

Abstract
Interstitial fluid pressure (IFP) presents a barrier to drug uptake in solid tumors, including the aggressive primary brain tumor glioblastoma multiforme (GBM). It remains unclear how fluid dynamics impacts tumor progression and can be targeted therapeutically. To address this issue, a novel telemetry-based approach was developed to measure changes in IFP during progression of GBM xenografts. Antisecretory factor (AF) is an endogenous protein that displays anti-secretory effects in animals and patients. Here, endogenous induction of AF protein or exogenous administration of AF peptide reduced IFP and increased drug uptake in GBM xenografts. AF inhibited cell volume regulation of GBM cells, an effect that was phenocopied in vitro by the sodium-potassium-chloride co-transporter 1 (SLC12A2/NKCC1) inhibitor bumetanide. As a result, AF induced apoptosis and increased survival in GBM models. In vitro, the ability AF to reduce GBM cell proliferation was phenocopied by bumetanide and NKKC1 knockdown. Next, AF’s ability to sensitize GBM cells to the alkylating agent temozolomide, standard of care in GBM patients, was evaluated. Importantly, combination of AF induction and temozolomide treatment blocked regrowth in GBM xenografts. Thus, AF-mediated inhibition of cell volume regulation represents a novel strategy to increase drug uptake and improve outcome in GBM.
Keywords: brain, glioma, interstitial fluid pressure
Introduction
Glioblastoma (GBM) is the most common and aggressive primary brain tumor with a median survival of 12-15 months (1). Growing in the confines of the skull, GBMs in patients produce elevated intracranial pressures (10-100 mmHg) compared to matched controls (0-5 mmHg) (2). In GBM, tumor cells displace non-neoplastic astrocytes, which, together with vascular abnormalities, lead to failure in barrier properties, inducing vessel permeability that allows plasma and fluid to leak into the tumor tissue, inducing cerebral edema and increasing IFP (3). Fluid accumulation compresses the tumor and surrounding normal tissue and reduces cellular uptake in solid tumors (3).
Although anti-angiogenic therapies effectively reduce IFP, they may also contribute to invasion and treatment-resistance (3,4). Changes in cell volume caused by water and ion transport across the cell membrane are necessary for proliferation and migration of GBM cells (5). Invading GBM cells maximally decrease their cell volume by 30-35%, a value that represents all free unbound cytoplasmic water (6). Any volume reduction beyond this value is expected to compromise the survival of GBM cells (7). Therefore, we hypothesized that IFP-reducing strategies that prevent osmotic adaptation of tumor cells should reduce tumor progression without driving invasiveness.
In the brain, transport of water and ions across the plasma membrane by aquaporins and the sodium-potassium-chloride co-transporter 1 (NKCC1) is necessary for glial cells to adapt to osmotic challenges (8,9). Although knockdown of aquaporins reduced migration and survival of GBM cells, therapeutic targeting of these integral membrane proteins remains a major challenge (10). Expression of NKCC1 is lower in the brain and increases with glioma grade (11), shown to regulate tumor cell invasion (12). We recently demonstrated that the FDA-approved NKCC1 inhibitor bumetanide abrogated regulatory volume increase and potentiated temozolomide-induced cytotoxicity of human GBM cells (13). However, poor brain penetrance and side-effects from the highly potent diuretic bumetanide limit its chronic use (14,15). Is there an alternative approach to regulate tumor cell volume that reduces IFP and produces anti-tumor activity in GBM?
Efforts to identify agents that prevent cholera toxin-induced hypersecretion from intestine lead to the identification of a protein in intestinal organs that demonstrated antisecretory properties (16). Purification of this antisecretory factor (AF) protein identified an eight-amino acid N-terminal sequence that displayed antisecretory properties in higher vertebrates and humans (17–20). Endogenous AF protein can be induced in animals and humans within days following treatment with a specially processed cereal (SPC) (21,22). Exogenous administration of AF protein through oral route (Salovum) or nasal uptake of AF peptide produced more immediate effects (23,24). Endogenous and exogenous induction of AF protein was well-tolerated in animals and patients (21,25). More recent studies show that AF can reverse fluid dysregulation, including intracranial pressure, in the brain and other organs (22–24,26–28). The mechanism of action has been linked to targets known to regulate fluid homeostasis, including chloride permeability across cell membranes (29), and lipid content, including physical interaction of AF with the lipid raft protein flottilin-1 (30,31). AF’s ability to reduce intracranial pressure in the injured brain, lower IFP in subcutaneous tumors, and increase drug uptake in solid subcutaneous tumors suggest that it might also be effective in GBM (22,23).
Here, we used a telemetry-based approach to measure IFP during GBM progression in patient-derived xenografts (PDXs). In contrast to previous methodology that measured IFP in tumors of anaesthetized animals for a few hours (32,33), this non-invasive approach enabled us to remotely and continuously measure IFP during tumor progression. Oral or nasal AF therapy reduced IFP levels and increased drug uptake in this PDX model. Oral administration of SPC diet (endogenous AF induction) inhibited tumor growth and extended survival in GBM xenografts. Upon closer examination, ~40% of tumor cells displayed reduced tumor cell volume along with increased apoptotic index following administration of SPC diet versus control diet. AF induction reduced NKCC1 expression in GBM xenografts and treatment with AF peptide, more potently than bumetanide, inhibited regulatory volume increase and chloride permeability in GBM tumorsphere cultures. Using a compression force bioreactor, we show that AF peptide, bumetanide, and NKCC1 knockdown blocked compression-induced proliferation of GBM tumorspheres. Importantly, a combination of SPC diet and temozolomide completely prevented GBM progression and regrowth of PDXs. Thus, AF therapy represents a novel IFP-reducing strategy that produces anti-tumor activity in GBM.
