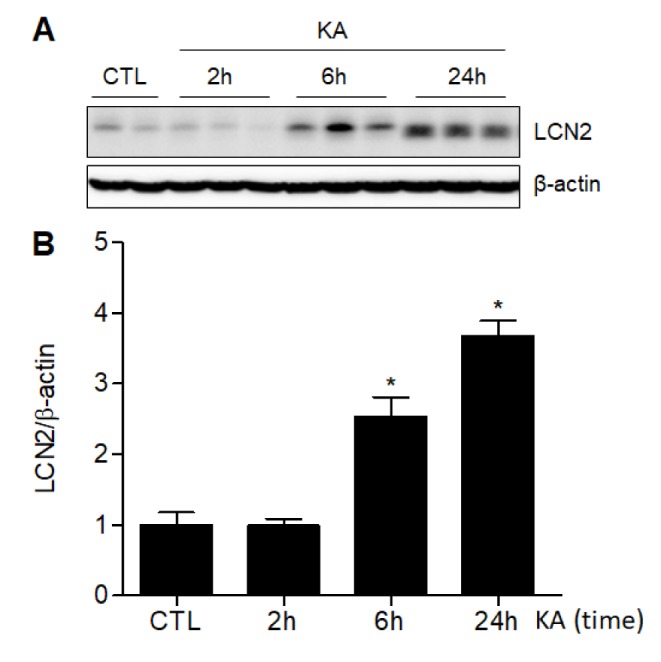
(A) Western blot showing altered LCN2 expression in KA-treated hippocampus. The hippocampal LCN2 expression appeared 6 h after KA treatment. (B) Quantitative expression of LCN2 protein from the western blot analysis. Densitometric values for LCN2 were normalized to β-actin levels. The mean values were obtained from two separate experiments (n=5-6 mice per group). Data are shown as mean±SEM. *p<0.05 vs. KA.
Effect of AS pretreatment on seizure activity in KA-treated mice
To assess the effect of pretreatment with AS on neuroprotection in mice with KA-induced seizures, we administered AS for 7 days at a dose of 10 mg/kg as previously reported [10,18,19]. We evaluated the anticonvulsant effects of AS in KA-induced seizures by monitoring the behavioral seizure activity and percent survival for 2 h after KA treatment (Fig. 2A and B). AS pretreatment reduced the seizure activity scores to >3 in KA-induced seizures mice. In addition, the KA group had significantly more severe seizures than the KA+AS group, beginning 40 min after KA-injection (p<0.001, Fig. 2A). Mice treated with KA+AS had a higher survival rate than those treated with KA alone (Fig. 2B).
Fig. 2. Effect of atorvastatin pretreatment on seizure activity and hippocampal cell death in KA-treated mice.

At 24 h after KA systemic injection, seizure activity was scored during the 2 h observation period. Behavioral seizure scores (A) and percentage of survival (B) in KA-treated mice with or without AS are shown. Data (n=15 mice per group) are shown as mean±SEM. *p<0.05 vs. KA. (C) Representative microphotographs of cresyl violet-stained sections and immunofluorescent images of TUNEL-stained sections in CA3 regions. The boxed area of cresyl violet-stained images is magnified in the center panel. Scale bar=50 mm.
Effect of AS pretreatment on neuroprotection in KA-treated mice
As hippocampal LCN2 expression levels were increased 24 h after KA treatment (Fig. 1), we sought to evaluate the neuroprotective effects of AS pretreatment. Mice were sacrificed 24 h after KA injection, and brain sections were examined with cresyl violet and TUNEL staining (Fig. 2C). Cresyl violet staining showed that KA-treated mice exhibited significant cell death in the hippocampal CA3 region, where the pyramidal neurons had typical pyknotic nuclei; however, these apoptotic nuclei did not appear in the hippocampus of the KA+AS mice. The KA mice displayed more TUNEL-positive nuclei than control mice, confirming that the observed cell death was apoptotic, and this robust TUNEL labeling in the CA3 neurons of KA mice declined after AS pretreatment. Specifically, the number of TUNEL-positive cells (38.0±4.0) in the KA-treated hippocampus was decreased by AS (1±0.6). These results suggest that AS pretreatment protects KA-induced hippocampal cell death.
