

ABSTRACT
1,8-Dihydroxynaphthalene (1,8-DHN) is a key intermediate in the biosynthesis of DHN melanin, which is specific to fungi. In this study, we characterized the enzymatic properties of the gene products of an operon consisting of soceCHS1, bdsA, and bdsB from the Gram-negative bacterium Sorangium cellulosum. Heterologous expression of soceCHS1, bdsA, and bdsB in Streptomyces coelicolor caused secretion of a dark-brown pigment into the broth. High-performance liquid chromatography (HPLC) analysis of the broth revealed that the recombinant strain produced 1,8-DHN, indicating that the operon encoded a novel enzymatic system for the synthesis of 1,8-DHN. Simultaneous incubation of the recombinant SoceCHS1, BdsA, and BdsB with malonyl-coenzyme A (malonyl-CoA) and NADPH resulted in the synthesis of 1,8-DHN. SoceCHS1, a type III polyketide synthase (PKS), catalyzed the synthesis of 1,3,6,8-tetrahydroxynaphthalene (T4HN) in vitro. T4HN was in turn converted to 1,8-DHN by successive steps of reduction and dehydration, which were catalyzed by BdsA and BdsB. BdsA, which is a member of the aldo-keto reductase (AKR) superfamily, catalyzed the reduction of T4HN and 1,3,8-tetrahydroxynaphthalene (T3HN) to scytalone and vermelone, respectively. The stereoselectivity of T4HN reduction by BdsA occurred on the si-face to give (R)-scytalone with more than 99% optical purity. BdsB, a SnoaL2-like protein, catalyzed the dehydration of scytalone and vermelone to T3HN and 1,8-DHN, respectively. The fungal pathway for the synthesis of 1,8-DHN is composed of a type I PKS, naphthol reductases of the short-chain dehydrogenase/reductase (SDR) superfamily, and scytalone dehydratase (SD). These findings demonstrated 1,8-DHN synthesis by novel enzymes of bacterial origin.
IMPORTANCE Although the DHN biosynthetic pathway was thought to be specific to fungi, we discovered novel DHN synthesis enzymes of bacterial origin. The biosynthesis of bacterial DHN utilized a type III PKS for polyketide synthesis, an AKR superfamily for reduction, and a SnoaL2-like NTF2 superfamily for dehydration, whereas the biosynthesis of fungal DHN utilized a type I PKS, SDR superfamily enzyme, and SD-like NTF2 superfamily. Surprisingly, the enzyme systems comprising the pathway were significantly different from each other, suggesting independent, parallel evolution leading to the same biosynthesis. DHN melanin plays roles in host invasion and adaptation to stress in pathogenic fungi and is therefore important to study. However, it is unclear whether DHN biosynthesis occurs in bacteria. Importantly, we did find that bacterial DHN biosynthetic enzymes were conserved among pathogenic bacteria.
KEYWORDS: 1,8-dihydroxynaphthalene; biosynthesis; melanin
INTRODUCTION
Dihydroxynaphthalene (DHN) melanin is a darkly pigmented polymer that is abundant in fungi. In fungal cells, melanins confer resistance to environmental stress and enhance the survival and competitive abilities of organisms in certain environments. In some pathogenic fungi, such as Colletotrichum and Magnaporthe, DHN melanin is essential for the development of melanized appressoria, which are required for host invasion (1). In contrast, in Rosellinia species, DHN melanin is not directly involved in pathogenesis but rather is associated with the development of pseudosclerotia, which are melanized hyphae required for survival under adverse conditions (2).
Similarly to the case in fungi, in some bacteria, melanization and pathogenesis are closely related (3). For example, an hmgA mutant of Vibrio cholerae overproduced pyomelanin and showed a 5-fold increase in the ability to colonize the intestines of infant mice (4). Similarly, pyomelanin hyperproduction and increased persistence in host cells were accompanied by hmgA disruption (5). In addition, Legionella pneumophila utilizes pyomelanin for iron uptake under iron-limiting conditions (6). Although pyomelanin and dihydroxyphenylalanine melanin, which is derived from the oxidation of tyrosine by tyrosinases or laccases (3), are distributed among bacteria and fungi, DHN melanin has not been discovered in bacteria.
1,8-DHN (compound 1) is a monomeric unit of DHN melanin that is synthesized via a polyketide pathway (7, 8) (Fig. 1A). A type I polyketide synthase (PKS) assembles five acetate units, derived from decarboxylation of malonyl-coenzyme A (malonyl-CoA), to produce 1,3,6,8-tetrahydroxynaphthalene (T4HN; compound 2). Compound 2 is reduced by tetrahydroxynaphthalene reductase (T4HNR) to (R)-scytalone [(R)-compound 3], which is readily dehydrated by scytalone dehydratase (SD) to 1,3,8-trihydroxynaphthalene (T3HN; compound 4). Compound 4 is then reduced by trihydroxynaphthalene reductase (T3HNR) to (R)-vermelone [(R)-compound 5] and aromatized by SD to compound 1. Compound 1 is polymerized into melanin by the action of laccases (9). In Magnaporthe grisea, mutants lacking T3HNR or SD failed to infect host plants (10); however, significant quantities of compound 3 were accumulated due to the presence of T4HNR (11). In contrast, a mutant lacking the T4HNR was found to have the wild-type phenotype (11).
FIG 1.

Biosynthesis of melanins from T4HN in bacteria and fungi. (A) The pathway of biosynthesis of 1,8-DHN by bacterial and fungal enzymes. The bacterial and fungal enzymes are represented in plain and italic letters, respectively. Under the enzyme description, family names of enzymes are given in brackets. (B) HPQ melanin and mompain biosynthesis by bacterial enzymes. Boldface numbers represent compound numbers.
Previously, we discovered a bacterial enzyme, RppA, that could synthesize compound 2 from an actinomycete, Streptomyces griseus (12) (Fig. 1B). The fungal system for the biosynthesis of compound 2 uses a type I PKS, whereas RppA is a type III PKS. The pivotal unit for synthesizing polyketides is a β-ketoacyl-acyl carrier protein synthase (KS) that condenses acetate units. Type I PKSs are composed of a KS and “modules,” which are catalytic domains with distinct catalytic properties. These modules are combined into a large polypeptide, consequently forming a large highly modular protein. The type III PKS is a homodimer of KS, and other enzymes are not directly/physically associated. In contrast, for RppA, modification enzymes, which act after the synthesis of compound 2, are organized as an operon with rppA. Instead of the double deoxygenation by naphthalene reductases and SD commonly found in fungi, actinomycetes utilize P-450 for dimerization of compound 2 to produce 1,4,6,7,9,12-hexahydroxyperylene-3,10-quinone (HPQ) (13) (Fig. 1B). HPQ readily autopolymerizes to generate HPQ melanin. In other cases, a monooxygenase, MomA, catalyzes the oxygenation of compound 2 to flaviolin (compound 6) (14) (Fig. 1B). However, the bacterial enzymes responsible for the double-deoxygenation steps from compound 2 to compound 1 have not yet been identified.
Sorangium cellulosum is a soil-dwelling Gram-negative bacterium of the group Myxobacteria. The genome size of S. cellulosum is 13 Mbp, and this organism has great potential for providing secondary products (15). In fact, S. cellulosum is a producer of the eukaryotic cytotoxic chivosazol, the antitumor epothilone, and the siderophore myxochelin. The heterologous expression of SoceCHS1, a type III PKS from S. cellulosum, shares 70% amino acid identity with RppA, resulting in the accumulation of compound 6, an auto-oxidized product of compound 2, in Pseudomonas putida KT2440 (16).
In this study, we focused on an operon consisting of soceCHS1, sce2134, and sce2135 from S. cellulosum. Because functionally related genes are often organized in operons, we expected that sce2134 and sce2135 would be involved in the metabolism of compound 2. Our results showed that sce2134, encoding a product designated bacterial DHN synthase A (BdsA), was a novel naphthol reductase gene involved in catalyzing the reduction of compound 2 and compound 4 to give compound 3 and compound 5, respectively. T4HNR and T3HNR are members of the short-chain dehydrogenase/reductase (SDR) superfamily, whereas BdsA was found to belong to the aldoketoredutase (AKR) superfamily. In addition, sce2135, encoding a product designated bacterial DHN synthase B (BdsB), catalyzed the dehydration of compound 3 and compound 5 to give compound 4 and compound 1, respectively. BdsB was found to be phylogenetically distinct from SD, indicating that BdsB was a novel scytalone/vermelone dehydratase.
