

Abstract
Background
Advanced biliary tract cancers (BTCs) have a poor prognosis and limited treatment options. This exploratory phase II study aimed to evaluate the activity of the mTOR inhibitor everolimus in advanced BTC and explore prognostic biomarkers.
Methods
Patients with advanced BTCs, who had not received chemotherapy for advanced disease, were enroled to receive everolimus (10 mg daily). The primary endpoint was disease control rate (DCR) at 12 weeks. Secondary endpoints included overall response rate, progression-free survival (PFS), overall survival (OS) and adverse events. Activation status of the RAS and phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR pathways was assessed by DNA sequencing and immunohistochemistry on archival tumour tissue.
Results
The study enroled 27 patients and the DCR at 12 weeks was 48%. Median PFS was 5.5 months (95% confidence interval (CI): 2.1–10.0 months) and median OS was 9.5 months (95% CI: 5.5–16.6 months). DCR at 12 weeks was significantly worse for gall bladder carcinoma compared to other anatomical sites, and there was a trend towards a worsened PFS and OS. Treatment was well tolerated. KRAS (12%) and PIK3CA mutations (12%) were uncommon. Immunohistochemical staining for PI3K/AKT/mTOR pathways did not significantly correlate with outcome.
Conclusion
In unselected patients, everolimus demonstrated clinical activity as first-line monotherapy in advanced BTC.
Subject terms: Targeted therapies, Prognostic markers
Introduction
Biliary tract cancers (BTCs) are a heterogenous group of tumours of the intrahepatic and extrahepatic biliary system and the gall bladder. Advanced or unresectable locally advanced disease has a poor prognosis with limited systemic treatment options.1 Combination platinum-gemcitabine chemotherapy is an active first-line treatment regimen;2 however, it is associated with low rates of radiologic response and short time to tumour progression. Effective new treatments are therefore urgently required.
Everolimus is a derivative of rapamycin that selectively inhibits mTORC1 (mammalian target of rapamycin complex 1), a key protein kinase complex which regulates cell growth, proliferation and survival. Activation of mTORC1 is mediated by the phosphatidylinositol 3-kinase (PI3K) pathway through activation of AKT/PKB and subsequent inhibition of the tuberous sclerosis complex 1/2 (TSC1/2).3 S6 kinase and 4EBP1 are key downstream effectors of this pathway and are critical regulators of protein synthesis.
Constitutive activation of the PI3K/AKT/mTOR pathway due to genetic alterations in PIK3CA,4,5 PTEN,6 PIK3R1,5 PI3KC2G,7 AKT1,6 AKT35 and TSC15 collectively occurs in ~10–20% BTCs. In addition, immunohistochemical (IHC) staining of PI3K pathway components phosphorylated (p)-AKT and p-mTOR suggests that up to 50% of BTCs may harbour activation of this pathway,8,9 which is associated with worse survival outcomes.10–12 Finally, constitutive activation of the PI3K pathway contributes to the development of BTC in genetic mouse models,13 making mTOR inhibition an attractive therapeutic target in BTC.
In this regard, we and others have recently demonstrated that mTOR inhibitors, including everolimus, inhibit the proliferation of BTC cell lines in vitro and in vivo.9,14–17 Biomarker studies in a panel of BTC cell lines revealed mutations and/or amplification of KRAS to be associated with resistance, and high basal p-AKT levels to be associated with response to everolimus.17 Activating KRAS mutations are present in 16–22% of BTCs.18–20
mTOR inhibitors are approved for the treatment of renal cell, neuroendocrine and hormone-positive/HER-2-negative advanced breast cancer.21–23 Recently, the phase II Italian Trials in Medical Oncology (ITMO) trial assessed everolimus activity in patients with chemorefractory advanced BTC, where a favourable toxicity profile and encouraging anti-tumour activity was reported.24
The objective of this exploratory single-arm phase II study, therefore, was to evaluate the clinical activity and safety of everolimus in advanced cholangiocarcinoma in the first-line setting. In addition, we sought to identify potential prognostic and predictive biomarkers of response.
Materials and methods
Eligibility
This study enroled patients of any age with metastatic or locally advanced (unresectable) adenocarcinoma of the gall bladder, intrahepatic or extrahepatic biliary tract, or ampulla of Vater, as confirmed by histology or cytology. Eligible patients had an Eastern Cooperative Oncology Group (ECOG) performance status of 0, 1 or 2; no prior chemotherapy for advanced BTC (prior adjuvant chemotherapy allowed), and adequate organ function (defined as creatinine ≤1.5 × ULN (upper limit of normal), bilirubin ≤1.5 x ULN, neutrophils ≥1.5, platelets ≥100). Measurable or non-measurable disease were allowed.
