

 This work is licensed under a
This work is licensed under a Summary
Granular cell tumours (GCT) are rare, slow-growing, benign neoplasms that are usually located in the head and neck. They are more frequent in the female gender and typically have an asymptomatic clinical course, being diagnosed only at autopsy. Symptomatic GCT of the neurohypophysis are exceedingly rare, being less than 70 cases described so far. The authors report on a case of a 28-year-old male that presented to the Endocrinology clinic with clinical and biochemical evidence of hypogonadism. He also reported minor headaches without any major visual symptoms. Further laboratory tests confirmed hypopituitarism (hypogonadotrophic hypogonadism, central hypothyroidism and hypocortisolism) and central nervous system imaging revealed a pituitary macroadenoma. The patient underwent transcranial pituitary adenoma resection and the pathology report described a GCT of the neurohypophysis with low mitotic index. The reported case is noteworthy for the rarity of the clinicopathological entity.
Learning points:
Symptomatic GCTs are rare CNS tumours whose cell of origin is not well defined that usually give rise to visual symptoms, headache and endocrine dysfunction.
Imaging is quite unspecific and diagnosis is difficult to establish preoperatively.
Surgical excision is challenging due to lesion’s high vascularity and propensity to adhere to adjacent structures.
The reported case is noteworthy for the rarity of the clinicopathological entity.
Background
Pituitary tumours are a frequent type of central nervous system neoplasm. They are present in up to 10.6% of the general population and have a wide spectrum of presentations and behavior (1). Adenomatous pituitary tumours are among the most frequent intracranial tumours, representing 15.5% of all primary central nervous system (CNS) neoplasms (2). Other extremely rare tumours may arise in the sellar region mimicking the clinical and radiological presentation of a pituitary adenoma. Although several post-mortem studies have documented incidental small granular cell tumours or tumourlettes in 6.5–17% of the autopsies, symptomatic granular cell tumours (GCT) are rare, with less than 70 cases described so far (3). These tumours are usually benign and may occur in different locations namely tongue, skin, gastrointestinal tract and central nervous system and have a predilection for the adult female population (4). Preoperative diagnosis is not easy, resembling other sellar lesions such as adenoma, meningioma, chordoma and teratoma. Surgery is the cornerstone of treatment; however, complete tumour excision is often unachievable due to tumour size, proximity to the optic chiasm and high vascularity resulting in a considerable rate of recurrences. The authors report on a case of a 28-year-old male that presented with a GCT and briefly review the topic.
Case presentation
The patient was a 28-year-old male with a past medical history of factor VII deficiency, that reported a history of headaches, lassitude, reduced libido, erectile dysfunction, orthostatic hypotension and weight loss of insidious onset and slow progression over two years. One week before the first endocrinology clinic, he presented to the emergency department with nausea, vomiting and slightly decreased visual acuity. Clinical examination was remarkable for decreased facial and body hair, slight muscle atrophy without any other features suggestive of pituitary dysfunction namely hypotension, polyuria, polydipsia or delayed puberty. Baseline ophthalmological examination revealed bilateral optic atrophy and absent visual field defects upon confrontation.
Investigation
Basal laboratory studies revealed slightly elevated prolactin (49 ng/mL,normal range (NR): 3.7–17.9), hypogonadotrophic hypogonadism (total testosterone <20 ng/dL; LH <0.22 U/L and FSH <0.66 U/L), low IGF-1 (39.3 ng/mL, NR: 115–340), low serum cortisol along with inadequately normal ACTH (cortisol 2.92 µg/dL, NR: 6.2–19.4 and ACTH of 12.1 pg/mL, NR: 7.2–63.3) and central hypothyroidism (TSH: 2.18 µU/mL, NR: 0.27–4.2; fT4: 9.22 pmol/L, NR: 12–22) despite no electrolyte imbalance, hypoglycaemia nor decreased urine density. Magnetic resonance imaging (MRI) showed a large suprasellar mass extending caudally to the sella turcica with 29 × 27 × 35 mm compressing the pituitary and optic chiasm. The lesion was isointense on T1 and hypointense on T2-weighted imaging (Fig. 1).
Figure 1.