Effect of AS pretreatment on LCN2 expression in KA-treated hippocampus
LCN2 expression was observed in the pyramidal cell layers of CA1-3 of the hippocampus, co-localizing with mostly the neuronal marker NeuN and to a lesser extent, with the astrocyte marker GFAP [22,23]. To determine whether the expression of LCN2 is affected by AS pretreatment in KA-induced seizures, we performed immunoblotting and immunofluorescence analysis with LCN2 antibody (Figs. 3A and B). KA-induced hippocampal LCN2 expression was significantly reduced by AS pretreatment (Fig. 3A). Double immunofluorescence analysis revealed that many LCN2-positive cells colocalized with GFAP expression around KA-induced neuronal death (neuronal loss) in the CA3 regions (Fig. 3B). However, few LCN2-positive astrocytes were detected in the KA+AS-treated hippocampus. As shown in Fig. 3B, KA-induced neuronal death or loss in the CA3 regions was evident. These lesions consisted of a few neurons and many activated glial cells including astrocytes and microglia. Compared to control mice, LCN2 expression was observed in many reactive astrocytes in CA3 of the hippocampus, co-localizing with mostly GFAP and to a lesser extent in the pyramidal cell layers of CA3. Thus, the representative microphotographs do not appear to completely support the western blot data (Figs. 3A and B). In addition, we found that few LCN2-positive cell was colocalized with Iba-1 (Fig. 3C). These findings indicate that reactive astrocyte-derived LCN2 may play an important role in KA-induced neuroinflammation.
Fig. 3. Effect of atorvastatin pretreatment on LCN2 expression in KA-treated hippocampus.

(A) Western blots show hippocampal LCN2 expression 24 h after KA treatment. Densitometric values of LCN2 expression from three separate experiments (n=6–7 mice per group) were normalized to β-actin levels. *p<0.05 vs. CTL. †p<0.05 vs. KA. Representative images of double-immunofluorescence staining for LCN2 (red) and GFAP (green) (B) and LCN2 (red) and Iba-1 (green) (C) in the CA3 region of CTL, KA, KA+AS, and AS mice. Scale bar=50 µm.
Effect of AS pretreatment on hippocampal inflammation in KA-treated mice
COX-2 expression is induced at regions of inflammation and localizes to pyramidal neurons in KA-treated hippocampus [24]. To determine whether AS attenuates KA-induced COX-2 expression in the hippocampus, we performed immunoblotting and immunofluorescence analyses with the COX-2 antibody (Figs. 4A and B). We observed that KA-induced hippocampal COX-2 expression was significantly reduced by AS pretreatment (Fig. 4A). Consistently, KA+AS mice had a significant decrease of COX-2 immunoreactivity in the hippocampus CA3 compared with KA-treated mice. In addition, we performed immunoblotting analysis of astrocyte-specific GFAP (Fig. 4C) and microglia-specific Iba-1 (Fig. 4D). These results, which are consistent with LCN2-associated glial inflammation, indicated that KA-induced glial activation was significantly inhibited by AS pretreatment.
Fig. 4. Effect of atorvastatin pretreatment on neuroinflammation in KA-treated hippocampus.

(A) Western blots show hippocampal COX-2 expression 24 h after KA treatment. Densitometric values of COX-2 expression from three separate experiments (n=6–7 mice per group) were normalized to β-actin levels. *p<0.05 vs. CTL. †p<0.05 vs. KA. (B) Representative images of immunostained COX-2 in the hippocampus of CTL, KA, KA+AS, and AS mice. The boxed area of each image is magnified in the lower panel. Scale bar=50 mm. (C) Western blots show hippocampal GFAP (C) and Iba-1 (D) expression 24 h after KA treatment. Densitometric values (n=6–7 mice per group) for GFAP and Iba1 were normalized to β-actin levels. *p<0.05 vs. CTL. †p<0.05 vs. KA-treated mice.
Effect of AS pretreatment on AKT phosphorylation in KA-treated hippocampus
The neuroprotective role of statins is associated with AKT phosphorylation in brain injury [25,26]. To evaluate whether AS pretreatment affects pAKT and total AKT expression levels in the hippocampus 24 h after KA treatment, we performed western blot analysis (Fig. 5A). AS significantly inhibited KA-induced upregulation of pAKT expression in KA-treated hippocampus, and we detected no changes in hippocampal pAKT or total AKT expression in mice treated with only AS (Fig. 5B).