RESULTS
Organization of the bds operon and homology search and phylogenetic analysis of BdsA and BdsB.
The gene organization of the 4.3-kb bds gene fragment that includes the bds operon is shown in Fig. 2. The bds operon was found to be 2.5 kb in length and consisted of soceCHS1, bdsA, and bdsB. The stop codon of soceCHS1 was 56 nucleotides upstream from the start codon of bdsA, and the TGA stop codon of bdsA overlapped the GTG start codon of bdsB. The organization of the bds operon suggested that these three formed an operon that was transcribed from a promoter upstream of soceCHS1. sce2132, encoding a 141-amino-acid protein with no apparent homology, appeared to not be functionally linked to the bds operon. sce2136, which was located 191 nucleotides downstream of bdsB, was found to contain four predicted transmembrane helices encoding 123 amino acid residues. Sce2136 is a member of the EamA-like transporter family, implying that Sce2136 may act as a transporter. Sce2132 and Sce2136 have not been studied to date because the bds operon is sufficient for the production of compound 1 (as described below).
FIG 2.
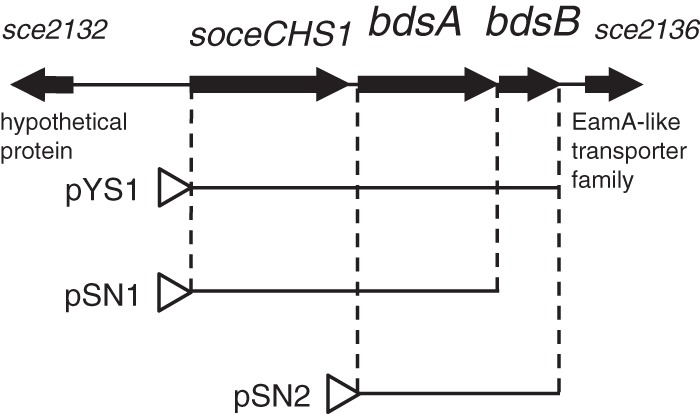
Genetic organization of the soceCHS1, bdsA, and bdsB genes in S. cellulosum. DNA fragments cloned for heterologous expression are presented. pYS1 contained soceCHS1, bdsA, and bdsB. pSN1 contained soceCHS1 and bdsA. pSN2 contained bdsA and bdsB.
A BLAST search revealed that BdsA, consisting of 320 amino acid residues, showed the highest (68% or higher) identity with several functionally unknown proteins of the AKR superfamily, the members of which typically reduce carbonyl substrates in an NAD(P)H-dependent manner (17). AKRs can be categorized into 16 families and are found in all phyla. Furthermore, AKRs have an (α/β)8-barrel motif and retain a conserved catalytic tetrad consisting of Asp, Tyr, Lys, and His. According to the phylogenetic tree analysis, BdsA was classified into the AKR13 family (see Fig. S1 in the supplemental material). Among the identified AKRs, BdsA shared 53% identity with XF1729 from Xylella fastidiosa 9a5c, which is a pathogen associated with crop disease. XF1729 catalyzes the reduction of (±)-glyceraldehyde and 2-nitrobenzaldehyde in the presence of NADPH (18). Sequence alignment of BdsA with AKRs revealed that the catalytic tetrad (Asp62, Tyr67, Lys92, and His142) was conserved in BdsA (Fig. S2). These in silico analyses led us to hypothesize that BdsA may be a reductase acting on compound 2.
BdsB, consisting of 144 amino acid residues, belongs to the NTF2-like superfamily, which is diverse in both sequence and function. By BLAST search, we were able to identify more than 100 NTF2-like proteins, which shared 30% or higher identity with BdsB; however, none of these proteins were functionally characterized. NTF2-like proteins include limonene-1,2-epoxide hydrolases (19), Δ5-3-ketosteroid isomerase (20), scytalone dehydratase (21), polyketide cyclase SnoaL (22), and polyketide monooxygenase SnoaL2 (23, 24), all of which have been functionally characterized. However, these enzymes did not have sequence or functional similarities to BdsB (Fig. S3 and S4). Moreover, BdsB and its homologous proteins formed a distinct clade among the NTF2-like proteins (Fig. S3).
Heterologous expression of bds proteins in Streptomyces coelicolor.
Streptomycetes are Gram-positive, soil-living bacteria that produce a wide variety of secondary metabolites, including aromatic polyketides. Over the past 2 decades, streptomycetes have been utilized as hosts for the heterologous production of aromatic polyketides (25). The key advantages of this approach include the posttranslational modification of carrier proteins of polyketide synthases, the availability of acyl-CoA substrates, and high tolerance of products due to abundant transporter proteins. S. coelicolor is a model species for the study of actinomycete genetics and biology, and many genetic tools are available to manipulate this organism (26). Therefore, to investigate the catalytic roles of these enzymes, we overexpressed bds proteins in S. coelicolor M1146 (27) using high-copy-number plasmid pIJ6021 (28). We constructed six plasmids (pYS1, pSN1, pSN2 [Fig. 2], pIJ6021-soceCHS1, pIJ6021-bdsA, and pIJ6021-bdsB), and S. coelicolor M1146 was transformed with these plasmids.
HPLC analysis of the culture extracts revealed that expression of SoceCHS1 in S. coelicolor M1146 resulted in the accumulation of compound 6 (Fig. 3A), consistent with previous expression studies in Pseudomonas (16). Interestingly, S. coelicolor M1146/pSN1, a recombinant strain carrying soceCHS1 and bdsA, produced compound 3, along with the side product compound 6 (Fig. 3B), which was identical to authentic flaviolin. The structure of compound 3 was characterized as scytalone by nuclear magnetic resonance (NMR; Fig. S5) and mass spectrometry (MS) analyses. The absolute configuration of compound 3 was shown to be the (R)-form, since the observed optical rotation value () and the reported value () (8) were nearly equal. For determination of the enantiomeric excess of compound 3, we prepared 3,8-dihydroxy-6-methoxy-tetralone (compound 7) by methylation of compound 3. Using chiral chromatography, the enantiomeric excess of compound 7 was found to be 92% (Fig. S6). These results strongly suggested that BdsA may be a novel AKR reductase that reduces compound 2 to (R)-compound 3.
FIG 3.
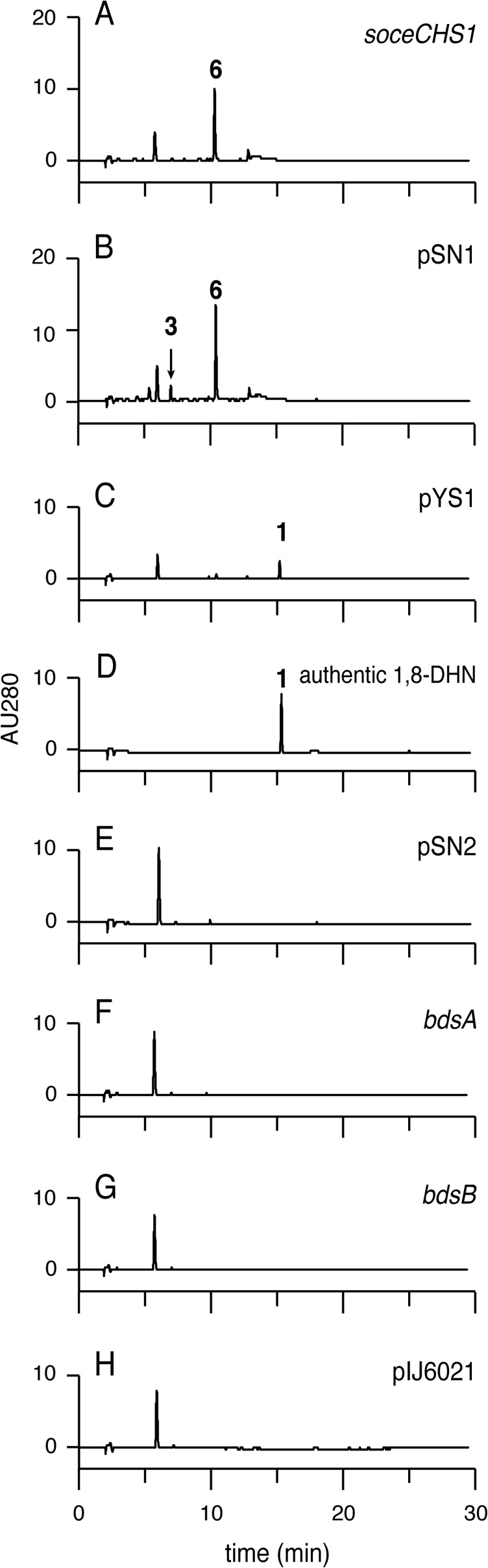
HPLC chromatograms of the recombinant Streptomyces strains. Chromatograms of strains expressing SoceCHS1 (A); SoceCHS1 and BdsA (B); SoceCHS1, BdsA, and BdsB (C); BdsA and BdsB (E); BdsA (F); and BdsB (G) are shown. (D) Chromatogram of authentic compound 1. (H) Chromatogram of the strain carrying an empty vector. AU, absorbance units.