Materials and Methods
Mice
All mouse experiments were approved by and performed according to the guidelines of the UCSF Institutional Animal Care and Use Committee. For PDX experiments, we purchased 4-6 week-old athymic nu/nu mice (Simonsen Laboratories).
Intracranial injections
We stereotactically injected 50,000 GBM43 cells, 300,000 GBM43 cells, or 300,000 GBM6 cells expressing luciferase (LUC) into the frontal cortex of adult athymic mice. We also injected 200,000 mouse GBM cells expressing human EGFRvIII (pEGFRlow), mCherry, and LUC (34) into adult FVBn mice (Charles River Laboratories, Wilmington, MA). Injections were done at a depth of 3 mm and the coordinates were 2-mm anterior and 1.5-mm lateral of the right hemisphere relative to Bregma. We measured body weight and bioluminescence every second day during progression. Bioluminescence was measured using a Xenogen IVIS imager and survival was plotted using Kaplan–Meier curves. Symptomatic mice were sacrificed according to ethical guidelines approved by the UCSF Institutional Animal Care and Use Committee (IACUC).
Treatment studies
For endogenous AF protein induction, mice were administered (as indicated in figures) SPC diet (10% SPC from POA Pharma cold-pressed with 90% Global Protein Rodent Diet, Envigo). Control mice were treated with 100% Global Protein Rodent Diet. For exogenous AF administration, nasal AF was injected acutely preceding a single tail-vein injection of doxorubicin as stated for uptake studies below. For long-term exogenous AF administration, mice were given 4% Salovum (egg yolk powder, POA Pharma) dissolved in 3% glucose-containing water. Mice were administered Erlotinib (100 mg/kg/day, Selleck Chemicals, five days on, two days off for three weeks). Temozolomide hydrochloride (50 mg/kg/day, Selleck Chemicals) was administered through oral gavage on days 10-11.
Implantation of IFP recording probes
Before transplantation, each pressure transducer (PA-C10, Data Sciences International) was calibrated and sterilized according to DSI instruction manual. An offset reading was recorded for each probe and was accounted for in pressure readings throughout the experiment. An opening (approximately 1 mm diameter) was drilled into the right side of the parietal skull 2-mm anterior and 1.5-mm lateral of the right hemisphere relative to Bregma. Sterilized, custom pan slot vented pure titanium screws (United Titanium Inc), were screwed in the drilled hole. Injections of GBM cells were done at a depth of 3 mm. Subsequently, the tip of the sensor was inserted into the same hole to a depth of 3-4 mm. Surgical glue was applied around and on the top of the screw to fixate the probe. The transducer device was then placed under the skin on the back of the mice. The mouse was put back in its cage for recovery.
Doxorubicin and Erlotinib uptake studies
As bioluminescence levels reached 108 p/s/cm2/sr, we administered 9 mg/kg doxorubicin hydrochloride (in 0.9% NaCl; Sigma-Aldrich) by tail-vein injection to SPC-treated mice followed by harvesting of tumor tissue 15 min later (35). We performed intranasal injections of AF-16 (9 μg in PBS/mouse, Phoenix Pharmaceuticals Inc.) peptide 20 minutes prior to doxorubicin injections in a separate cohort of xenografted mice. Mice were perfused and brains were harvested and sectioned as 15 μm sections on glass slides. The slides were counterstained with DAPI and mounted with ProLong Gold antifade reagent (Thermofisher Scientific). Images were acquired using the Zeiss M1 AxioImager and the Zen software. For quantification of doxorubicin, tumor regions were identified and the mean fluorescence intensity of the red channel (emission at 590 nm) was determined using the image analysis tool in Zen. The background was removed on all images to the same level until only nuclear doxorubicin could be found. Three mice in each group (Control, AF, SPC) were analyzed with three brain sections for each mouse and four regions in each brain section. Results were normalized to a sample with no doxorubicin administration.
Erlotinib was dissolved in OraPlus and administered (100 mg/kg) through oral gave to mice followed by dissection of tumor tissue one hour later. We applied previous protocols using LC-MS/MS to quantify Erlotinib (Selleck Chemicals) in tumor isolates (36). Erlotinib-D6 hydrochloride (Toronto Research Chemicals) was used as an internal standard.
Measurement of IFP
Each cage (containing one mouse with an implanted device) was placed on top of a receiver (RPC-1, Data Sciences International), which was connected to a computer equipped with Ponemah Software System (Data Sciences International). Continuous IFP could then be measured in awake and freely moving mice. The devices could be turned on and off when being held close to a magnet (provided by Data Sciences International). Each mouse was recorded for at least two hours each day at the same time. The collected data was analyzed using the Ponemah Software System following protocol provided by DSI.