Patients were excluded if they had received any prior mTOR inhibitor therapy or had intolerance or hypersensitivity to rapalogs; severe and/or uncontrolled diabetes mellitus, hyperlipidemia, liver disease, infection or impaired pulmonary function and/or prior history of another malignancy (with the exception of non-melanoma skin cancer, carcinoma in situ of the uterine cervix or any other cancer treated with curative intent without evidence of relapse for more than 2 years).
Study protocol
All patients received oral everolimus at a starting dose of 10 mg per day until tumour progression, unacceptable toxicity or at the discretion of the investigator.
Clinical review and assessment for toxicity was undertaken every 3 weeks. Fasting glucose and lipid profile and computerised tomography (CT) was performed every 6 weeks. Tumour response was assessed according to Response Evaluation Criteria in Solid Tumours (RECIST) 1.0 criteria.
Two dose reductions, 5 mg daily, then 5 mg/s daily, were allowed in the event of drug toxicity or treatment interruption. Patients requiring a third dose reduction were required to discontinue study treatment.
The primary endpoint was disease control rate (DCR) at 12 weeks, which was defined as the proportion of patients with complete response, partial response or stable disease in the intention-to-treat population. Secondary endpoints included objective response rate (ORR), progression-free survival (PFS), overall survival (OS) and grade 3 or 4 adverse events according to NCI Common Terminology Criteria for Adverse Event v3.0. PFS was defined as the time to disease progression, the occurrence of new disease or death from any cause. Exploratory endpoints included evaluation of intra-tumoural expression of components of the PI3K/AKT/mTOR signalling pathway, KRAS and PIK3CA mutation status and KRAS amplification status, and correlation with DCR, PFS and OS.
Novartis provided the study drug and an unrestricted grant but was not involved in the analysis or interpretation of the data or the writing of the manuscript. The RADiChol study (ClinicalTrials.gov identifier: NCT00973713) was performed in accordance with the NHMRC Statement on Ethical Conduct in Research Involving Humans 1999 (© Commonwealth of Australia 1999) and the principles laid down by the World Medical Assembly in the Declaration of Helsinki and complied with ICH GCP Guidelines. The protocol was approved by the Human Research and Ethics Committee at both study sites. Patients provided written informed consent for study participation and donation of tumour tissue for exploratory biomarker analysis prior to enrolment.
Biomarker analysis
Formalin-fixed, paraffin-embedded (FFPE) archival tumour tissue was available from 21 of 27 study participants for IHC staining analyses. Briefly, 4 μm sections were cut onto charged glass slides. Slides underwent microwave heat-induced epitope retrieval in sodium citrate buffer (pH 6.0) or Tris-EDTA (pH 9.0), followed by incubation with primary antibodies. Sections were incubated with Real Envision-HRP secondary antibody, diaminobenzidine substrate (DAKO) and counterstained with haematoxylin. Positive and negative controls from a BTC cell line microarray were used for each antibody to validate the staining protocol. Scoring was based on the strongest cytoplasmic staining intensity in each tumour section, scored as 0 (absent), 1+ (weak), 2+ (moderate) or 3+ (strong). Scoring was performed by two investigators who were blinded to the clinical outcomes. For clinical correlation, expression was categorised into low (0 or 1+) and high expression (2+ or 3+). Primary antibodies against phospho-S6 Ser 235/236 (cat. no. 4858), phospho-S6 Ser 240/244 (5364), phospho-mTOR Ser 2448 (2976), phospho-AKT Ser 473 (4060) and phospho-4EBP1 T37/46 (2855) were obtained from Cell Signalling Technology (Beverly, MA, USA). Anti-PTEN (6H2.1) was purchased from Cascade Bioscience (Winchester, MA, USA).