S. coelicolor M1146/pYS1, a recombinant strain carrying soceCHS1, bdsA, and bdsB, secreted a dark-brown pigment into the broth. HPLC analysis revealed that S. coelicolor M1146/pYS1 produced a single compound, which appeared to be identical to authentic compound 1 (Fig. 3C and D). In contrast, recombinants expressing BdsA and BdsB (Fig. 3E), BdsA (Fig. 3F), or BdsB (Fig. 3G) did not produce any of the specific products compound 1, compound 3, and compound 6, indicating that expression of SoceCHS1 was crucial for the production of polyketides. The contaminant migrating at a retention time of 6 min was derived from the host strain (Fig. 3H). These results indicated that SoceCHS1, BdsA, and BdsB were necessary and sufficient for the production of compound 1 in a heterologous host.
In vitro reconstitution of 1,8-DHN synthesis.
Simultaneous incubation of recombinant SoceCHS1, BdsA, and BdsB with malonyl-CoA and NADPH resulted in the reconstitution of compound 1 synthesis in vitro (Fig. S7). This result was consistent with our in vivo data and indicated that endogenous enzymes of the host were not involved in the formation of compound 1. In addition, SoceCHS1 gave compound 2 from malonyl-CoA (Fig. S7), suggesting that the substrate of BdsA was not compound 6, which could be obtained from air oxidation of compound 2. These observations also indicated that BdsA and BdsB could catalyze both of the deoxygenation steps, i.e., deoxygenation of compound 2 to compound 4 and deoxygenation of compound 4 to compound 1.
Catalytic properties of BdsA.
In order to investigate the properties of BdsA, reactions with BdsA were performed in vitro. When BdsA was incubated with compound 2 and NADPH, compound 3 was observed by HPLC analysis (Fig. 4A). For the determination of the stereoselectivity in reduction by BdsA, the in vitro product was methylated to compound 7, and compound 7 was analyzed by chiral HPLC analysis (Fig. S6). The methylated derivative of compound 3 from the in vitro reaction comigrated with that of (R)-compound 7 prepared from the recombinant strain, indicating that compound 3 synthesized in vitro was a (R)-isomer (Fig. S6). The reaction did not proceed when boiled BdsA was used instead of active enzyme or when NADPH was not added to the reaction (Fig. 4A). The reaction followed Michaelis-Menten kinetics (Fig. 4B), and the apparent Km and Vmax values of compound 2 for the synthesis of compound 3 were 95.4 ± 13 μM and 0.050 ± 0.003 μM s−1 (means ± standard errors [SE], n = 3), respectively. These results clearly showed that BdsA was a novel naphthol reductase of bacterial origin. We also examined the reverse reaction for BdsA (Fig. S8). Incubation of BdsA, (R)-compound 3, and NADP+ gave compound 2, with a small amount of compound 6 and contaminants which could be derived from compound 2, showing that BdsA could also catalyze the oxidation of (R)-compound 3 to give compound 2. Consistent with this, T4HNR and T3HNR of Magnaporthe grisea catalyze the reverse reactions of compound 3 to compound 2 and compound 5 to compound 4, respectively (11).
FIG 4.
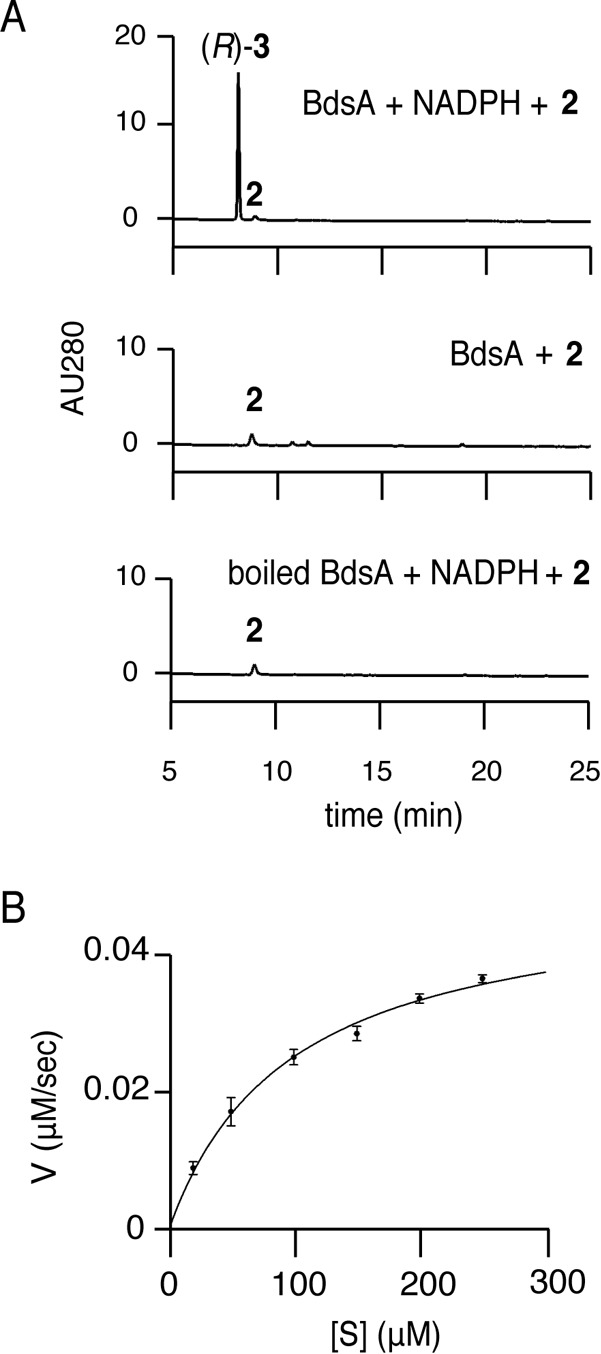
Catalytic properties of BdsA. (A) HPLC chromatograms representing BdsA reactions. The enzyme, substrate, and cofactor used are shown at the top right of the chromatograms. (B) Michaelis-Menten saturation curve of BdsA.
To ascertain whether BdsA was involved in the second deoxygenation step, BdsA was incubated with compound 4 and NADPH (Fig. S9). HPLC analysis confirmed that compound 4 was converted to the product compound 5, the structure of which was deduced from liquid chromatography–high-resolution MS (LC-HRMS) and 1H-NMR analyses (Fig. S10). In contrast, inactive BdsA did not convert compound 4 to compound 5. These results clearly showed that BdsA was involved in the first and second reduction steps of the deoxygenation events. Note that compound 4 was readily auto-oxidized to 2-hydroxyjuglone (compound 8) (29) due to its instability in air (Fig. S9). The limited availability of compound 5 prevented our performing further analysis of this reaction. Then, we chose 1,3-DHN (compound 9), a compound that was structurally analogous to compound 4 and more stable than compound 4, as an alternative substrate for investigation of the second deoxygenation step (Fig. S11). Similarly to compound 4, compound 9 is subject to oxidation via atmospheric O2 to give a corresponding oxygen adduct, lawsone (compound 10) (Fig. S11), but with a somewhat lower rate than the decomposition of compound 4. Incubation of BdsA with compound 9 and NADPH gave 3-hydroxy-1-tetralone (compound 11) as a single product (Fig. S11). The optical rotation of compound 11 was . The chiral chromatography of compound 11 revealed that the product was almost completely optically pure (Fig. S11). This reaction was also catalyzed by naphthol reductase, which was partially purified from the brm-1 mutant of Verticillium dahliae (30). In addition, T4HNR from Magnaporthe grisea also catalyzes this reaction, albeit with a low conversion rate (8). However, those studies did not report the optical density or the absolute configuration of compound 11. Unfortunately, because of the extreme instability of the Mosher ester of compound 11, which undergoes further elimination and tautomerization to 1-naphthol, we could not determine the absolute configuration of compound 11.