In vivo MRI
Magnetic resonance imaging (MRI) was performed with a 7T preclinical horizontal bore magnet interfaced with Agilent imaging console. Animals were anaesthetized by inhalation of 2% isoflurane with 98% oxygen and placed into a costume made linear polarized birdcage coil with 21 mm ID. The body temperature of the animals was kept at 35°C during imaging experiments. Monitoring of the heartbeat, breathing and temperature of the animals was done with MRI compatible small animal life support system (SA Instruments Inc, Stony Brook, NY). To identify tumor location in the mouse brain, T2-weighted spin echo MRI was performed with following parameters: Repetition Time (TR)=3 s, Echo Time (TE)=42 ms, 10 slices with 1 mm thickness and ~120 μm in plane resolution, 2 repetitions.
Since diffusion is inversely proportional to water pressure, we hypothesized that high intra-tumoral pressure can be correlated with apparent diffusion coefficient (ADC) of water molecules in the tissue. Measurements of ADC were performed with diffusion weighted spin echo pulse sequence and the following parameters: TR=3 s, TE=42 ms, b=0, 1,027 s/mm2, diffusion gradients were applied along readout direction, 10 slices with 1 mm thickness and ~120 μm in plane resolution. To minimize motion artifacts cardiac and breathing gating was employed. At the end of the imaging experiment ADC map was reconstructed and average ADC values were calculated for the normal and tumor brain tissues. Color ADC maps were created using ImageJ software (NIH image). A conversion of grey-scale into color images was performed with “spectrum” color map.
High intra-tumor pressure can potentially prevent extravasation of a contrast agent into the tumor tissue. To test this hypothesis we performed Dynamic Contrast Enhancement (DCE) experiments. We administered 100 μl 83 mM Gd-DTPA (Magnevist, Bayer, USA) via a catheter in the tail vein. Images were obtained from 4 central slices of tumor tissue with temporal resolution of ~ 14.9 s. Gradient echo pulse sequence was utilized with the same resolution as diffusion images (~120 μm). Parameters of the sequence were TR=29 ms TE=3.6 ms, flip angle=500, total acquisition time ~25 min.
Microscopy
Images were acquired using Zeiss M1 AxioImager or Zeiss 780 confocal microscopes and Zen software. For quantification, tumor regions were identified and region of interest was determined, avoiding the edge of tumor/section and necrotic regions. The mean fluorescence intensity was determined using the image analysis tool in Zen.
Tissue preparation
Brains were fixed for 24h in 10% formalin buffered solution, paraffin-embedded, sectioned at 5 μm, and dehydrated. Frozen tissue sections (20μm) were collected onto glass slides using a cryostat (Leica). Mice were perfused transcardially using PBS followed by 4% paraformaldehyde (PFA). Brains were harvested and incubated overnight in 4% PFA at 4°C, and transferred to 30% sucrose at 4°C. Brains were frozen embedded in O.C.T. compound (Tissue-Tek), and sectioned using a cryostat (Leica) as 15 or 30 μm sections on glass slides (SuperFrost Plus, Fisher Scientific).
Immunohistochemistry
Paraffin-embedded sections were stained for GFAP (Agilent) and Iba1 (Wako) using the Ventana system at the UCSF Neurosurgery Tissue Core. Glass slides with PFA-fixed frozen sections were washed 3×10 min in Tris-buffered saline (TBS, pH 7.4). The sections were then incubated in blocking solution (5% donkey serum, 1% BSA, 0.3% TritonX-100) for 1 h at room temperature. Primary antibodies were diluted in blocking solution and incubated on sections overnight at 4°C. Antibodies used: Cleaved Caspase3 (1:500, Cell Signaling), NKCC1, (1:200, Santa Cruz Technologies), pNKCC1 (1:500, MRC PPU Reagents), CD31 (1:250, BD Biosciences), CD45 (1:500, BD Biosciences), GFAP (1:1000; Thermofisher Scientific), Ki67 (1:100, Novus), AF (1:200, Phoenix Pharmaceuticals Inc.) and Phalloidin (1:100, Thermofisher Scientific). Sections were washed in TBS, and incubated with the appropriate Alexa488 (1:500), 555 (1:500), 647 (1:500) secondary antibodies (Thermofisher Scientific) in blocking solution for 2 h at room temperature. The sections were washed in TBS 3×10 min (DAPI was included in the second wash), following by mounting with ProLong Gold antifade reagent (ThermoFisher Scientific).
Western blotting
For tissue specimen, samples were frozen in dry ice and stored at -80°C until processed. Samples were homogenized and sonicated in cell lysis buffer (Cell Signaling). For cells, samples were washed in ice-cold PBS before lysis with cell lysis buffer (Cell Signaling). All samples were cleared by centrifugation at 16,000 rpm for 10 min at 4°C and quantified using the Pierce BCA protein assay kit (ThermoFisher Scientific). Equal amounts of total protein lysates were loaded and resolved on a 4-12% Bis-Tris Gel with MOPS running buffer and transferred to PVDF membranes. Membranes were blotted with antibodies against NKCC1 (Santa Cruz Technologies), phistone H3, pAKT, and AKT (Cell Signaling), β-ACTIN (BD Biosciences), and GAPDH (Millipore), all used at 1 μg/ml dilution. Antibodies were detected with horseradish peroxidase-linked antibody against mouse (Calbiochem), rabbit (Calbiochem) or goat (Santa Cruz Technology) immunoglobulin G, followed by ECL (Amersham).