Mutation analysis
Genomic DNA was macrodissected from FFPE tissue blocked and extracted using the QIAmp DNA FFPE Tissue Kit (Qiagen, Germany). KRAS and PIK3CA genotyping were determined by amplification of KRAS and PIK3CA using Platinum Taq DNA Polymerase High Fidelity (Invitrogen) and conventional Sanger sequencing at the Australian Genomic Research Facility. The primer sequences were as follows: KRAS exon 2F: 5′-AGTCACATTTTCATTATTTT-3′, R: 5′-AGAAACCTTTATCTGTATCAAAGAATG-3′; KRAS exon 3F: 5′-CACTGTAATAATCCAGACTGTG-3′, R: 5′-CTATAATTACTCCTTAATGTCAGC-3′; PIK3CA exon 9F: 5′-TGTGAATCCAGAGGGGAAAA-3′, R: 5′-AAATTCAGTTATTTTTTCTG-3′; exon 20F: 5′-GCTCCAAACTGACCAAACTG-3′, R: 5′-CTGTTTAATTGTGTGGAAGA-3′.
Fluorescent in situ hybridisation
Fluorescent dual probes for KRAS and CEN12 were applied to FFPE tissue with ZytoLight Fluorescent in situ hybridisation (FISH)-Tissue Implementation Kit (Zytovision, Germany) as per the manufacturer’s instructions and visualised with a Carl-Zeiss AXISKOP2 Microscope.
Statistical analysis
Based on pre-clinical findings, it was anticipated that cytostatic treatments such as mTOR inhibitors would have the greatest impact on disease control (i.e., inhibition of tumour progression). Therefore, the primary endpoint was DCR at 12 weeks. It was assumed that a DCR of 60% at 12 weeks would be of interest and that a DCR of 35% would be of little interest. The sample size was based upon a Simon’s two-stage design to reject the null hypothesis with 80% power. The study would proceed past the first stage if >3 out of 9 patients had disease control at 12 weeks, and would be declared active if >13 out of 27 patients had disease control at 12 weeks.
The final efficacy analysis was by intention to treat. PFS and OS were analysed by the Kaplan–Meier method. DCR confidence intervals were determined by the Clopper–Pearson method. Toxicity was analysed by treatment received. Fisher’s exact test was used to determine the relationship between specific biomarkers and DCR, and a log-rank test was used to determine the relationship with PFS and OS. All P values were two-sided and a P value <0.05 was considered statistically significant. Statistical analysis was performed on R-studio version 0.99.447.
Results
Patient characteristics
A total of 27 patients were enroled between September 2009 and December 2011 at two institutions—Austin Health, Melbourne (n = 26) and Queen Elizabeth Hospital, Adelaide (n = 1), and all patients received at least one dose of everolimus at a starting dose of 10 mg per day.
Baseline patient characteristics are outlined in Table 1. The median age was 64 years, 41% of patients were male (n = 11) and 93% had an ECOG performance status of 0–1. Two patients had received adjuvant chemotherapy with single-agent gemcitabine or fluorouracil.
Table 1.
Baseline characteristics of study participants
| n (%) | |
|---|---|
| Number of patients | 27 |
| Age, years (range) | 64 (34–84) |
| Male | 11 (41%) |
| Performance status (ECOG) | |
| 0 | 9 (33%) |
| 1 | 16 (59%) |
| 2 | 2 (7%) |
| Tumour location | |
| Intrahepatic | 6 (22%) |
| Extrahepatic/hilar | 6 (22%) |
| Gall bladder | 12 (44%) |
| Ampulla of Vater | 2 (7%) |
| Unknown | 1 (4%) |
| Prior therapies | |
| Curative surgery | 4 (15%) |
| Palliative surgery | 6 (22%) |
| Adjuvant chemotherapy | 2 (7%) |
| Biliary stent | 8 (30%) |
| Radiotherapy | 0 (0%) |
ECOG Eastern Cooperative Oncology Group
The median time on study was 3.8 months (range 0.6–21.0). Nineteen patients discontinued the study drug due to tumour progression. Four patients ceased treatment due to toxicity, including one patient within 12 weeks of enrolment. Two patients withdrew consent and two patients died due to tumour progression on study.
Efficacy analysis
The first part of the study was passed with 3/9 patients demonstrating disease control at 12 weeks. The study therefore proceeded to the second stage, enroling a total of 27 patients.
The primary endpoint, DCR at 12 weeks was 48% (n = 13/27, 95% CI: 28.7–68.0). Twenty-five patients had evaluable disease by RECIST 1.0. There were three partial responses and no complete responses (ORR 12%, 95% CI: 3–31%). There were 15 (60%) patients who achieved stable disease; however, five of these patients had clinically progressed at 12 weeks. Seven patients (28%) had progressive disease (Fig. 1).