Catalytic properties of BdsB.
Next, the dehydration reaction catalyzed by BdsB was investigated in vitro. BdsB readily dehydrated (R)-compound 3 to compound 4, whereas in the reaction using boiled BdsB, (R)-compound 3 remained intact (Fig. 5A). BdsB displayed Michaelis-Menten kinetics (Fig. 5B), and the apparent Km and Vmax values of (R)-compound 3 for compound 4 synthesis were 5.5 ± 1.7 μM and 0.20 ± 0.02 μM s−1 (means ± SE, n = 3), respectively. In addition, the second deoxygenation step was reconstituted by incubating compound 5 with BdsB. The reaction smoothly gave compound 1 as a single product, whereas in the reaction using boiled BdsB, compound 5 remained intact (Fig. S12). These results clearly showed that BdsB was a novel scytalone/vermelone dehydratase of bacterial origin.
FIG 5.
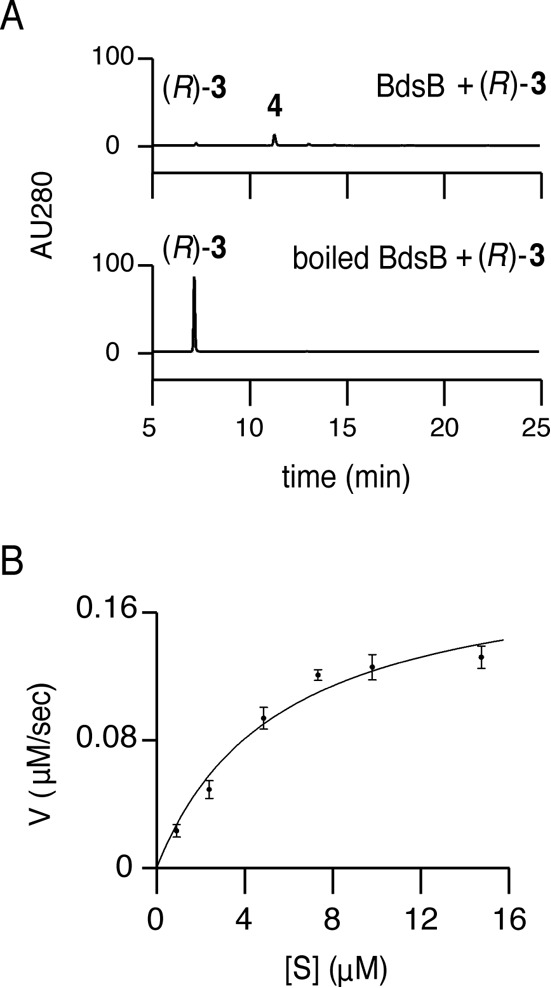
Catalytic properties of BdsB. (A) HPLC chromatograms representing BdsB reactions. The enzyme and substrate used are shown at the top right of the chromatograms. (B) Michaelis-Menten saturation curves of BdsB.
DISCUSSION
In this study, we discovered DHN (compound 1) biosynthetic enzymes of bacterial origin. The biosynthetic route of compound 1 was composed of three classes of reactions, including synthesis of polyketides, reduction of the carbonyl group, and dehydration of the phenolic hydroxyl group. For polyketide synthesis, the bacterial DHN synthetic route adopts a type III PKS, SoceCHS1, whereas the fungal route utilizes a type I PKS, such as PKS1, which synthesizes compound 2 from malonyl-CoA (31). Type III PKSs are thought to have evolved from a common ancestral protein, such as FabH, which initiates the elongation of fatty acyl intermediates by condensing acetyl-CoA and malonyl-ACP to form acetoacetyl-ACP (32). In contrast, the KS domain of type I PKS is homologous to FabF, which is responsible for the repetitive condensation of malonyl-ACP to fatty acyl intermediates to accomplish the extension of saturated and unsaturated fatty acids of 16 to 18 carbons (32). Type I PKSs are widely distributed among bacteria, although PKS1 and its orthologous enzymes have not been characterized. Moreover, type III PKSs for compound 2 have not been found in fungi. These facts, together with the dissimilarity in the double-deoxygenation steps (see below), suggested that the bacterial and fungal DHN synthesis routes may have evolved independently.
The reduction of aldehydes and ketones to primary and secondary alcohols, respectively, is often catalyzed by proteins that belong to two protein superfamilies, i.e., SDRs and AKRs (17). The catalytic mechanisms of SDRs and AKRs are similar, although the protein folds are unrelated (17). We have shown that BdsA, a novel member of the AKR superfamily, catalyzes the reduction of compound 2 and compound 4 to compound 3 and compound 5. In contrast, fungal naphthol reductases, i.e., T4HNR and T3HNR, belong to the SDR superfamily, indicating that BdsA may be a novel naphthol reductase of bacterial origin. The reactions of BdsA and T4HNR were found to share a feature in that the stereoselectivity in the reduction of compound 2 to give (R)-compound 3 was the same in both. In addition, both BdsA and T4HNR catalyzed the reverse reaction, e.g., oxidation of (R)-compound 3 to compound 2.
We could not determine the stereochemistry of compound 5 or compound 11 because of the limited availability of the compounds as well as their instability. Therefore, the facial selectivity of the reduction of compound 4 remains unknown. We assumed that the reduction of compound 4 could occur on the si face to give (R)-compound 5 because compound 2 and compound 4 were structurally related compounds. The facial selectivity of the reduction by T4HNR depends on the relatively fixed position of the carbonyl oxygen atom of substrate naphthols (8). The correlation between the substrate structure and the facial selectivity of the reduction by BdsA remains to be clarified.
We showed that BdsB catalyzed the dehydration of (R)-compound 3 and compound 5 to compound 4 and compound 1. BdsB and fungal SD are both members of the NTF2-like superfamily and catalyze the dehydration of (R)-compound 3 and compound 5. However, BdsB and SD were found to be phylogenetically distinct. BdsB contained a conserved SnoaL_2 (pfam12680)-like domain, which is thought to be a hydroxylase of the aromatic polyketide nogalamycin (24). Siitonen et al. showed that SnoaW and SnoaL2 made up a two-component monooxygenase (23). SnoaW reduces quinone to dihydroquinone, which enables the activation of oxygen and the formation of a hydroperoxy intermediate. Then, SnoaL2 catalyzes the protonation of the hydroperoxy intermediate to yield the hydroxylated product. The protonation step may also be involved in the dehydration of secondary alcohol by BdsB, since protonation of the alcoholic oxygen is critical to improve the leaving group, a neutral water. Indeed, aspartic acid and glutamic acid and other proposed catalytic residues of SnoaL2 are conserved among BdsB enzymes and SnoaL2 (24). Although SnoaL2 family proteins are often found in type II PKS clusters, the reaction and the amino acid sequence of BdsB are distinct from those of type II PKS.
Although the DHN biosynthetic pathway was thought to be specific to fungi, we discovered novel DHN synthesis enzymes of bacterial origin. Interestingly, the same biosynthetic pathway of a secondary metabolite was found to be conserved between bacterium and eukaryote domains. DHN biosynthesis involved synthesis of polyketides, reduction of the carbonyl group, and dehydration of the phenolic hydroxyl group. Moreover, bacterial DHN biosynthesis utilized a type III PKS for polyketide synthesis, an AKR superfamily member for reduction, and a SnoaL2-like NTF2 superfamily member for dehydration, whereas fungal DHN biosynthesis utilized a type I PKS, a SDR superfamily enzyme, and an SD-like NTF2 superfamily member. Surprisingly, the enzyme systems comprising the pathway were significantly different from each other, suggesting independent, parallel evolution leading to the same biosynthesis pathway.