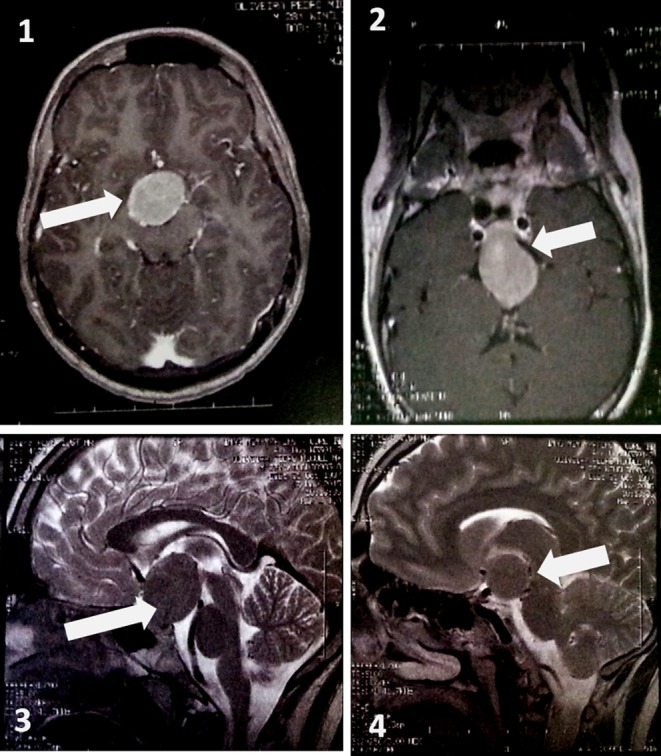
Large suprasellar mass extending caudally to the sella turcica with 29 × 27 × 35 mm compressing the pituitary gland and optic chiasm. The lesion was isointense on T1 (panels 1 and 2) and hypointense on T2-weighted imaging (Panels 3 and 4). It is responsible for the enlargement of the sella turcica, marked compression of the infundibulum, optic chiasm, third ventricle pavement (causing ventricular deformation) and right cerebral peduncle.
Semen analysis was not performed because fertility was no longer an issue to the patient and his partner that already had one daughter.
Treatment
An initial trans-sphenoidal endoscopic approach was attempted with only partial tumour removal being achieved due to abundant intraoperative hemorrhage. A second surgery was performed with a transcranial approach (fronto-orbitary) making subtotal tumour removal and chiasmatic decompression possible. Light microscopy revealed a neoplasm composed of densely packed nests of polygonal and fusiform cells with abundant granular eosinophilic PAS-positive cytoplasm and small, round nuclei. No mitosis, atypia or necrosis was documented. Immunohistochemistry for S100, CD68, vimentin and TTF-1 (nuclear) were positive and GFAP and pituitary hormones were all negative. Ki-67 proliferation index was extremely low (~1%). Ultrastructurally, numerous phagolysosomes filled the cytoplasm and the presence of neurosecretory granules or mitochondria were excluded, determining the diagnosis of GCT (Figs 2 and 3).
Figure 2.
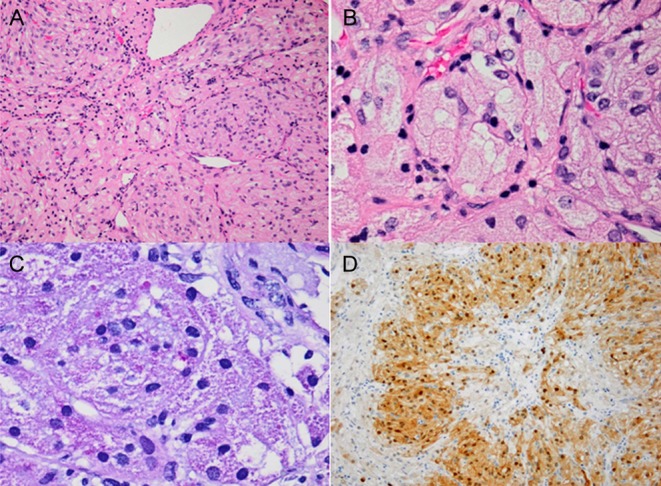
(A) Hematoxylin and eosin, 200×; Highly cellular neoplasm, composed of nests of fusiform and polygonal cells with abundant eosinophilic cytoplasm. The nuclei are small, round and monotonous, without mitotic figures. Several large vessels can be appreciated. (B) Hematoxylin and eosin, 400×; In higher magnification, a nest of cells showing granular cytoplasm and eccentrically located nuclei. (C) Periodic acid-Schiff diastase, 400×; Cytoplasmic granules are PAS-D positive, revealing that the granular is not due to accumulation of glycogen. (D) S100, 100×; Immunohistochemistry for S100 was positive.