Fig. 1.
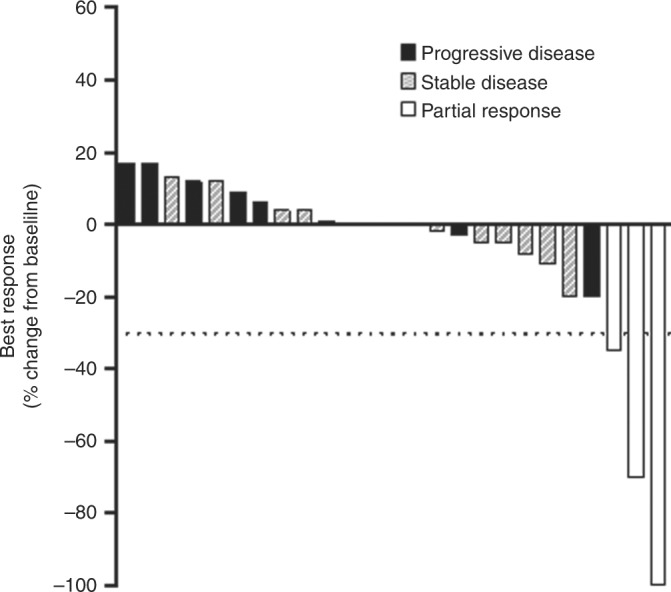
Objective tumour responses with everolimus. Waterfall plot of the maximum percentage change in target lesions compared with baseline measurements. Three patients achieved a partial response. Four patients had a 0% change in tumour burden. The dotted line denotes a 30% reduction in target lesions
Median PFS was 5.5 months (95% CI: 2.0–10.0 months) and median OS was 9.5 months (95% CI: 5.5–16.6 months) (Fig. 2). On progression, 10 patients (37%) proceeded to second-line therapy with cytotoxic chemotherapy.
Fig. 2.

Survival outcomes in patients with advanced biliary tract cancer who received everolimus. Kaplan–Meier estimates of progression-free survival (a) and overall survival (b). CI confidence interval
DCR at 12 weeks for gall bladder carcinoma was 25% versus non-gall bladder 71% (P = 0.047). Median PFS for gall bladder carcinoma was 2.1 months (95% CI: 1.4–6.0) and for non-gall bladder carcinoma 8.4 months (95% CI: 1.4–12.7) but was not significantly different (P = 0.395). Median OS for gall bladder and non-gall bladder carcinoma was 5.6 months (95% CI: 1.8–17.1) and 15.5 months (95% CI: 6.7–19.9) respectively (P = 0.092).
Adverse events
The most common drug-related toxicity was oral mucositis/stomatitis (63%). Expected drug-related toxicities of any grade were rash (52%), thrombocytopenia (7%), neutropenia (7%), anaemia (19%), hyperglycaemia (22%), hypercholesterolaemia (19%) and pneumonitis (15%) (Table 2).
Table 2.
Adverse events according to Common Terminology Criteria for Adverse Events (CTCAE) v3.0
| All grades | Grade 3 or 4 | |
|---|---|---|
| Adverse event | n (%) | n (%) |
| Stomatitis/oral mucositis | 17 (63%) | 2 (7%) |
| Pain | 16 (59%) | 4 (15%) |
| Rash | 14 (52%) | 1 (4%) |
| Nausea | 12 (44%) | 0 (0%) |
| Diarrhoea | 12 (44%) | 1 (4%) |
| Infections | 11 (41%) | 7 (26%) |
| Fatigue | 10 (37%) | 1 (4%) |
| Cough | 9 (33%) | 0 (0%) |
| Anorexia | 8 (30%) | 1 (4%) |
| Vomiting | 8 (30%) | 0 (0%) |
| Pruritus | 7 (26%) | 0 (0%) |
| Oedema | 7 (26%) | 0 (0%) |
| Hyperglycaemia | 6 (22%) | 3 (11%) |
| Weight loss | 6 (22%) | 0 (0%) |
| Epistaxis | 6 (22%) | 0 (0%) |
| Anaemia | 5 (19%) | 3 (11%) |
| Hypercholesterolaemia | 5 (19%) | 0 (0%) |
| Hypokalaemia | 5 (19%) | 1 (4%) |
| Taste alteration | 4 (15%) | 0 (0%) |
| Pneumonitis | 4 (15%) | 1 (4%) |
| Dry skin | 4 (15%) | 0 (0%) |
| Nail changes | 4 (15%) | 0 (0%) |
| Insomnia | 4 (15%) | 0 (0%) |
| Constipation | 3 (11%) | 1 (4%) |
| Headache | 3 (11%) | 0 (0%) |
| Neutropenia | 2 (7%) | 0 (0%) |
| Thrombocytopenia | 2 (7%) | 1 (4%) |
| Flu-like syndrome | 2 (7%) | 0 (0%) |
| Thromboembolism | 2 (7%) | 2 (7%) |
| Ocular toxicity | 2 (7%) | 0 (0%) |
The most frequently reported grade 3/4 adverse events were infection (26%), pain (15%), hyperglycaemia (11%) and anaemia (11%). Of the 70.4% of patients with any grade 3/4 adverse events, most were attributed to underlying disease, not drug toxicity. No unexpected treatment-related toxicities or deaths were reported.