Fig. 5. Effect of atorvastatin pretreatment on AKT phosphorylation in KA-treated hippocampus.
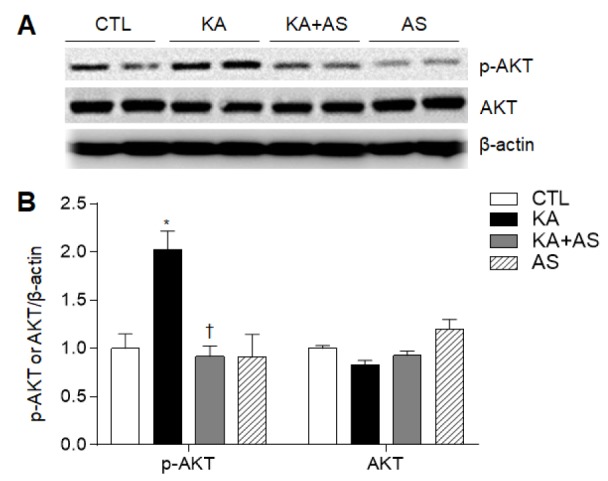
(A) Western blot shows p-AKT and AKT expression in KA-treated hippocampus. (B) Quantitative expression of p-AKT and AKT protein from the western blot analysis. Densitometric values of expression of each protein from three separate experiments (n=6–7 mice per group) were normalized to β-actin levels. Data are shown as mean±SEM. *p<0.05 vs. CTL. †p<0.05 vs. KA-treated mice.
DISCUSSION
KA treatment in mice induces progressive limbic seizures, leading to neuronal death and an inflammatory response in the hippocampus [2]. Here, we found that AS pretreatment attenuated KA-induced seizure activity, hippocampal neuronal damage, and inflammation as indicated by reduction of enhanced LCN2 and COX-2 expression. Our results were consistent with a previous study, which reported that KA-induced neuronal cell death was less prominent in mice that were treated with statin than in non-treated mice [10]. Our data indicate that AS pretreatment attenuates KA-induced glial cell activation, neuroinflammation, and neuronal cell death. Therefore, we suggest that AS may exert beneficial effects to control seizures and other neurological disorders associated with excitotoxicity.
Statins inhibit HMG-CoA reductase and are used to prevent secondary ischemic stroke and coronary heart disease [27,28]. Recently, statins have been recognized as neuroprotective and anti-inflammatory drugs as well [29,30]. A previous study showed that both lovastatin and simvastatin, but not AS, have potential for the treatment of neurodegenerative diseases [31]. By contrast, another study reported that AS inhibits KA-induced seizures, neuroinflammation, and hippocampal cell death [10]. In our study of mice with KA-induced seizures, we confirmed that AS reduced neuronal cell death and attenuated neuroinflammation including COX-2 expression and glial activation. These results were consistent with the previously reported neuroprotective role of AS and other statins (simvastatin and lovastatin) in KA-induced brain damage [10,31,32,33]. A previous study reported that the observed neuroprotective effect of AS was not accompanied by changes in cholesterol levels in plasma or cerebral cortex or by changes in BBB permeability from pentylenetetrazol-induced seizures [19]. In the present study, however, we did not evaluate whether AS pretreatment was accompanied by changes in cholesterol levels in plasma or hippocampus in mice with KA-induced seizures. These parameters could be measured in future studies in order to identify the relationship between cholesterol levels and the anti-inflammatory effects of statins and to elucidate the molecular mechanisms underlying these effects in hippocampal neurons.
LCN2 is a siderophore-binding protein. In vivo studies demonstrated that LCN2 expression is associated with excitotoxic brain injury and reactive astrogliosis [34,35]. In rats, KA treatment activates many proinflammatory cytokines such as interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and inducible nitric oxide synthase (iNOS), ultimately leadings to astrocyte activation and induction of neuronal cell apoptosis [10]. Our findings are consistent with a previous report that hippocampal LCN2 expression levels in rats were increased 24 h after LPS or KA treatment [34]. Furthermore, Chia et al. [34] demonstrated that increased LCN2 expression is maintained for 3 d to 2 weeks after KA treatment. In addition, our findings are consistent with our previous report that hippocampal COX-2 expression is significantly increased 6 h after KA treatment [24]. These data indicate that LCN2 acts as a chemokine inducer in KA-induced neuronal damage and are also consistent with the notion that LCN2 plays a key role in regulating astrocyte activation [36], as this protein is a well-characterized marker of reactive astrocytes under neuroinflammatory conditions [17]. LCN2-positive cells colocalize with NeuN-positive neurons in rat brains during ischemia [37], and this factor is released by damaged neurons as a damage signal that elicits further reactive astrocytosis. We found that LCN2 staining was particularly evident in KA-treated hippocampus and colocalized with GFAP-positive astrocytes. Importantly, these increases were reversed by AS pretreatment. Finally, our findings indicate that KA-induced COX-2 expression and glial activation were inhibited by AS pretreatment. These results suggest that AS inhibits KA-induced neuroinflammation and protects neuronal cell death in mice with KA-induced seizures via two mechanisms. First, AS could attenuate proinflammatory cytokines, which activate astrocytes. Second, this factor could attenuate reactive astrocytes via regulation of LNC2 expression.