Cell volume measurements
To measure approximate values of total cell volume and nucleus volume in GBM xenografts, we used DAPI and Phalloidin488-stained sections and performed z-stacks using a Zeiss 780 confocal microscope. Cell and nuclei volumes were measured using a script in the Imaris 8.0.1 software (Bitplane Inc.). In brief, parameters were set to ‘Detect Nucleus and Cell.’ DAPI was set as the source channel and ‘Nucleus Diameter’ was set to 5.00 μM in order to distinguish individual cell nuclei. ‘Cell Detection Type’ was set to ‘Cell Body Detection’ and the source channel was set to the Phalloidin488 channel. Cell membrane width was set to 0.700 μM. Mitotic cells and apoptotic cell bodies were excluded from analysis.
Proliferation assay
To assess proliferation of human GBM tumorspheres, we plated 0.2 × 104 GBM43 cells per well in NBE media on polyornithine/laminin coated 96-well plates and analyzed the DNA content 0-8 days using the Cyquant NF proliferation assay (Thermofisher Scientific) (37). In brief, cells were lysed and simultaneously incubated at 37 °C with a fluorescent probe to label non-fragmented DNA as an indirect measure of the number of cells. The fluorescence intensity was measured at Ex/Em: 485/530 nm using a Tecan microplate reader. A standard curve was generated by plotting number of plated cells (1,000-40,000) against corresponding fluorescent values, resulting in the equation y=x+150, R2=1. The number of cells in each sample was calculated using this equation. Experiments were done in triplicates.
3D hyaluronic acid gel culture
Cells were plated on non-adherent plates for 4 days to allow formation of tumorspheres. Tumorspheres were then collected, spun down and resuspended in 2% solution of 30% methacrylated 65 kDa hyaluronic acid (generous gift of Dr. Jason Burdick) in complete neurobasal media (-A, Invitrogen; supplemented with 1% v/v B27 supplement (Gibco), 0.5% v/v N2 supplement (Gibco), 20 ng/ml FGF-2 (Peprotech), 20 ng/ml EGF (Sigma-Aldrich), 2 mM L-glutamine, Pencillin/streptavidin). Gels were photo-crosslinked with 0.05% Irgacure 2959 (Sigma-Aldrich) with 365 nm UV light (10 min at a distance of 1.5 inches away from the top of the constructs).
Compression bioreactor
Custom-built compression bioreactor (Tissue Growth Technologies) with 12 individual compression wells operating under closed loop position/force feedback with computer control and custom software was utilized for compression studies. For all experiments, trapezoid input was sent to the servo controller with force ramping from 0 to 30% at a rate of 0.0005 N/s, held at 30% for 48 hours, then ramped down to 0% at a rate of 0.005N/s; at this point the constructs were extracted from the bioreactor and immediately subjected to fixation or RNA/protein sample collection.
RNA/protein isolation from 3D hyaluronic acid gels
Immediately upon completion of compression studies, hyaluronic acid gels were removed from the bioreactor assembly and the triplicates combined in single Eppendorf tubes with either RNA (700 μl) or protein (300 μl) lysis buffers (Cell Signaling). Gels were then crushed mechanically, sonicated briefly, and RNA/proteins were allowed to diffuse out into the supernatant on ice for 2 hours. Subsequently, hyaluronic acid gel-RNA/protein solution was sonicated at maximum speed at 4°C for 10 min; supernatant was then aliquoted into new tubes. Protein samples were treated identically to standard western blot protocol; RNA was isolated using RNAeasy mini kit (Qiagen) and further purified to remove any remaining glycosaminoglycans (Zymo Research); all samples were then flash-frozen on dry ice and stored in -80°C.
Microarray and bioinformatics
Human GBM tumorspheres (GBM5, GBM14, GBM34, and GBM43) embedded in HA-gels were subjected to no compression or compression (48h, 30%) using a compression force bioreactor (Tissue Growth Technologies) with/without incubation with 1 ng/ml AF peptide. We isolated GBM cells and total RNA was prepared as mentioned in qRT-PCR section (Qiagen). The concentration and quality of RNA was determined using Nanodrop followed by use of a RNA 6000 Nano Kit (Agilent) and bioanalyzer chips (Applied Biosystems) to determine RIN values. We then sent 300 ng total RNA (50 ng/ml) to the UCSF Institute for Human Genetics for further processing. In brief, 150 ng was used in the first and second strand cDNA synthesis and IVT cRNA synthesis. We then performed cRNA purification and samples were normalized to 13.33 μg cRNA in 24 μl volume. Samples then go through a second cycle of cDNA synthesis and RNase H hydrolysis. The cDNA was purified and normalized to 5.5 μg cDNA in 31.2 μl volume. We then performed cDNA fragmentation, labeling, and hybridized 90 μl of the sample in the Affymetrix GeneTitan Hybridization Oven instrument for 16 h at 48°C. The arrays were processed with the Affymetrix GeneTitan Fluidics Station instrument, using the HuGene-2_1-st fluidics script and GeneTitan Hybridization, Wash, and Stain Kit for WT Array Plates. Then, the arrays were scanned in the Affymetrix GeneTitan Multi-Channel (MC) instrument with the AGCC GeneTitan Instrument Control. Quality control of samples was performed using the Affymetrix Expression console and data was initially analyzed using Affymetrix Transcriptome Analysis Console. CEL files were normalized by RMA using the oligo package in R (38). Differential expression was assessed by paired t-test using the samr package with a 5% False Discovery Rate threshold (39), and Gene Ontology enrichment analysis was assessed using BiNGO (40). Raw microarray data for this study has been deposited at the Gene Expression Omnibus (GEO) repository under accession GSE81473.