Biomarker analyses
Analysis of the PI3K/AKT/mTOR pathway
As everolimus targets the PI3K/AKT/mTOR pathway, we performed an exploratory biomarker analysis to determine whether the activation status of the pathway correlated with drug efficacy. Tumour tissue was available from 21 of the 27 participants (78%) for biomarker analysis by immunohistochemistry, and sufficient DNA for mutation status analysis was obtained from 16 patients.
Two patients harbouring activating hotspot mutations in PIK3CA were identified. One of these patients who had a previously resected T2N0 ampullary carcinoma with intestinal differentiation and received adjuvant gemcitabine 2 years before enrolment in this study had a prolonged PFS on everolimus (16.7 months).
Activation status of the PI3K/AKT/mTOR pathway was also determined immunohistochemically by staining for basal expression of the target of rapamycin, p-mTOR, the upstream pathway components PTEN and p-AKT (Ser 473) and the downstream components p-S6 (Ser 235/236), p-S6 (240/244) and p-4EBP1 (T37/46) (Fig. 3)
Fig. 3.

Immunohistochemistry staining of biliary tract cancer tissue from the RADICHOL trial. Representative low and high expression of p-AKT (a, b), p-mTOR (c, d), p-S6 Ser 235/236 (e, f), p-S6 Ser 240/244 (g, h), p-4EBP1 (i, j) and PTEN (k, l); 10× magnification
We first assessed whether the pathway is activated in a linear manner in BTCs by examining the correlation between markers. While overexpression of p-S6 (Ser 235/236) was elevated in the two patients harbouring PIK3CA mutations, staining intensity of p-AKT, p-mTOR, p-S6 (Ser 235/236), p-S6 (Ser 240/244) and p-4EBP1 did not correlate across the tumours, indicating a complex mode of pathway activation.
Assessment of the association between marker expression and everolimus response identified a trend towards worse OS in patients with high tumour p-4EBP1 staining (P = 0.052) (Table 3). There was no statistically significant correlation between staining intensity of p-AKT, p-mTOR, p-S6 (Ser 235/236) or p-S6 (Ser 240/244) and efficacy outcomes.
Table 3.
Association of markers of PI3K/AKT/mTOR pathway activity with clinical outcome
| Basal biomarker | n (%) | DCR at 12 weeks (%) | P value | Median PFS (95% CI), months | P value | Median OS (95% CI), months | P value |
|---|---|---|---|---|---|---|---|
| p-S6 Ser 235/236 | |||||||
| Low | 14 (67) | 6 (43) | 0.659 | 3.0 (1.4–12.7) | 0.792 | 8.5 (2.5–16.6) | 0.281 |
| High | 7 (33) | 4 (57) | 6.0 (1.1–NR) | 9.5 (1.1–20.5) | |||
| p-S6 Ser 240/244 | |||||||
| Low | 13 (62) | 7 (54) | 0.659 | 5.8 (1.4–12.7) | 0.471 | 14.4 (2.47–18.6) | 0.378 |
| High | 8 (38) | 3 (37) | 3.0 (1.1–NR) | 6.6 (1.09–16.6) | |||
| p-mTOR | |||||||
| Low | 15 (68) | 6 (40) | 0.685 | 5.5 (1.4–12.7) | 0.295 | 9.5 (5.5–16.6) | 0.851 |
| High | 7 (32) | 3 (43) | 2.5 (1.1–8.4) | 6.7 (1.1–17.1) | |||
| p-AKT Ser 473 | |||||||
| Low | 14 (67) | 7 (50) | 1 | 5.5 (1.4–11.2) | 0.409 | 8.1 (1.81–16.6) | 0.853 |
| High | 7 (33) | 3 (43) | 6.0 (1.41–NR) | 9.5 (4.38–19.6) | |||
| p-4EBP1 | |||||||
| Low | 13 (62) | 8 (61) | 0.183 | 6.0 (1.4–16.8) | 0.143 | 14.4 (2.5–19.9) | 0.052 |
| High | 8 (38) | 2 (25) | 2.6 (1.09–NR) | 6.6 (1.09–16.5) | |||
| PTEN | |||||||
| Low | 5 (24) | 3 (60) | 0.635 | 7.1 (2.8–NR) | 0.844 | 16.6 (5.6–NR) | 0.621 |
| High | 16 (76) | 7 (44) | 5.5 (1.4–16.8) | 7.2 (2.5–17.1) | |||
CI confidence interval, NR not reached
Analysis of KRAS amplification and mutation status