Based on the discovery of DHN biosynthetic enzymes, it is unclear whether DHN melanin may be produced in bacteria. Gross et al. assumed that soceCHS1 in S. cellulosum So ce56 was not expressed and did not function under laboratory conditions because no changes in secondary metabolite patterns were observed in soceCHS1-inactivated strains (16). It is sometimes difficult to ascertain correlations between stress (hyperthermia, hyperosmotic medium, starvation) and melanization because the response to stress is assumed to be acquired individually via divergent evolution. Recently, Vijayan et al. isolated a Gram-negative, bioluminescent marine bacterium, Vibrio harveyi MMRF546, from a darkly pigmented sponge, Fasciospongia cavernosa (33). Surprisingly, V. harveyi MMRF546 produced a melanin molecule whose synthesis was inhibited by tricyclazole, an inhibitor of T3HNR. Although it is possible that tricyclazole cross-inhibited an unknown melanin pathway, this observation implied that the melanin produced by V. harveyi MMRF546 was synthesized via the DHN pathway. However, it is unclear whether DHN biosynthesis occurs in bacteria, although bacterial DHN biosynthetic enzymes are conserved among pathogenic bacteria (see Fig. S13 in the supplemental material), such as human-pathogenic Inquilinus limosus DSM 16000 (34) and plant-pathogenic Ralstonia solanacearum CMR15 (35), Pseudomonas cichorii JBC1 (36), and Nocardia casuarinae BMG51109 (37). Further studies are needed to assess melanization by these pathogenic organisms.
MATERIALS AND METHODS
Materials.
1,8-DHN (compound 1) and malonyl-CoA were purchased from Sigma-Aldrich (St. Louis, MO, USA). NADPH and NADP+ were purchased from Oriental Yeast (Tokyo, Japan). 1,3-DHN (compound 9) and trimethylsilyldiazomethane were purchased from Tokyo Chemical Industry (Tokyo, Japan). T4HN (compound 2) was synthesized according to the method of Ichinose et al. (38). (±)-Scytalone (compound 3) and (±)-3-hydroxy-1-tetralone (compound 11) were synthesized according to the methods of Schätzle et al. (8). Flaviolin (compound 6) was prepared from a recombinant S. coelicolor M1146 strain harboring pNF2 (13). Luria-Bertani (LB) Lennox medium was purchased from Sigma-Aldrich. S. cellulosum So ce56 was purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany). Escherichia coli HST08, E. coli HST04, the pColdI plasmid, restriction enzymes, and other DNA-modifying enzymes used for DNA manipulation were purchased from TaKaRa Bio Inc. (Shiga, Japan). E. coli BL21 was purchased from the National BioResource Project (National Institute of Genetics of Japan).
Construction of plasmids.
Medium, growth conditions, and general recombinant DNA techniques for E. coli and Streptomyces were previously described by Sambrook et al. (39) and Kieser et al. (26), respectively. The chromosomal DNA of S. cellulosum So ce56 was used as a template for PCR. An NdeI site was introduced at the start codon of soceCHS1 by PCR with primer I (5′-GCGGAATTCATATGGCCACACTGTGCAGG-3′; the EcoRI site is underlined, and the NdeI site is italicized) and primer II (5′-GCGGGATCCGCAGGAGCGACAGGGC-3′; the BamHI site is italicized). The amplified fragment was cloned between the EcoRI and BamHI sites of pUC19, yielding pUC19-soceCHS1. The NdeI-BamHI fragment excised from pUC19-soceCHS1 was cloned between the NdeI and BamHI sites of pIJ6021 and pIJ4123, resulting in pIJ6021-soceCHS1 and pIJ4123-soceCHS1, respectively.
An NdeI site was introduced at the start codon of bdsA (sce2134) by PCR with primer III (5′-CGGAATTATATGACCAACGCATCGCTCAA-3′C; the EcoRI site is underlined, and the NdeI site is italicized) and primer IV (5′-GCGGGATCCTCACGGCGTCTCCTTCGTG-3′; the BamHI site is italicized). The amplified fragment was cloned between the EcoRI and BamHI sites of pUC19, resulting in pUC19-bdsA. The NdeI-BamHI fragment excised from pUC19-bdsA was cloned between the NdeI and BamHI sites of pIJ6021 and pIJ4123, yielding pIJ6021-bdsA and pIJ4123-bdsA, respectively.
An NdeI site was introduced at the start codon of bdsB (sce2135) by PCR with primer V (5′-CGGAATTCATATGAGCACCCAGCAGCAAACCA-3′; the EcoRI site is underlined, and the NdeI site is italicized) and primer VI (5′-GCGGGATCCGGATGGCGGGAACGACAA-3′; the BamHI site is italicized). The amplified fragment was cloned between the EcoRI and BamHI sites of pUC19, yielding pUC19-bdsB. The NdeI-BamHI fragment excised from pUC19-bdsB was cloned between the NdeI and BamHI sites of pColdI, yielding pColdI-bdsB.
A 2.5-kb gene fragment containing soceCHS1, bdsA, and bdsB was amplified by PCR with primer I and primer VI. The amplified fragment was cloned between the EcoRI and BamHI sites of pUC19, yielding pUC19-YS1. The NdeI-BamHI fragment excised from pUC19-YS1 was cloned between the NdeI and BamHI sites of pIJ6021, yielding pSY1.
A 2.1-kb gene fragment containing soceCHS1and bdsA was amplified by PCR with primer I and primer IV. The amplified fragment was cloned between the EcoRI and BamHI sites of pUC19, yielding pUC19-YS1. The NdeI-BamHI fragment excised from pUC19-SN1 was cloned between the NdeI and BamHI sites of pIJ6021, yielding pSN1.
A 1.4-kb gene fragment containing bdsA and bdsB was amplified by PCR with primer III and primer IV. The amplified fragment was cloned between the EcoRI and BamHI sites of pUC19, yielding pUC19-SN2. The NdeI-BamHI fragment excised from pUC19-SN2 was cloned between the NdeI and BamHI sites of pIJ6021, yielding pSN2.
HPLC analysis of polyketides produced by recombinant Streptomyces.
S. coelicolor M1146 was transformed with pIJ6021, pIJ6021-soceCHS1, pIJ6021-bdsA, pIJ6021-bdsB, pSY1, pSN1, and pSN2. The recombinant strains were individually inoculated into 50 ml of yeast extract-malt extract liquid medium (26) containing 5 μg/ml kanamycin, and the resulting cultures were grown at 30°C. After 26 h, 5 μg/ml thiostrepton was added to induce the tip promoter, and the cultures were incubated for an additional 48 h. Next, 1-ml aliquots of the supernatants from the cultures were acidified with 30 μl of 6 M HCl and extracted with ethyl acetate. The ethyl acetate phases were evaporated, and the residues were dissolved in a small amount of methanol. Reverse-phase HPLC–photodiode array detector (HPLC-PDA) (Jasco, Tokyo, Japan) analysis was carried out using a Cosmosil 5C18 AR-II column (Nacalai Tesque, Kyoto, Japan) (4.6 by 150 mm), and the analytes were subjected to a gradient of 10% to 100% acetonitrile–water (both containing 0.1% trifluoroacetic acid) at a flow rate of 1 ml/min at 40°C for 30 min. UV absorbance was detected at 280 nm.
Isolation of scytalone (compound 3) from S. coelicolor M1146/pSN1.
S. coelicolor M1146/pSN1 was used to inoculate 8 × 0.5 liters of yeast extract-malt extract liquid medium containing 5 μg/ml kanamycin and was grown at 30°C. After 24 h, 5 μg/ml thiostrepton was added, and the cultures were incubated for an additional 72 h. The pH of the culture supernatant was adjusted to 2 with 6 M HCl, and extraction was then carried out using ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and evaporated until dry. The crude materials were dissolved in a small amount of benzene-acetone (7:3 [vol/vol]) and were then subjected to flash chromatography on a silica gel using benzene-acetone (7:3 [vol/vol]) as an eluent. The eluate was evaporated and dissolved in methanol for reverse-phase preparative HPLC. The crude materials were purified using a reverse-phase preparative HPLC apparatus equipped with a 5C18-AR-II column (Nacalai Tesque) (10 by 250 mm) by elution with 30% methanol–water–0.1% trifluoroacetic acid at a flow rate of 3 ml/min at ambient temperature. The collected fractions were lyophilized to give 30 mg of (R)-compound 3 as a white solid. NMR data were collected on a Bruker BioSpin AV400N FT-NMR spectrometer (Bruker, Billerica, MA, USA). LC-electrospray ionization-HRMS (LC-ESI/HRMS) analysis was carried out using a Q-exactive spectrometer (Thermo Fisher Scientific, MA, USA). Optical rotations were measured on a Jasco P-2200 digital polarimeter (Jasco, Tokyo, Japan). (R)-compound 3 measurements were as follows: for 1H-NMR (400 MHz, CD3OD), δ 2.64 (dd, 1H, J = 7.6, 16.8 Hz, H-2b), 2.86 to 2.91 (m, 2H, H-2a and H-4b), 3.12 (dd, 1H, J = 2.4, 16.0 Hz, H-4a), 4.28 (m, 1H, H-3), 6.13 (s, 1H, H-7), 6.25 (s, 1H, H-5); for 13C-NMR (100 MHz, CD3OD), δ 40.0 (C-4), 48.3 (C-2), 67.8 (C-3), 102.5 (C-7), 110.2 (C-5), 112.5 (C-9), 146.3 (C-10), 167.4 (C-8), 167.5 (C-6), 203.3 (C-1); for negative-mode LC-ESI/HRMS, [M–H]- = 193.0500 (calculated for C10H9O4-, 0.6 millimass units [mmu] error); (c 0.36, 95% ethyl alcohol [EtOH]).