Volume measurements of tumorspheres
We first diluted 2× concentrated 70 μm hyaluronic acid (HA) with neurobasal media, added igracure, and mixed 1:1 with 20-30 human GBM tumorspheres (100-200 μm diameter spheres four days after plating) preceding polymerization for seven minutes. Whole cell-embedded hyaluronic acid gels were stained with DAPI or Alexa Fluor 488 Phalloidin (ThermoFisher Scientific) and imaged (50 μm confocal z stacks with 0.2 μm z-steps) on a Nikon Eclipse Ti-E base microscope equipped with the Yokogawa CSU-X1 confocal scanner and Andor’s iXon3 EMCCD camera. Cell volume was subsequently quantified using 3D Suite of Image J Software (NIH).
Calcein-AM-based cell volume measurements
Cell volume change was determined using calcein as a marker of intracellular water volume, which was established previously (41). Briefly, cells on coverslips either coated with poly-L-lysine (50 μg/ml) (GC22) or polyornithine (50 μg/ml) and laminin (5 μg/ml) (GBM43) were incubated with 0.5 μM calcein-AM for 30 min at 37°C. The cells were placed in a heated (37°C) imaging chamber (Warner Instruments) on a Nikon Ti Eclipse inverted epifluorescence microscope equipped with perfect focus, a 40× Super Fluor oil immersion objective lens, and a Princeton Instruments MicroMax CCD camera. Calcein fluorescence was monitored using a FITC filter set (excitation 480 nm, emission 535 nm, Chroma Technology). Images were collected every 60 s with MetaFluor image-acquisition software (Molecular Devices). Regions of interest (0.5 μm in diameter) were selected in the cytoplasm of 5~15 cells. Baseline drift resulting from photobleaching and dye leakage was corrected as described before. The fluorescence change was plotted as a function of the reciprocal of the relative osmotic pressure and the resulting calibration curve applied to all subsequent experiments as previously described (41). The HEPES buffered isotonic solution contained (in mM, pH 7.4): 100 NaCl, 5.4 KCl, 1.3 CaCl2, 0.8 MgSO4, 20 HEPES, 5.5 glucose, 0.4 NaHC03, and 70 sucrose with 310 mOsm determined using an osmometer (Advanced Instruments). Anisosmotic solutions (262 and 432 mOsm) were prepared by removal or addition of sucrose.
Use of Clophensor for chloride imaging
ClopHensor is based on a chloride-sensitive GFP and can be combined with real-time optical detection of chloride fluxes in living cells. Clophensor can uniquely compensate for pH changes that are known to influence chloride levels (42). Electroporation of the ClopHensor plasmid was used to induce ClopHensor expression into cultured GBM cells. Cells were maintained in a humidified atmosphere at 37°C containing 5% CO2 during microscopy analysis. Wide field fluorescence images were acquired using a customized iMIC (Till Photonics) microscope equipped with a 0.65 NA plan-N 40× air objective (Olympus), a high stability xenon lamp and a 14-bit CCD camera (Stingray F145B-30fps, Allied Vision Technologies). The excitation filters 458/10 (Chroma), 482/18 (BrightLine HC) and 563/9 (Semrock) were mounted on the lamp fast filter switching device, and a dual channel filter set (dichroic: HC-Beamsplitter BS R488/561, emitter: Brightline HC 523/610, Semrock) was used in the filter cube. Thus, cyan, green and red channels were respectively acquired with excitation (emission) wavelengths at 458 ± 10 (525 ± 25), 482 ± 18 (525± 25), and 563 ± 9 (610 ± 30) nm. Intracellular chloride concentration was measured in time-lapse from cells exposed to hypertonic stress in the following sequence: 5 min isotonic HEPES-MEM (317 mOsm), 20 min hypertonic HEPES-MEM (432 mOsm), and 10 min isotonic HEPES-MEM. Cells were pre-incubated for 10 min in isotonic condition before time-lapse acquisition. BMT (10 μM) and AF peptide (5 μg/ml) were pre-incubated and present throughout the experiment. Osmolarity was measured using a cryoscopic osmometer (Osmomat 030, Gonotec GmbH). Three-channel images were acquired every 60 s and ratio analysis was performed as previously described (42), where chloride concentration and pH were calculated from the cyan-over-red (chloride) and the green-over-cyan (pH) ratios.
Statistical analysis
Statistical tests were performed using GraphPad Prism v6.0 software. Statistical analyses for experiments with two groups were performed using Student’s t-test. Statistical analyses for experiments with more than two groups were performed using one-way ANOVA followed by posthoc tests (for in vitro proliferation and therapy studies). Kaplan Meier survival analysis was conducted and the p value of the comparison between survival curves was determined to be significant by the log-rank (Mantel-Cox) test.