Mutations in KRAS have been associated with resistance to everolimus in BTC cell lines,17 and in patients with colorectal cancer who are treated with this agent.25 Sanger sequencing of KRAS (exons 2 and 3) identified only two patients harbouring KRAS mutations, of whom one was the patient with ampullary carcinoma with intestinal differentiation who also harboured a PIK3CA (E545K) mutation. KRAS amplification has previously been reported in a small subset of BTC patients;20 however, no patients with KRAS amplification using FISH were identified in the current study. Collectively, the low prevalence of KRAS mutations and amplification precluded investigation of associations with outcome.
Discussion
While this study narrowly failed to meet its primary endpoint of >13/27 patients experiencing disease control at 12 weeks, everolimus monotherapy did have clinical activity in a subset of patients with advanced BTC in the first-line setting.
The progression-free and OS outcomes observed in this study (5.5 and 9.5 months, respectively) were comparable to results from the ITMO phase II single-arm trial of everolimus monotherapy in BTC following progression on chemotherapy, which reported an 8-week DCR of 44.4%, PFS of 3.2 months, and OS 7.7 months.24 Nevertheless, platinum-gemcitabine doublet chemotherapy remains the most active regimen in an unselected population of advanced BTC (median PFS 8.0 months, median OS 11.7 months).2
DCR at 12 weeks was significantly worse for gall bladder carcinoma, and there was a trend towards a worsened PFS and OS. When compared to other anatomical sites, gall bladder carcinoma is not regarded as an adverse prognostic marker in BTC,26,27 suggesting that gall bladder carcinoma may be resistant to everolimus. While our study lacked statistical power, this result warrants further investigation.
The adverse event profile for everolimus was also similar to that previously reported in other studies.21–23 Oral mucositis/stomatitis was the most common drug-related toxicity (63%), which was most frequently grades 1 and 2. Conservative measures with a non-alcoholic mouthwash or topical analgesic mouth treatment can successfully manage this oral toxicity. One event of G3 pneumonitis was reported, necessitating treatment interruption. The favourable overall toxicity profile of everolimus suggests that it can be safely administered in the outpatient setting for advanced BTC.
An important feature of this study was an exploratory analysis of potential biomarkers of everolimus response. In this regard, we recently observed that BTC cell lines with high basal p-AKT expression respond preferentially to everolimus in vitro.28 Similarly, renal and gastric cancers harbouring higher basal activation of the PI3K/AKT/mTOR pathway have also been reported to show improved response to everolimus.29,30
In comparison, in the current study we failed to observe an association between the levels of a number of IHC readouts of PI3K/AKT/mTOR pathway activity and benefit to everolimus treatment. One limitation of this approach was the lack of concordance between the different markers of PI3K/AKT/mTOR pathway activity. This may reflect the complex biological regulation of this pathway in BTC, such as possible activation by multiple upstream effectors and crosstalk with other pathways including mitogen-activated protein kinase. Continued investigation of more robust biomarkers of everolimus is therefore required. However, we also acknowledge that the study lacked sufficient statistical power to definitively determine the predictive power of these markers and their potential to predict everolimus sensitivity should not be excluded.