Synthesis of 3,8-dihydroxy-6-methoxy-tetralone (compound 7).
A solution of (R)-compound 3 (6.7 mg, 35 μmol)–300 μl toluene-methanol (3:1 [vol/vol]) was added to 200 μl of 0.6 M trimethylsilyldiazomethane–hexane and stirred for 2 h at room temperature. The reaction was quenched with acetic acid, and the reaction mixture was evaporated under reduced pressure. The crude materials were purified using a reverse-phase preparative HPLC apparatus equipped with a 5C18-AR-II column (Nacalai Tesque) (20 by 250 mm) by elution with 40% acetonitrile–water–0.1% trifluoroacetic acid at a flow rate of 5 ml/min at ambient temperature. The collected fractions were evaporated to give 5.7 mg (27.4 μmol, 79%) of (R)-compound 7 as a pale brownish solid. Direct analysis in real time/time of flight MS (DART/TOF-MS) was performed using a JEOL AccuTOF-DART mass spectrometer (JEOL, Tokyo, Japan). (R)-compound 7 measurements were as follows: for 1H-NMR (400 MHz, CDCl3), δ 2.71 (dd, 1H, J = 7.9, 17.1 Hz, H-2b), 2.89 to 2.96 (m, 2H, H-2a and H-4b), 3.14 (dd, 1H, J = 3.6, 15.2 Hz, H-4a), 3.83 (s, 3H, -OCH3), 4.40 (m, 1H, H-3), 6.29 (d, 2H, H-5, and H-7), 12.70 (s, 1H, OH); for 13C-NMR (100 MHz, CD3OD), δ 38.63 (C-4), 46.87 (C-2), 55.71 (-OCH3), 66.39 (C-3), 99.31 (C-7), 108.11 (C-5), 111.36 (C-9), 143.08 (C-10), 165.71 (C-8), 166.49 (C-6), 200.57 (C-1); for positive-mode DART/TOF-MS, [M+H]+ = 209.08158 (calculated for C11H13O4+, 0.20 mmu error).
Chiral cell HPLC analysis.
Chiral cell HPLC analysis was carried out using a Chiralpak AS-H column (Daicel, Osaka, Japan) (4.6 by 250 mm), and the analytes were subjected to elution with 7% isopropyl alcohol–hexane at a flow rate of 1 ml/min at 30°C for 40 min. UV absorbance was detected at 280 nm.
Expression and purification of recombinant SoceCHS1, BdsA, and BdsB.
S. coelicolor M1146 harboring pIJ4123-soceCHS1 or pIJ4123-bdsA was used to inoculate 50 ml of yeast extract-malt extract liquid medium containing 5 μg/ml kanamycin, and the cultures were grown at 30°C. After 26 h, 5 μg/ml thiostrepton was added to induce the tip promoter, and the cultures were incubated further for an additional 48 h. Cells were harvested by centrifugation and resuspended in a mixture consisting of 50 mM NaH2PO4 (pH 8.0), 300 mM NaCl, 10 mM imidazole, and 10% glycerol. One tablet of cOmplete mini EDTA-free inhibitor cocktail (Sigma-Aldrich) was dissolved in the cell suspension. A crude cell lysate was prepared by sonication, and cell debris was removed by centrifugation at 20,000 × g for 20 min. The cleared lysate was applied to nickel-nitrilotriacetic acid (Ni-NTA) columns (Qiagen, Hilden, Germany) and then washed twice with a mixture consisting of 50 mM NaH2PO4 (pH 8.0), 300 mM NaCl, 20 mM imidazole, and 10% glycerol. The purified histidine-tagged protein was eluted with a mixture consisting of 50 mM NaH2PO4 (pH 8.0), 300 mM NaCl, 250 mM imidazole, and 10% glycerol and was dialyzed twice against 2 liters of 10 mM Tris-HCl (pH 7.5) containing 10% glycerol and 50 mM NaCl.
E. coli BL21 cells harboring pColdI-bdsB were grown at 37°C in 50 ml LB broth containing 50 μg/ml ampicillin. When the optical density at 600 nm reached 0.5, the cells were incubated for 30 min at 15°C and then induced by addition of 0.1 mM isopropyl thio-β-d-galactoside. The cells were then cultured for an additional 24 h at 15°C. The crude protein was purified using Ni-NTA as described above.
The protein concentration was estimated using Bradford assays with a Bio-Rad protein assay kit (Bio-Rad Laboratories, CA, USA) with bovine serum albumin as the standard. The purities of recombinant SoceCHS1, BdsA, and BdsB were verified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (see Fig. S14 in the supplemental material).
In vitro reactions of SoceCHS1, BdsA, and BdsB.
All reactions were performed in 100 μl of 100 mM sodium phosphate (pH 7.5). For the SoceCHS1 reactions, 200 μM malonyl-CoA and 2 μM SoceCHS1 were incubated in the buffer. For the forward BdsA reactions, 400 μM compound 2, compound 4, or compound 9; 1 mM NADPH; and 2 μM BdsA were incubated in the buffer. For the reverse BdsA reactions, (R)-compound 3, 1 mM NADP+, and 2 μM BdsA were incubated in the buffer. For the BdsB reactions, 400 μM (R)-compound 3 and 2 μM BdsB or 400 μM compound 5 and 1 μM BdsB were incubated in the buffer. All reaction mixtures were incubated at 30°C for 60 min and were acidified with 20 μl of 6 M HCl to quench the reactions. The mixture was then extracted with 200 μl ethyl acetate, and the ethyl acetate phase was evaporated to dryness. The residue was dissolved in 20 μl methanol for HPLC-PDA analysis as described above.
Determination of the kinetic parameters of BdsA.
The reactions, which used reaction mixtures containing 100 mM sodium phosphate (pH 7.5), compound 2, 1 mM NADPH, and 0.5 μM BdsA, were performed in a total volume of 100 μl. The concentration of compound 2 was adjusted to between 20 and 250 μM. The reactions were initiated by adding compound 2, continued for 7 min, and stopped with 20 μl of 6 M HCl. The consumption rates of the substrate were 19.7% (20 μM), 14.9% (50 μM), 10.8% (100 μM), 8.2% (150 μM), 7.2% (200 μM), and 6.3% (250 μM). The material in the mixture was then extracted with ethyl acetate. The organic layer was collected and evaporated, and the residual material was dissolved in 20 μl methanol for quantification of (R)-compound 3 by HPLC analysis, as described above. (R)-compound 3 was used to generate the standard curve for the quantification of the product. Each measurement was repeated three times independently. Steady-state parameters were determined by Sigma plot analysis (Systat Software, CA, USA).
Determination of the kinetic parameters of BdsB.
The reactions, which used reaction mixtures containing 100 mM sodium phosphate (pH 7.5), (R)-compound 3, and 0.05 μM BdsB, were performed in a total volume of 100 μl. The concentration of (R)-compound 3 was adjusted to between 1 and 15 μM. The reactions were initiated by adding (R)-compound 3, continued for 20 s, and stopped with 20 μl of 6 M HCl. The consumption rates of the substrate were 17.8% (1 μM), 7.9% (2.5 μM), 7.3% (5 μM), 8.7% (7.5 μM), 5.8% (10 μM), and 10.0% (15 μM). The material in the mixture was then extracted with ethyl acetate, and the unreacted (R)-compound 3 was quantified by HPLC, as described above, to measure the reaction rate. Each measurement was repeated three times independently. Steady-state parameters were determined by Sigma plot analysis (Systat Software).
Enzymatic preparation of T3HN (compound 4).