Figure 3.

Electron microscopy (performed in PEFF tissue). Numerous vesicular structures account for the cytoplasmic granularity. The vesicular structures consist of secondary lysosomes.
Outcome and follow-up
The patient was discharged and revaluated in Endocrinology consult. During follow-up, tumour regrowth has been documented, which has driven three additional surgical interventions and recently a ventriculoperitoneal shunt placement due to intractable headaches. Complete surgical excision has so far been unachievable due to the tumour hypervascularity and possibly patient’s hemorrhagic diatheses, despite adequate preoperative coagulation factor replacement. After two years of follow-up and four surgical interventions, the lesion’s current dimensions are 37 × 32 × 26 mm (Fig. 4). Visual symptoms and ophthalmological findings remain similar to baseline.
Figure 4.

Current tumour status, measuring 37 × 32 × 26 mm, isodense in T1 and T2 with intense homogeneous contrast enhancement.
Regarding pituitary function, treatment with testosterone, thyroxine and hydrocortisone has rendered the patient stable from the endocrinological standpoint with no evidence of diabetes insipidus.
Due to disease progression, the multidisciplinary team plans additional surgical interventions preceded by tumour embolization if feasible.
Discussion
GCTs were first described in 1893 by Boyce and Beadles (5). Since then, many autopsy series have revealed a high prevalence of granular cell tumourlettes in the general population, particularly in the elderly (6.7–17%) (4, 6). Although these small tumours are generally benign, non-secreting, slow growing and arising in the sellar region, malignant variants have also been described (7). In the majority of cases, they are incidental findings at the time of autopsy, being symptomatic cases extremely rare. In the literature, there have been less than 70 symptomatic cases described so far. Women are more frequently affected than men (2:1) with a peak incidence in the 5th decade of life. Presenting symptoms include insidious onset of visual disturbances in up to 90% of the cases, headaches and endocrine changes in approximately 50%: amenorrhea, galactorrhea, infertility, impotence and weight fluctuations (4). Signs of hypopituitarism or hyperprolactinaemia are the most frequent findings; however, a case report of acromegaly has been described in the literature (7). Radiological findings are rather nonspecific and may share features of other CNS lesions such as meningioma, pituitary adenoma, chordoma or teratoma. GCTs are usually iso or slightly hyperdense on CT scans before contrast infusion and show moderate contrast enhancement. On MRI are hypoisointense on both T1- and T2-weighted imaging and reveal marked, usually homogeneous, contrast enhancement after i.v. gadolinium administration (8). Accurate preoperative diagnosis is not possible and therefore there are no specific treatment guidelines for GCT. Diagnosis can only be reliably made after surgical excision.
On histopathological examination, the neoplastic cells showed both spindle and polygonal cells with eosinophilic granular cytoplasm, diffusely positive for PAS, S100, CD68, vimentin and TTF1 and negative for GFAP and pituitary hormones expression. Although these features excluded the possibility of a pituicytoma (that is GFAP positive), they did not establish a definitive diagnosis of other oncocytic tumours of the sellar region such as spindle cell oncocytoma (SCO), GCT and null pituitary adenoma. SCO, a tumour derived from folliculostellate cells of adenohypophysis differs from GCT, which can have both fusiform and polygonal cells, by the nature of the cytoplasmic granules, which are due to mitochondria accumulation and phagolysosomes, respectively. In our case, the definitive diagnosis relied on electron microscopy, which confirmed the presence of numerous cytoplasmic secondary lysosomes, leading to the diagnosis of a GCT (9, 10).
Treatment is surgical and should be based on clinical judgment and pituitary adenoma guidelines. Surgical removal can be performed via a transnasoseptal-sphenoidal approach (microscopic or endoscopic technique) or by pterional (frontotemporal) craniotomy depending on the tumour size and extension. These tumours are difficult to resect due to their tough and highly vascular structure and tendency to adhere to adjacent brain structures (optic pathway, hypothalamus, among others).