AKT phosphorylation serves an important function in the neuronal survival pathway via NF-κB, Bad, and caspase 9 [38]. Increased p-AKT (Ser473) has been reported to prevent quinolinic acid-induced neuronal death [26], and immunohistochemical analysis revealed an increase of hippocampal expression of p-AKT (Ser473) in the CA3 region 24 h following KA-induced seizures [39]. Noshita et al. [40] reported that dephosphorylation of AKT (Ser473) occurred at the ischemic core while undamaged areas displayed an upregulation of p-AKT. In addition, a global ischemia-induced transient increase of p-AKT in the hippocampus has been described in delayed neuronal death [41]. Thus our findings suggest that increased p-AKT expression in areas adjacent to damaged areas may contribute to these areas remaining undamaged in KA-treated hippocampus. These data support a role for p-AKT (Ser473) in neuronal survival following excitotoxicity-induced brain injury. Contrary to our observations, AKT phosphorylation was reduced in KA-treated hippocampus 24 h following KA treatment in another study [42]. The reasons for this contradictory results are not clear, but may be related to a different dose of KA or a different age and species of mouse. Nonetheless, the present study unequivocally demonstrated that AS pretreatment significantly reverses KA-induced AKT phosphorylation.
In conclusion, we confirmed the protective effect of AS pretreatment against neuronal cell death and on neuronal and astrocytic LCN2 expression. Recent work reported that statin treatment reduces the risk of post-stroke early onset seizures and prevents progression into chronic epilepsy in patients [43]. We suggest that the anti-inflammatory activity of statins will be an effective therapeutic strategy for patients with epilepsy. Future work will focus on the molecular genetic basis of these interactions.
ACKNOWLEDGEMENTS
The authors would also like to thank all of the participants who have contributed to this research. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2015R1A5A2008833).
Footnotes
Author contributions: Z.J., Y.J., C.Y., J.Y.L., and E.A.J. performed animal experiments and analyzed data. Z.J. and J.Y.L evaluated seizure activity. E.J.L., K.J.P., O.Y.K., and B.H.L. interpreted data and edited the manuscript. N.C.C. and G.S.R. supervised and coordinated the study. Z.J., Y.J., and G.S.R. wrote the manuscript.
CONFLICTS OF INTEREST: The authors declare no conflicts of interest.