Ten milliliters of solution containing 1 μM BdsB, 1 mM (R)-compound 3, and 100 mM sodium phosphate (pH 6.5) was incubated at 30°C for 60 min. The reaction was acidified with 6 M HCl and extracted with ethyl acetate. The ethyl acetate phase was evaporated to dryness, and the remaining residue was dissolved in a small amount of methanol and purified using a reverse-phase preparative HPLC apparatus equipped with a 5C18-AR-II column (Nacalai Tesque) (10 by 250 mm) by elution with 30% methanol–water–0.1% trifluoroacetic acid at a flow rate of 3 ml/min at ambient temperature. The collected fractions were lyophilized to give 2 mg T3HN (compound 4) as a white solid. 1H-NMR data are shown in Fig. S15. Compound 4 measurements were as follows: for 1H-NMR (400 MHz, acetone-d6), δ 6.47 (d, J = 2.1 Hz, H-2), 6.56 (d, J = 7.2 Hz, H-7), 6.65 (d, J = 1.9 Hz, H-4), 7.06 (d, J = 8.0 Hz, H-5), 7.13 (t, J = 7.6, 8.0 Hz, H-6); for negative-mode LC-ESI/HRMS, [M–H]- = 175.0392 (calculated for C10H7O3-, 0.9 mmu error).
Enzymatic preparation of (+)-3-hydroxy-1-tetralone (compound 11).
Next, 35 ml of a solution containing 1 μM BdsA, 1 mM compound 9, 1.5 mM NADPH, and 100 mM sodium phosphate (pH 6.5) was incubated at 30°C for 60 min. Compound 11 was extracted and purified as described above using a reverse-phase preparative HPLC apparatus equipped with a 5C18-AR-II column by elution with 60% methanol–water at a flow rate of 3 ml/min at ambient temperature. The yield of compound 11 was 0.4 mg. Compound 11 measurements were as follows: for 1H NMR (400 MHz, CDCl3), δ 2.79 (dd, 1H, J = 17.8, 8.0 Hz, H-2b), 2.95 to 3.04 (m, 2H, H-2a and H-4b), 3.23 (dd, 1H, J = 16.1, 3.9 Hz, H-4a), 4.45 (septet, 1H, H-3), 6.75 (dd, 1H, J = 7.4, 0.8 Hz, H-5), 6.84 (d, 1H, J = 8.4 Hz, H-7), 7.41 (t, 1H, J = 8.0 Hz, H-6), 12.26 (s, 1H, Ar-OH); for negative-mode LC-ESI/HRMS, [M–H]- = 179.0700 (calculated for C10H11O3+, 179.07027); for positive-mode LC-ESI/HRMS, [M+H]+ = 163.0753 (calculated for C10H11O2+, 0.1 mmu error); (c 0.05, 95% EtOH).
Enzymatic preparation of vermelone (compound 5).
Twenty milliliters of a solution containing 1 μM BdsA, 1 mM compound 4, 1.5 mM NADPH, and 100 mM sodium phosphate (pH 6.5) was incubated at 30°C for 3 h. Compound 5 was extracted as described above. The yield of compound 5 was 1 mg. Compound 5 measurements were as follows: for 1H NMR (400 MHz, CDCl3), δ 2.79 (dd, 1H, J = 8.0, 17.8 Hz, H-2a), 2.95 to 3.04 (m, 2H, H-2b and H-4a), 3.23 (dd, 1H, J = 3.9, 16.1 Hz, H-4b), 4.45 (m, 1H, H-3), 6.75 (dd, 1H, J = 0.8, 7.4 Hz, H-5), 6.84 (d, 1H, J = 8.4 Hz, H-7), 7.41 (dd, 1H, J = 0.8, 8.4 Hz, H-6), 12.26 (s, 1H, Ar-OH); for negative-mode LC-ESI/HRMS, [M–H]- = 179.0700 (calculated for C10H11O3+, 0.3 mmu error).
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to M. J. Bibb for providing us with S. lividans TK21 and S. coelicolor M1146. We are also grateful to E. Takano for providing us with the pIJ6021 and pIJ4123 expression vectors.
This work was supported in part by grants 23108521 and 17H05447 (Grant-in-Aid for Scientific Research on Innovative Areas) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00258-18.
REFERENCES
- 1.Giraldo MC, Valent B. 2013. Filamentous plant pathogen effectors in action. Nat Rev Microbiol 11:800–814. doi: 10.1038/nrmicro3119. [DOI] [PubMed] [Google Scholar]
- 2.Shimizu T, Ito T, Kanematsu S. 2014. Functional analysis of a melanin biosynthetic gene using RNAi-mediated gene silencing in Rosellinia necatrix. Fungal Biol 118:413–421. doi: 10.1016/j.funbio.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 3.Nosanchuk JD, Casadevall A. 2003. The contribution of melanin to microbial pathogenesis. Cell Microbiol 5:203–223. doi: 10.1046/j.1462-5814.2003.00268.x. [DOI] [PubMed] [Google Scholar]
- 4.Valeru SP, Rompikuntal PK, Ishikawa T, Vaitkevicius K, Sjöling A, Dolganov N, Zhu J, Schoolnik G, Wai SN. 2009. Role of melanin pigment in expression of Vibrio cholerae virulence factors. Infect Immun 77:935–942. doi: 10.1128/IAI.00929-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodríguez-Rojas A, Mena A, Martín S, Borrell N, Oliver A, Blázquez J. 2009. Inactivation of the hmgA gene of Pseudomonas aeruginosa leads to pyomelanin hyperproduction, stress resistance and increased persistence in chronic lung infection. Microbiology 155:1050–1057. doi: 10.1099/mic.0.024745-0. [DOI] [PubMed] [Google Scholar]
- 6.Zheng H, Chatfield CH, Liles MR, Cianciotto NP. 2013. Secreted pyomelanin of Legionella pneumophila promotes bacterial iron uptake and growth under iron-limiting conditions. Infect Immun 81:4182–4191. doi: 10.1128/IAI.00858-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Langfelder K, Streibel M, Jahn B, Haase G, Brakhage AA. 2003. Biosynthesis of fungal melanins and their importance for human pathogenic fungi. Fungal Genet Biol 38:143–158. doi: 10.1016/S1087-1845(02)00526-1. [DOI] [PubMed] [Google Scholar]
- 8.Schätzle MA, Flemming S, Husain SM, Richter M, Günther S, Müller M. 2012. Tetrahydroxynaphthalene reductase: catalytic properties of an enzyme involved in reductive asymmetric naphthol dearomatization. Angew Chem Int Ed Engl 51:2643–2646. doi: 10.1002/anie.201107695. [DOI] [PubMed] [Google Scholar]
- 9.Tsai HF, Wheeler MH, Chang YC, Kwon-Chung KJ. 1999. A developmentally regulated gene cluster involved in conidial pigment biosynthesis in Aspergillus fumigatus. J Bacteriol 181:6469–6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chumley FG, Valent B. 1990. Genetic analysis of melanin-deficient, nonpathogenic mutants of Magnaporthe grisea. Mol Plant Microbe Interact 3:135–143. doi: 10.1094/MPMI-3-135. [DOI] [Google Scholar]
- 11.Thompson JE, Fahnestock S, Farrall L, Liao DI, Valent B, Jordan DB. 2000. The second naphthol reductase of fungal melanin biosynthesis in Magnaporthe grisea: tetrahydroxynaphthalene reductase. J Biol Chem 275:34867–34872. doi: 10.1074/jbc.M006659200. [DOI] [PubMed] [Google Scholar]
- 12.Funa N, Ohnishi Y, Fujii I, Shibuya M, Ebizuka Y, Horinouchi S. 1999. A new pathway for polyketide synthesis in microorganisms. Nature 400:897–899. doi: 10.1038/23748. [DOI] [PubMed] [Google Scholar]
- 13.Funa N, Funabashi M, Ohnishi Y, Horinouchi S. 2005. Biosynthesis of hexahydroxyperylenequinone melanin via oxidative aryl coupling by cytochrome P-450 in Streptomyces griseus. J Bacteriol 187:8149–8155. doi: 10.1128/JB.187.23.8149-8155.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Funa N, Funabashi M, Yoshimura E, Horinouchi S. 2005. A novel quinone-forming monooxygenase family involved in modification of aromatic polyketides. J Biol Chem 280:14514–14123. doi: 10.1074/jbc.M500190200. [DOI] [PubMed] [Google Scholar]