Results
AF reduces IFP and increases MRI-based ADC levels in GBM xenografts
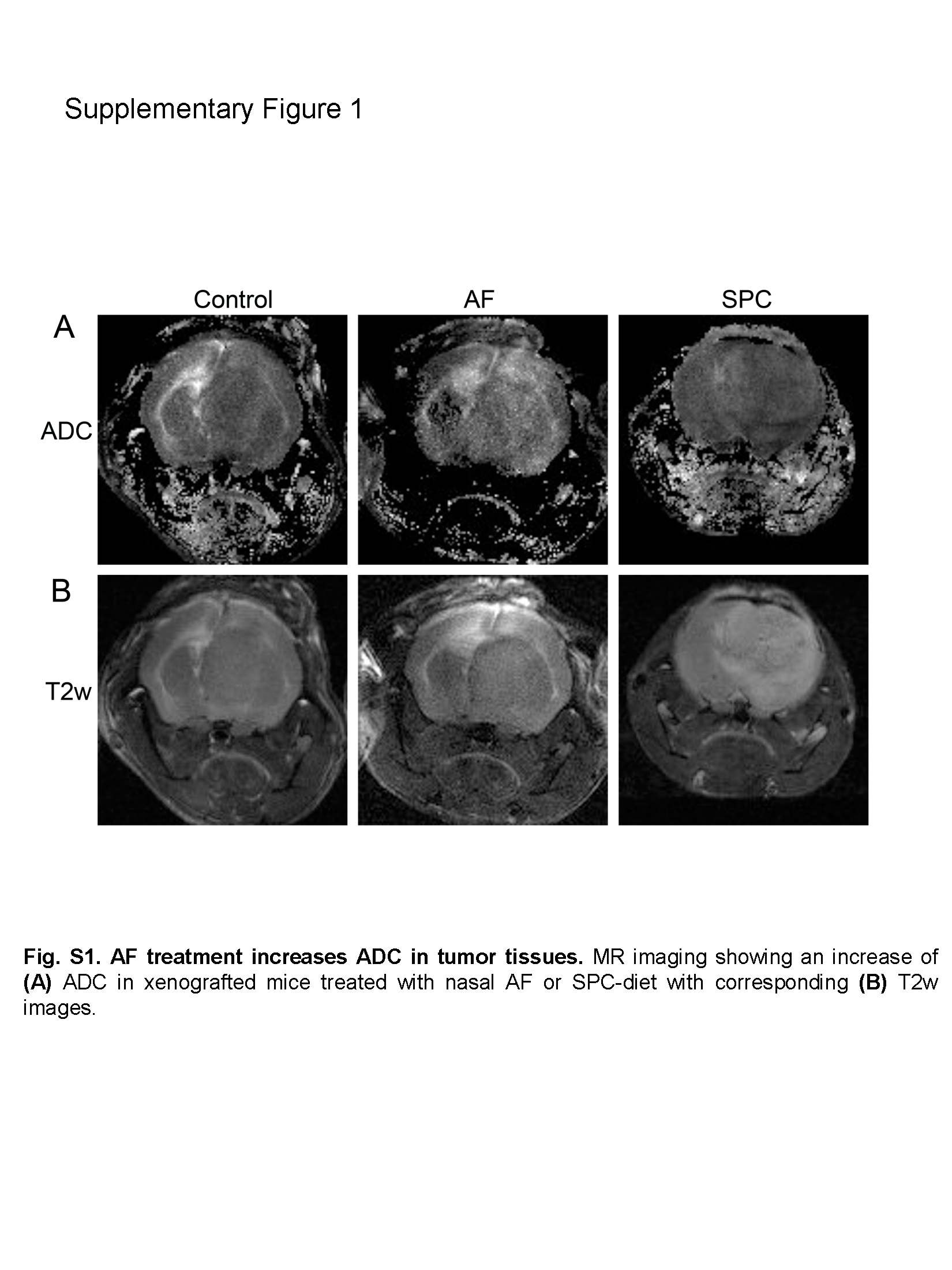
To assess how AF treatment impacted fluid dynamics, we administered a diet containing 10% SPC (to induce AF) beginning six days following orthotopically engraftment of patient-derived GBM43 cells into athymic mice, and then analyzed effects by MRI. Treatment with SPC diet significantly increased apparent diffusion coefficient (ADC) levels in tumors, a measure of fluid movement on MRI (Fig. 1A and B, Supplementary Fig. S1A and B). To confirm that increased ADC levels following SPC treatment were mediated by AF, we next attempted nasal delivery of AF peptide, a preclinical method for delivery of therapeutics (43). We next repurposed telemetry-based methodology (44) to measure IFP during tumor progression by injecting luciferase (LUC)- expressing GBM43 cells into the frontal cortex followed by immediate implantation of a pressure sensor (Fig. 1C). A radiofrequency signal emitted from the pressure sensor transmitter placed within intracranial human GBM43 xenografts enabled parallel measurements of IFP and LUC during tumor progression (Fig. 1D and E). We observed no changes in the numbers of tumor-associated macrophages or reactive astrocytes following insertion of the pressure sensor (Supplementary Fig. S2A to D). Immunostainings using an antibody recognizing the antisecretory region of the AF protein showed that SPC diet increased expression AF protein levels in tumor cells but not in the surrounding brain (Fig. 1F). We observed no induction of AF protein in CD31+ tumor vasculature and GFAP+ reactive astrocytes, and low levels in CD45+ tumor-associated immune cells (Fig. 1F). Administration of SPC diet suppressed IFP levels compared to animals administered control diet (Fig. 1G). Similarly, IFP levels were normalized for up to 30 min after nasal administration of AF peptide (Fig. 1H). Thus, AF therapy represents a novel IFP-reducing strategy.