References
- 1.McCord MC, Lorenzana A, Bloom CS, Chancer ZO, Schauwecker PE. Effect of age on kainate-induced seizure severity and cell death. Neuroscience. 2008;154:1143–1153. doi: 10.1016/j.neuroscience.2008.03.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carta M, Fièvre S, Gorlewicz A, Mulle C. Kainate receptors in the hippocampus. Eur J Neurosci. 2014;39:1835–1844. doi: 10.1111/ejn.12590. [DOI] [PubMed] [Google Scholar]
- 3.Ravizza T, Rizzi M, Perego C, Richichi C, Velísková J, Moshé SL, De Simoni MG, Vezzani A. Inflammatory response and glia activation in developing rat hippocampus after status epilepticus. Epilepsia. 2005;46(Suppl 5):113–117. doi: 10.1111/j.1528-1167.2005.01006.x. [DOI] [PubMed] [Google Scholar]
- 4.Kim JE, Ryu HJ, Choi SY, Kang TC. Tumor necrosis factor-α-mediated threonine 435 phosphorylation of p65 nuclear factor-κB subunit in endothelial cells induces vasogenic edema and neutrophil infiltration in the rat piriform cortex following status epilepticus. J Neuroinflammation. 2012;9:6. doi: 10.1186/1742-2094-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shin HJ, Jeon BT, Kim J, Jeong EA, Kim MJ, Lee DH, Kim HJ, Kang SS, Cho GJ, Choi WS, Roh GS. Effect of the calcineurin inhibitor FK506 on K+-Cl-cotransporter 2 expression in the mouse hippocampus after kainic acid-induced status epilepticus. J Neural Transm (Vienna) 2012;119:669–677. doi: 10.1007/s00702-011-0746-y. [DOI] [PubMed] [Google Scholar]
- 6.Xie WJ, Dong M, Liu Q, Meng HM. Early predictors and prevention for post-stroke epilepsy: changes in neurotransmitter levels. Transl Neurosci. 2016;7:1–5. doi: 10.1515/tnsci-2016-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ní Chróinín D, Asplund K, Åsberg S, Callaly E, Cuadrado-Godia E, Díez-Tejedor E, Di Napoli M, Engelter ST, Furie KL, Giannopoulos S, Gotto AM, Jr, Hannon N, Jonsson F, Kapral MK, Martí-Fàbregas J, Martínez-Sánchez P, Milionis HJ, Montaner J, Muscari A, Pikija S, Probstfield J, Rost NS, Thrift AG, Vemmos K, Kelly PJ. Statin therapy and outcome after ischemic stroke: systematic review and meta-analysis of observational studies and randomized trials. Stroke. 2013;44:448–456. doi: 10.1161/STROKEAHA.112.668277. [DOI] [PubMed] [Google Scholar]
- 8.Li Q, Zhuang QK, Yang JN, Zhang YY. Statins excert neuroprotection on cerebral ischemia independent of their lipid-lowering action: the potential molecular mechanisms. Eur Rev Med Pharmacol Sci. 2014;18:1113–1126. [PubMed] [Google Scholar]
- 9.Pannu R, Christie DK, Barbosa E, Singh I, Singh AK. Post-trauma Lipitor treatment prevents endothelial dysfunction, facilitates neuroprotection, and promotes locomotor recovery following spinal cord injury. J Neurochem. 2007;101:182–200. doi: 10.1111/j.1471-4159.2006.04354.x. [DOI] [PubMed] [Google Scholar]
- 10.Lee JK, Won JS, Singh AK, Singh I. Statin inhibits kainic acid-induced seizure and associated inflammation and hippocampal cell death. Neurosci Lett. 2008;440:260–264. doi: 10.1016/j.neulet.2008.05.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang L, Zhang X, Liu L, Yang R, Cui L, Li M. Atorvastatin protects rat brains against permanent focal ischemia and downregulates HMGB1, HMGB1 receptors (RAGE and TLR4), NF-kappaB expression. Neurosci Lett. 2010;471:152–156. doi: 10.1016/j.neulet.2010.01.030. [DOI] [PubMed] [Google Scholar]
- 12.Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell. 2002;10:1033–1043. doi: 10.1016/s1097-2765(02)00708-6. [DOI] [PubMed] [Google Scholar]
- 13.Devireddy LR, Gazin C, Zhu X, Green MR. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell. 2005;123:1293–1305. doi: 10.1016/j.cell.2005.10.027. [DOI] [PubMed] [Google Scholar]