Nevertheless, the biomarker analysis did reveal two potentially interesting findings. First, although the relatively low frequency of KRAS and PIK3CA mutations in this cohort (12.5%) precluded assessment of an association with outcome, we noted that one patient with ampullary carcinoma with intestinal differentiation who had a concurrent PIK3CA and KRAS mutation had a prolonged PFS of 16.7 months. In comparison to other BTCs, ampullary carcinomas have a higher rate of PIK3CA (13%) and KRAS (54%) mutations, and these mutations have been reported to co-exist.31 Whether the response of this patient to everolimus is a consequence of their PIK3CA mutation status, or reflective of other biological features unique to ampullary carcinoma requires additional investigation.
Second, we observed that high tumour expression of p-4EBP1 was associated with a worse outcome. However, the lack of a control arm on the current study does not allow for distinction between whether high p-4EBP is predictive of resistance to everolimus or prognostic of poor outcome. In this regard, it is notable that several previous studies have linked hyperactivation of the PI3K/AKT/mTOR pathway as assessed by IHC measurement of p-mTOR, PTEN and p-4EBP1 with a poorer prognosis in BTC.10–12
In this exploratory phase II trial, although single-agent everolimus narrowly failed to meet its primary endpoint, favourable anti-tumour activity was observed in a subset of patients with advanced BTCs. Further studies of everolimus in BTC are warranted with ongoing focus on identifying predictive biomarkers.
Acknowledgements
We thank all the patients, their families and investigators who participated in this study.
Funding
This work was supported by an unrestricted educational grant from Novartis with additional funding from the Victorian Cancer Agency (TRP-13083). D.K.L. received financial support La Trobe University (Australian Postgraduate Award) and Pancare/RMA Network. F.C. received financial support from National Health and Medical Research Council (Medical Postgraduate Scholarship, 1017737). Y.H.Y. was supported by a Fellowship from the ANZ Trustees Foundation—Brian Smith Endowment and J.M. is supported by a NH&MRC Senior Research Fellowship (1046092). The Olivia Newton-John Cancer Research Institute acknowledges the support of Ludwig Cancer Research, and the Operational Infrastructure Support Programme of the Victorian State Government.
Conflict of interest
The authors declare that they have no conflict of interest
Footnotes
Note: This work is published under the standard licence to publish agreement. After 12 months the work will become freely available.
These authors contributed equally: John M. Mariadason and Niall C. Tebbutt.
Contributor Information
John M. Mariadason, Phone: +61 3 9496 3068, Email: john.mariadason@onjcri.org.au
Niall C. Tebbutt, Phone: +61 3 9496 5763, Email: niall.tebbutt@onjcri.org.au
References
- 1.Eckel F, Schmid RM. Chemotherapy and targeted therapy in advanced biliary tract carcinoma: a pooled analysis of clinical trials. Chemotherapy. 2014;60:13–23. doi: 10.1159/000365781. [DOI] [PubMed] [Google Scholar]
- 2.Valle J, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010;362:1273–1281. doi: 10.1056/NEJMoa0908721. [DOI] [PubMed] [Google Scholar]
- 3.O’Donnell A, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J. Clin. Oncol. 2008;26:1588–1595. doi: 10.1200/JCO.2007.14.0988. [DOI] [PubMed] [Google Scholar]
- 4.Li M, et al. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat. Genet. 2014;46:872–876. doi: 10.1038/ng.3030. [DOI] [PubMed] [Google Scholar]
- 5.Nakamura H, et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015;47:1003–1010. doi: 10.1038/ng.3375. [DOI] [PubMed] [Google Scholar]
- 6.Borger DR, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist. 2012;17:72–79. doi: 10.1634/theoncologist.2011-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiao Y, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet. 2013;45:1470–1473. doi: 10.1038/ng.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee H, et al. Comprehensive genomic profiling of extrahepatic cholangiocarcinoma reveals a long tail of therapeutic targets. J. Clin. Pathol. 2016;69:403–408. doi: 10.1136/jclinpath-2015-203394. [DOI] [PubMed] [Google Scholar]
- 9.Simbolo M, et al. Multigene mutational profiling of cholangiocarcinomas identifies actionable molecular subgroups. Oncotarget. 2014;5:2839–2852. doi: 10.18632/oncotarget.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herberger B, et al. Activated mammalian target of rapamycin is an adverse prognostic factor in patients with biliary tract adenocarcinoma. Clin. Cancer Res. 2007;13:4795–4799. doi: 10.1158/1078-0432.CCR-07-0738. [DOI] [PubMed] [Google Scholar]