Fig. 1. AF increases MRI-based ADC levels and lowers IFP in GBM xenografts.
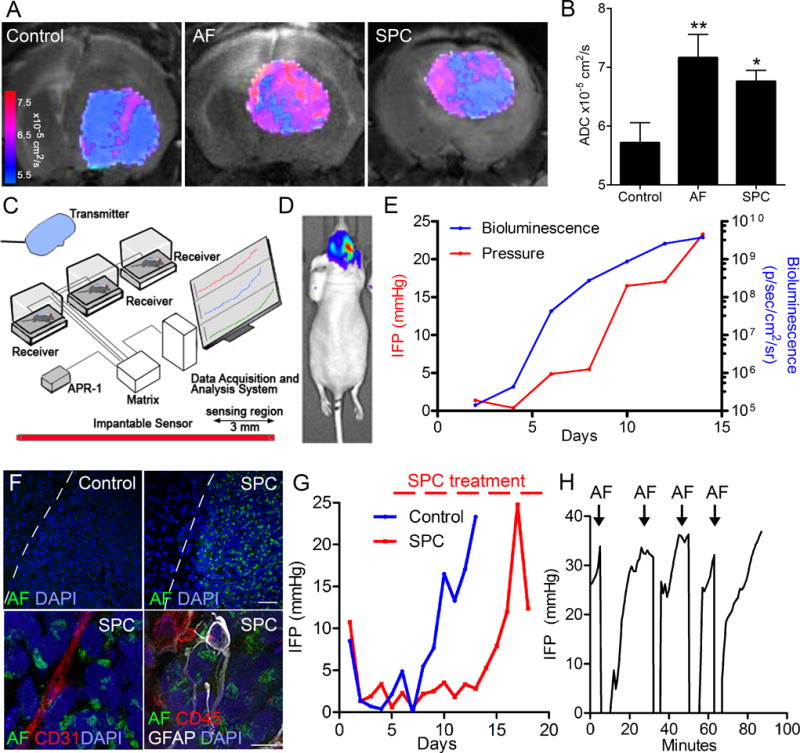
(A–B) MRI-based measurements of ADC levels following acute nasal injections of AF peptide or chronic induction of AF protein (SPC diet) showed increased ADC levels in GBM43 xenografts versus control. (C) Setup for telemetry-based recordings of IFP in GBM xenografts. (D–E) Parallel measurements of tumor growth (bioluminescence) and IFP (mmHg) in GBM43 xenografts. (F) Immunoreactivity for AF protein ten days following administration of SPC diet to GBM43 xenografts showing high expression in tumor cells (dotted line; tumor border) but not in CD31+endothelial cells, GFAP+ reactive astrocytes, or CD45+ immune cells. Scale bar: top panel 50 μm, bottom panel 10 μm. (G) IFP measurements in GBM43 xenografts. SPC diet lowered IFP levels versus control. (H) Nasal administration of AF peptide effectively reduced IFP levels for 10-30 minutes. *P < 0.05, **P < 0.01.
AF increases drug uptake in GBM
Administration of SPC diet increased uptake of the contrast agent gadolinium and reduced clearance in tumor tissue of GBM43 xenografts compared to controls (Supplementary Fig. S3A and B), suggesting that reduced IFP levels promote drug uptake. The uptake of doxorubicin, a topoisomerase II inhibitor can be assayed based on its fluorescence (35). Doxorubicin (IV injection) demonstrated increased (seven-fold) uptake in nuclei of tumor cells following administration of SPC or nasal injections of AF peptide in GBM43 xenografts (Fig. 2A to E), resulting in increased survival of mice (Fig. 2F). Moreover, we observed increased uptake of the EGFR inhibitor Erlotinib along with extended survival following SPC treatment in GBM43 tumors (Supplementary Fig. S3C to E). As a consequence of AF’s ability to lower IFP levels, AF represents a novel therapeutic to promote drug uptake in GBM.
Fig. 2. AF treatment increases doxorubicin uptake in GBM xenografts.