- 15.Schneiker S, Perlova O, Kaiser O, Gerth K, Alici A, Altmeyer MO, Bartels D, Bekel T, Beyer S, Bode E, Bode HB, Bolten CJ, Choudhuri JV, Doss S, Elnakady YA, Frank B, Gaigalat L, Goesmann A, Groeger C, Gross F, Jelsbak L, Jelsbak L, Kalinowski J, Kegler C, Knauber T, Konietzny S, Kopp M, Krause L, Krug D, Linke B, Mahmud T, Martinez-Arias R, McHardy AC, Merai M, Meyer F, Mormann S, Muñoz-Dorado J, Perez J, Pradella S, Rachid S, Raddatz G, Rosenau F, Rückert C, Sasse F, Scharfe M, Schuster SC, Suen G, Treuner-Lange A, Velicer GJ, Vorhölter FJ, et al. . 2007. Complete genome sequence of the myxobacterium Sorangium cellulosum. Nat Biotechnol 25:1281–1289. doi: 10.1038/nbt1354. [DOI] [PubMed] [Google Scholar]
- 16.Gross F, Luniak N, Perlova O, Gaitatzis N, Jenke-Kodama H, Gerth K, Gottschalk D, Dittmann E, Müller R. 2006. Bacterial type III polyketide synthases: phylogenetic analysis and potential for the production of novel secondary metabolites by heterologous expression in pseudomonads. Arch Microbiol 185:28–38. doi: 10.1007/s00203-005-0059-3. [DOI] [PubMed] [Google Scholar]
- 17.Penning TM. 2015. The aldo-keto reductases (AKRs): overview. Chem Biol Interact 234:236–246. doi: 10.1016/j.cbi.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosselli LK, Oliveira CL, Azzoni AR, Tada SF, Catani CF, Saraiva AM, Soares JS, Medrano FJ, Torriani IL, Souza AP. 2006. A new member of the aldo-keto reductase family from the plant pathogen Xylella fastidiosa. Arch Biochem Biophys 453:143–150. doi: 10.1016/j.abb.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 19.Arand M, Hallberg BM, Zou J, Bergfors T, Oesch F, van der Werf MJ, de Bont JA, Jones TA, Mowbray SL. 2003. Structure of Rhodococcus erythropolis limonene-1,2-epoxide hydrolase reveals a novel active site. EMBO J 22:2583–2592. doi: 10.1093/emboj/cdg275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cho HS, Ha NC, Choi G, Kim HJ, Lee D, Oh KS, Kim KS, Lee W, Choi KY, Oh BH. 1999. Crystal structure of Δ5-3-ketosteroid isomerase from Pseudomonas testosteroni in complex with equilenin settles the correct hydrogen bonding scheme for transition state stabilization. J Biol Chem 274:32863–32868. doi: 10.1074/jbc.274.46.32863. [DOI] [PubMed] [Google Scholar]
- 21.Lundqvist T, Rice J, Hodge CN, Basarab GS, Pierce J, Lindqvist Y. 1994. Crystal structure of scytalone dehydratase-a disease determinant of the rice pathogen, Magnaporthe grisea. Structure 2:937–944. doi: 10.1016/S0969-2126(94)00095-6. [DOI] [PubMed] [Google Scholar]
- 22.Sultana A, Kallio P, Jansson A, Wang JS, Niemi J, Mäntsälä P, Schneider G. 2004. Structure of the polyketide cyclase SnoaL reveals a novel mechanism for enzymatic aldol condensation. EMBO J 23:1911–1921. doi: 10.1038/sj.emboj.7600201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siitonen V, Blauenburg B, Kallio P, Mäntsälä P, Metsä-Ketelä M. 2012. Discovery of a two-component monooxygenase SnoaW/SnoaL2 involved in nogalamycin biosynthesis. Chem Biol 19:638–646. doi: 10.1016/j.chembiol.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 24.Beinker P, Lohkamp B, Peltonen T, Niemi J, Mäntsälä P, Schneider G. 2006. Crystal structures of SnoaL2 and AclR: two putative hydroxylases in the biosynthesis of aromatic polyketide antibiotics. J Mol Biol 359:728–740. doi: 10.1016/j.jmb.2006.03.060. [DOI] [PubMed] [Google Scholar]
- 25.Pfeifer BA, Khosla C. 2001. Biosynthesis of polyketides in heterologous hosts. Microbiol Mol Biol Rev 65:106–118. doi: 10.1128/MMBR.65.1.106-118.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical Streptomyces genetics. The John Innes Foundation, Norwich, England. [Google Scholar]
- 27.Gomez-Escribano JP, Bibb MJ. 2011. Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb Biotechnol 4:207–215. doi: 10.1111/j.1751-7915.2010.00219.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takano E, White J, Thompson CJ, Bibb MJ. 1995. Construction of thiostrepton-inducible, high-copy-number expression vectors for use in Streptomyces spp. Gene 166:133–137. doi: 10.1016/0378-1119(95)00545-2. [DOI] [PubMed] [Google Scholar]
- 29.Bell AA, Wheeler MH. 1986. Biosynthesis and functions of fungal melanin. Annu Rev Phytopathol 24:411–451. doi: 10.1146/annurev.py.24.090186.002211. [DOI] [Google Scholar]
- 30.Simpson TJ, Bandumathie Weerasooriya MK. 2000. NMR studies of tautomerism in the fungal melanin biosynthesis intermediate 1,3,8-trihydroxynaphthalene. J Chem Soc Perkin 1 2000:2771–2775. doi: 10.1039/b002030n. [DOI] [Google Scholar]
- 31.Fujii I, Mori Y, Watanabe A, Kubo Y, Tsuji G, Ebizuka Y. 2000. Enzymatic synthesis of 1,3,6,8-tetrahydroxynaphthalene solely from malonyl coenzyme A by a fungal iterative type I polyketide synthase PKS1. Biochemistry 39:8853–8858. doi: 10.1021/bi000644j. [DOI] [PubMed] [Google Scholar]
- 32.Cronan JE, Thomas J. 2009. Bacterial fatty acid synthesis and its relationships with polyketide synthetic pathways. Methods Enzymol 459:395–433. doi: 10.1016/S0076-6879(09)04617-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vijayan V, Jasmin C, Anas A, Parakkaparambil Kuttan S, Vinothkumar S, Perunninakulath Subrayan P, Nair S. 2017. Sponge-associated bacteria produce non-cytotoxic melanin which protects animal cells from photo-toxicity. Appl Biochem Biotechnol 183:396–411. doi: 10.1007/s12010-017-2453-0. [DOI] [PubMed] [Google Scholar]
- 34.Coenye T, Goris J, Spilker T, Vandamme P, LiPuma JJ. 2002. Characterization of unusual bacteria isolated from respiratory secretions of cystic fibrosis patients and description of Inquilinus limosus gen. nov., sp. nov. J Clin Microbiol 40:2062–2069. doi: 10.1128/JCM.40.6.2062-2069.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Remenant B, Coupat-Goutaland B, Guidot A, Cellier G, Wicker E, Allen C, Fegan M, Pruvost O, Elbaz M, Calteau A, Salvignol G, Mornico D, Mangenot S, Barbe V, Médigue C, Prior P. 2010. Genomes of three tomato pathogens within the Ralstonia solanacearum species complex reveal significant evolutionary divergence. BMC Genomics 11:379. doi: 10.1186/1471-2164-11-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hung NB, Ramkumar G, Lee YH. 2014. An effector gene hopA1 influences on virulence, host specificity, and lifestyles of Pseudomonas cichorii JBC1. Res Microbiol 165:620–629. doi: 10.1016/j.resmic.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 37.Ghodhbane-Gtari F, Beauchemin N, Louati M1, Nouioui I, Ktari A, Hezbri K, Gueddou A, Chen A, Huntemann M, Ivanova N, Kyrpides N, Markowitz V, Mavrommatis K, Pagani I, Sen A, Wall L, Woyke T, Gtari M, Tisa LS. 2016. Permanent improved high-quality draft genome sequence of Nocardia casuarinae strain BMG51109, an endophyte of actinorhizal root nodules of Casuarina glauca. Genome Announc 4:e00799-16. doi: 10.1128/genomeA.00799-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ichinose K, Ebizuka Y, Sankawa U. 2001. Mechanistic studies on the biomimetic reduction of tetrahydroxynaphthalene, a key intermediate in melanin biosynthesis. Chem Pharm Bull 49:192–196. doi: 10.1248/cpb.49.192. [DOI] [PubMed] [Google Scholar]
- 39.Sambrook J, Maniatis T, Fritsch EF. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.