Surgical outcomes are variable and usually only subtotal removal is possible if preservation of adherent normal brain structures is desired. In order to achieve complete resection, sacrifice or damage of the pituitary stalk and other structures is frequent and carries a higher risk of endocrine sequelae such as diabetes insipidus and hypopituitarism. Radiotherapy as a sole or adjuvant treatment strategy is controversial and cannot be routinely recommended. Cases with anaplastic features, progressive clinical course or inoperable recurrence may constitute indications for adjuvant radiotherapy. Prognosis is strictly dependent on the surgical removal (4).
The reported case is remarkable for the younger age at disease presentation (symptoms began at 26 years of age) and minimal visual symptoms despite the large tumour size. Panhypopituitarism was present at the first endocrinology clinic visit and surgery was an effective measure towards the minor visual changes. Diagnosis was not suspected preoperatively and trans-sphenoidal followed by a transcranial approach was required for significant tumour removal. Reporting this clinico-pathological entity attempts to increase medical awareness and knowledge in what regards to such an uncommon disease.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Patient consent
Written informed consent for publication of their clinical details and/or clinical images was obtained from the patient/parent/guardian/relative of the patient.
Author contribution statement
Carlos Tavares Bello, major article writing. Patricia Cipriano, article revision and clinical disease diagnosis. Vanessa Henriques, article writing and revision and pathological diagnosis. João Sequeira Duarte, article revision and patient’s Endocrinology attending physician. Conceição Canas Marques, article revision and patient’s Neurosurgery attending physician.
References
- 1.Molitch ME. Nonfunctioning pituitary tumors and pituitary incidentalomas. Endocrinology Metabolism Clinics of North America 2008. 37 151–171. ( 10.1016/j.ecl.2007.10.011) [DOI] [PubMed] [Google Scholar]
- 2.Ostrom QT, Gittleman H, Fulop J, Liu M, Blanda R, Kromer C, Wolinsky Y, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical Report: primary brain and central nervous system tumors diagnosed in the United States in 2008–2012. Neuro-Oncology 2015. 17 (Supplement 4) iv1 ( 10.1093/neuonc/nov189) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cohen-Gadol AA, Pichelmann MA, Link MJ, Scheithauer BW, Krecke KN, Young WF, Giannini C. Granular cell tumor of the sellar and suprasellar region: clinicopathologic study of 11 cases and literature review. Mayo Clinic Proceedings 2003. 78 567–573. ( 10.4065/78.5.567) [DOI] [PubMed] [Google Scholar]
- 4.Latini F, Ambrosio MR, Guerra A, Uberti ED, Cavallo MA, Lapparelli M. Pituitary granular cell tumor: single-center experience and comprehensive update. Contemporary Neurosurgery 2014. 36 1–7. ( 10.1097/01.CNE.0000455825.70290.92) [DOI] [Google Scholar]
- 5.Boyce R, Beadles CF. A further contribution to the study of the pathology of the hypophysis cerebri. Journal of Pathology and Bacteriology 1893. 1 359–383. ( 10.1002/path.1700010310) [DOI] [Google Scholar]
- 6.Luse S, Kernohan J. Granular cell tumors of the stalk and posterior lobe of the pituitary gland. Cancer 1955. 8 616–622. () [DOI] [PubMed] [Google Scholar]
- 7.Losa M, Saeger W, Mortini P, olfi C, Terreni MR, Taccagni G, Giovanelli M. Acromegaly associated with a granular cell tumor of the neurohypophysis: a clinical and histological study. Case report. Journal of Neurosurgery 2000. 93 121–126. ( 10.3171/jns.2000.93.1.0121) [DOI] [PubMed] [Google Scholar]
- 8.Gregoire A, Godfraind C. Granular cell tumor of the pituitary stalk: a rare and benign entity. Journal of the Belgian Society of Radiology 2015. 99 79–81. ( 10.5334/jbr-btr.841) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vaquero J, Leunda G, Cabezudo J. Granular pituicytomas of the pituitary stalk. Acta Neurochirurgica 1981. 59 209–215. ( 10.1007/BF01406350) [DOI] [PubMed] [Google Scholar]
- 10.Nishio S, Takeshita I, Yoshimoto K, Yamaguchi T. Granular cell tumor of the pituitary stalk. Clinical Neurology and Neurosurgery 1998. 100 144–147. ( 10.1016/S0303-8467(98)00017-1) [DOI] [PubMed] [Google Scholar]