- 11.Wang Z, et al. Immunohistochemical analysis of the mTOR pathway in intrahepatic cholangiocarcinoma. Neoplasma. 2012;59:137–141. doi: 10.4149/neo_2012_018. [DOI] [PubMed] [Google Scholar]
- 12.Chung JY, et al. The expression of phospho-AKT, phospho-mTOR, and PTEN in extrahepatic cholangiocarcinoma. Clin. Cancer Res. 2009;15:660–667. doi: 10.1158/1078-0432.CCR-08-1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu X, et al. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption of Smad4 and Pten in mice. J. Clin. Invest. 2006;116:1843–1852. doi: 10.1172/JCI27282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansel DE, et al. Identification of novel cellular targets in biliary tract cancers using global gene expression technology. Am. J. Pathol. 2003;163:217–229. doi: 10.1016/S0002-9440(10)63645-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rizell M, et al. Effects of the mTOR inhibitor sirolimus in patients with hepatocellular and cholangiocellular cancer. Int. J. Clin. Oncol. 2008;13:66–70. doi: 10.1007/s10147-007-0733-3. [DOI] [PubMed] [Google Scholar]
- 16.Wu Q, et al. Therapeutic effect of rapamycin on gallbladder cancer in a transgenic mouse model. Cancer Res. 2007;67:3794–3800. doi: 10.1158/0008-5472.CAN-06-3214. [DOI] [PubMed] [Google Scholar]
- 17.Yeung Y, et al. K-Ras mutation and amplification status is predictive of resistance and high basal pAKT is predictive of sensitivity to everolimus in biliary tract cancer cell lines. Mol. Oncol. 2017;11:1130–1142. doi: 10.1002/1878-0261.12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahn DH, et al. Next-generation sequencing survey of biliary tract cancer reveals the association between tumor somatic variants and chemotherapy resistance. Cancer. 2016;122:3657–3666. doi: 10.1002/cncr.30247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jusakul A, et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov. 2017;7:1116–1135. doi: 10.1158/2159-8290.CD-17-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakamura H, et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015;47:1003–1010. doi: 10.1038/ng.3375. [DOI] [PubMed] [Google Scholar]
- 21.Baselga J, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N. Engl. J. Med. 2012;366:520–529. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.She QB, et al. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell. 2010;18:39–51. doi: 10.1016/j.ccr.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao JC, et al. Everolimus for advanced pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011;364:514–523. doi: 10.1056/NEJMoa1009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buzzoni R, et al. Activity and safety of RAD001 (everolimus) in patients affected by biliary tract cancer progressing after prior chemotherapy: a phase II ITMO study. Ann. Oncol. 2014;25:1597–1603. doi: 10.1093/annonc/mdu175. [DOI] [PubMed] [Google Scholar]
- 25.Di Nicolantonio F, et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J. Clin. Invest. 2010;120:2858–2866. doi: 10.1172/JCI37539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miyakawa S, et al. Biliary tract cancer treatment: 5,584 results from the Biliary Tract Cancer Statistics Registry from 1998 to 2004 in Japan. J. Hepatobiliary. Pancreat. Surg. 2009;16:1–7. doi: 10.1007/s00534-008-0015-0. [DOI] [PubMed] [Google Scholar]
- 27.Bridgewater J, et al. Prognostic factors for progression-free and overall survival in advanced biliary tract cancer. Ann. Oncol. 2016;27:134–140. doi: 10.1093/annonc/mdv483. [DOI] [PubMed] [Google Scholar]
- 28.Yeung Y, et al. K-Ras mutation and amplification status is predictive of resistance and high basal pAKT is predictive of sensitivity to everolimus in biliary tract cancer cell lines. Mol Oncol. 2017;11:1130–1142. doi: 10.1002/1878-0261.12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li S, et al. Phosphorylation of mTOR and S6RP predicts the efficacy of everolimus in patients with metastatic renal cell carcinoma. BMC Cancer. 2014;14:1471–2407. doi: 10.1186/1471-2407-14-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoon DH, et al. Phase II study of everolimus with biomarker exploration in patients with advanced gastric cancer refractory to chemotherapy including fluoropyrimidine and platinum. Br. J. Cancer. 2012;106:1039–1044. doi: 10.1038/bjc.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gingras MC, et al. Ampullary cancers harbor ELF3 tumor suppressor gene mutations and exhibit frequent WNT dysregulation. Cell Rep. 2016;14:907–919. doi: 10.1016/j.celrep.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]