- 14.Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- 15.Lee S, Kim JH, Kim JH, Seo JW, Han HS, Lee WH, Mori K, Nakao K, Barasch J, Suk K. Lipocalin-2 is a chemokine inducer in the central nervous system: role of chemokine ligand 10 (CXCL10) in lipocalin-2-induced cell migration. J Biol Chem. 2011;286:43855–43870. doi: 10.1074/jbc.M111.299248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin M, Kim JH, Jang E, Lee YM, Soo Han H, Woo DK, Park DH, Kook H, Suk K. Lipocalin-2 deficiency attenuates neuroinflammation and brain injury after transient middle cerebral artery occlusion in mice. J Cereb Blood Flow Metab. 2014;34:1306–1314. doi: 10.1038/jcbfm.2014.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee S, Park JY, Lee WH, Kim H, Park HC, Mori K, Suk K. Lipocalin-2 is an autocrine mediator of reactive astrocytosis. J Neurosci. 2009;29:234–249. doi: 10.1523/JNEUROSCI.5273-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sehar N, Agarwal NB, Vohora D, Raisuddin S. Atorvastatin prevents development of kindling by modulating hippocampal levels of dopamine, glutamate, and GABA in mice. Epilepsy Behav. 2015;42:48–53. doi: 10.1016/j.yebeh.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 19.Funck VR, de Oliveira CV, Pereira LM, Rambo LM, Ribeiro LR, Royes LF, Ferreira J, Guerra GP, Furian AF, Oliveira MS, Mallmann CA, de Mello CF, Oliveira MS. Differential effects of atorvastatin treatment and withdrawal on pentylenetetrazol-induced seizures. Epilepsia. 2011;52:2094–2104. doi: 10.1111/j.1528-1167.2011.03261.x. [DOI] [PubMed] [Google Scholar]
- 20.Hu RQ, Koh S, Torgerson T, Cole AJ. Neuronal stress and injury in C57/BL mice after systemic kainic acid administration. Brain Res. 1998;810:229–240. doi: 10.1016/s0006-8993(98)00863-4. [DOI] [PubMed] [Google Scholar]
- 21.Kang DH, Heo RW, Yi CO, Kim H, Choi CH, Roh GS. High-fat diet-induced obesity exacerbates kainic acid-induced hippocampal cell death. BMC Neurosci. 2015;16:72. doi: 10.1186/s12868-015-0202-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mucha M, Skrzypiec AE, Schiavon E, Attwood BK, Kucerova E, Pawlak R. Lipocalin-2 controls neuronal excitability and anxiety by regulating dendritic spine formation and maturation. Proc Natl Acad Sci U S A. 2011;108:18436–18441. doi: 10.1073/pnas.1107936108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skrzypiec AE, Shah RS, Schiavon E, Baker E, Skene N, Pawlak R, Mucha M. Stress-induced lipocalin-2 controls dendritic spine formation and neuronal activity in the amygdala. PLoS One. 2013;8:e61046. doi: 10.1371/journal.pone.0061046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeong EA, Jeon BT, Shin HJ, Kim N, Lee DH, Kim HJ, Kang SS, Cho GJ, Choi WS, Roh GS. Ketogenic diet-induced peroxisome proliferator-activated receptor-γ activation decreases neuroinflammation in the mouse hippocampus after kainic acid-induced seizures. Exp Neurol. 2011;232:195–202. doi: 10.1016/j.expneurol.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 25.Chen J, Zhang ZG, Li Y, Wang Y, Wang L, Jiang H, Zhang C, Lu M, Katakowski M, Feldkamp CS, Chopp M. Statins induce angiogenesis, neurogenesis, and synaptogenesis after stroke. Ann Neurol. 2003;53:743–751. doi: 10.1002/ana.10555. [DOI] [PubMed] [Google Scholar]
- 26.Piermartiri TC, Vandresen-Filho S, de Araújo Herculano B, Martins WC, Dal'agnolo D, Stroeh E, Carqueja CL, Boeck CR, Tasca CI. Atorvastatin prevents hippocampal cell death due to quinolinic acid-induced seizures in mice by increasing Akt phosphorylation and glutamate uptake. Neurotox Res. 2009;16:106–115. doi: 10.1007/s12640-009-9057-6. [DOI] [PubMed] [Google Scholar]
- 27.Pursnani A, Massaro JM, D'Agostino RB, Sr, O'Donnell CJ, Hoffmann U. Guideline-based statin eligibility, coronary artery calcification, and cardiovascular events. JAMA. 2015;314:134–141. doi: 10.1001/jama.2015.7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Castilla-Guerra L, Fernández-Moreno Mdel C, López-Chozas JM. Statins in the secondary prevention of stroke: New evidence from the SPARCL Study. Clin Investig Arterioscler. 2016;28:202–208. doi: 10.1016/j.arteri.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 29.Takemoto M, Yokote K. Prevention, treatment and management of inflammation in atherosclerosis. Nihon Rinsho. 2011;69:18–24. [PubMed] [Google Scholar]
- 30.Wood WG, Mΰller WE, Eckert GP. Statins and neuroprotection: basic pharmacology needed. Mol Neurobiol. 2014;50:214–220. doi: 10.1007/s12035-014-8647-3. [DOI] [PubMed] [Google Scholar]
- 31.Ramirez C, Tercero I, Pineda A, Burgos JS. Simvastatin is the statin that most efficiently protects against kainate-induced excitotoxicity and memory impairment. J Alzheimers Dis. 2011;24:161–174. doi: 10.3233/JAD-2010-101653. [DOI] [PubMed] [Google Scholar]
- 32.Gouveia TL, Scorza FA, Silva MJ, Bandeira Tde A, Perosa SR, Argañaraz GA, Silva Mde P, Araujo TR, Frangiotti MI, Amado D, Cavalheiro EA, Silva JA, Jr, Naffah-Mazzacoratti Mda G. Lovastatin decreases the synthesis of inflammatory mediators in the hippocampus and blocks the hyperthermia of rats submitted to longlasting status epilepticus. Epilepsy Behav. 2011;20:1–5. doi: 10.1016/j.yebeh.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 33.Saito T, Nito C, Ueda M, Inaba T, Kamiya F, Muraga K, Katsura K, Katayama Y. Continuous oral administration of atorvastatin ameliorates brain damage after transient focal ischemia in rats. Life Sci. 2014;94:106–114. doi: 10.1016/j.lfs.2013.11.018. [DOI] [PubMed] [Google Scholar]
- 34.Chia WJ, Dawe GS, Ong WY. Expression and localization of the iron-siderophore binding protein lipocalin 2 in the normal rat brain and after kainate-induced excitotoxicity. Neurochem Int. 2011;59:591–599. doi: 10.1016/j.neuint.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 35.Jang E, Lee S, Kim JH, Kim JH, Seo JW, Lee WH, Mori K, Nakao K, Suk K. Secreted protein lipocalin-2 promotes microglial M1 polarization. FASEB J. 2013;27:1176–1190. doi: 10.1096/fj.12-222257. [DOI] [PubMed] [Google Scholar]
- 36.Lee S, Lee J, Kim S, Park JY, Lee WH, Mori K, Kim SH, Kim IK, Suk K. A dual role of lipocalin 2 in the apoptosis and deramification of activated microglia. J Immunol. 2007;179:3231–3241. doi: 10.4049/jimmunol.179.5.3231. [DOI] [PubMed] [Google Scholar]
- 37.Xing C, Wang X, Cheng C, Montaner J, Mandeville E, Leung W, van Leyen K, Lok J, Wang X, Lo EH. Neuronal production of lipocalin-2 as a help-me signal for glial activation. Stroke. 2014;45:2085–2092. doi: 10.1161/STROKEAHA.114.005733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fukunaga K, Kawano T. Akt is a molecular target for signal transduction therapy in brain ischemic insult. J Pharmacol Sci. 2003;92:317–327. doi: 10.1254/jphs.92.317. [DOI] [PubMed] [Google Scholar]
- 39.Dunleavy M, Provenzano G, Henshall DC, Bozzi Y. Kainic acid-induced seizures modulate Akt (SER473) phosphorylation in the hippocampus of dopamine D2 receptor knockout mice. J Mol Neurosci. 2013;49:202–210. doi: 10.1007/s12031-012-9927-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Noshita N, Lewén A, Sugawara T, Chan PH. Akt phosphorylation and neuronal survival after traumatic brain injury in mice. Neurobiol Dis. 2002;9:294–304. doi: 10.1006/nbdi.2002.0482. [DOI] [PubMed] [Google Scholar]
- 41.Kawano T, Fukunaga K, Takeuchi Y, Morioka M, Yano S, Hamada J, Ushio Y, Miyamoto E. Neuroprotective effect of sodium orthovanadate on delayed neuronal death after transient forebrain ischemia in gerbil hippocampus. J Cereb Blood Flow Metab. 2001;21:1268–1280. doi: 10.1097/00004647-200111000-00003. [DOI] [PubMed] [Google Scholar]
- 42.Kim YS, Choi MY, Lee DH, Jeon BT, Roh GS, Kim HJ, Kang SS, Cho GJ, Choi WS. Decreased interaction between FoxO3a and Akt correlates with seizure-induced neuronal death. Epilepsy Res. 2014;108:367–378. doi: 10.1016/j.eplepsyres.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 43.Guo J, Guo J, Li J, Zhou M, Qin F, Zhang S, Wu B, He L, Zhou D. Statin treatment reduces the risk of poststroke seizures. Neurology. 2015;85:701–707. doi: 10.1212/WNL.0000000000001814. [DOI] [PubMed] [Google Scholar]