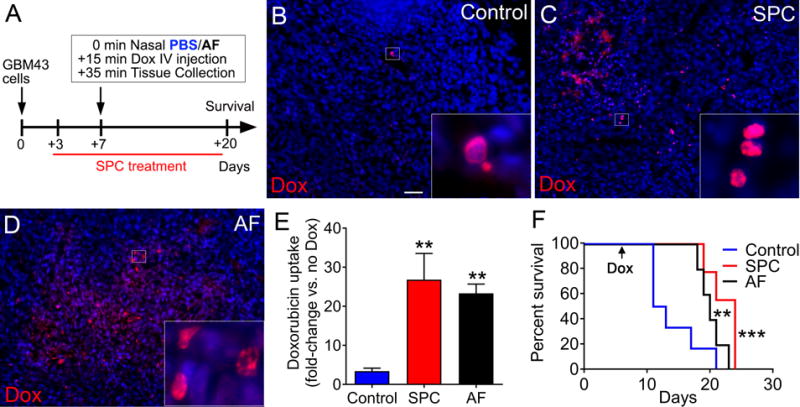
(A) Mice xenografted with GBM43 cells were given a combination of doxorubicin (Dox) and SPC diet or nasal AF. (B–D) Doxorubicin (red) and DAPI (blue) in tumors 20 min after tail-vein injection of doxorubicin. Scale bar: 40 μm. (E) Quantification of fluorescence (red) shows increased uptake of doxorubicin in tumor tissues of mice treated with SPC diet or nasal AF (n=3, **P < 0.01, one-way ANOVA). Kaplan-Meier (n=6 for control, n=5 for AF, and n=9 for SPC, **P < 0.01, ***P < 0.001; one-way ANOVA for control v/s AF and SPC, respectively) curve showing survival benefit following combination treatments with doxorubicin and SPC diet or nasal AF.
AF treatment inhibits GBM tumor growth
GBM xenografts typically showed rapid elevation of IFP during late stages of tumor development (Fig. 3A). In contrast, mice administered SPC diet delayed increases in bioluminescence and maintained low IFP in GBM43 xenografts (Fig. 3A). As tumor growth and IFP eventually increased in SPC-treated mice, we observed a characteristic drop in IFP at terminal stages of tumor development. Tumor tissue from SPC-treated mice showed reduced proliferation (Ki67) and induction of apoptosis (cleaved caspase 3) compared to control tumors (Fig. 3B and C). Endogenous AF protein induction through SPC treatment extended overall survival in orthotopic GBM43 xenografts (Fig. 3D). Exogenous administration of AF protein was achieved by dissolving Salovum (500-fold enrichment of AF protein in sterile egg yolk powder) in the drinking water. Similarly to SPC diet, Salovum treatment increased survival in GBM43 xenografts (Fig. 3D). SPC diet and Salovum treatment had no effect on body weight (Supplementary Fig. 4A and B). In contrast to mesenchymal GBM43 tumors that display NF1 mutations, proliferative GBM6 tumors express EGFRvIII. SPC diet and Salovum treatment significantly increased survival in GBM6 xenografts (Fig. 3D). To test whether AF therapy shows anti-tumor activity in an immuno-competent GBM model, we administered SPC diet to mice allografted with Ink4a/Arf null neural stem cells expressing human EGFRvIII, mCherry, and LUC (Fig. 3E) (34). Treatment with SPC diet induced AF protein in mCherry+ tumors and resulted in massive apoptosis (Fig. 3F and G). Complete tumor regression was observed in 5/6 allografted mice following treatment with SPC diet (Fig 3H and Supplementary Fig. 4C). Thus, AF treatment reduced proliferation and induced apoptosis of GBM cells, ultimately leading to reduced tumor growth and extended survival.
Fig. 3. AF treatment reduces tumor growth in GBM models.
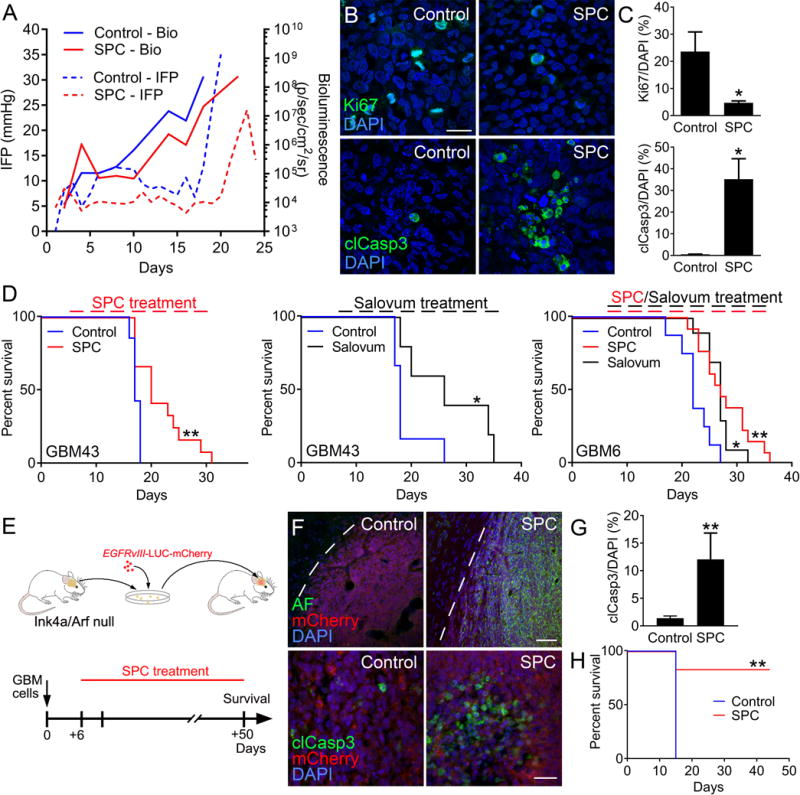
(A) Parallel IFP and bioluminescence measurements showed that treatment with SPC diet reduced IFP levels and tumor growth compared to control GBM43 xenografts. (B–C) SPC treatment reduced proliferation (Ki67) and induced apoptosis (cleaved caspase 3) in GBM43 xenografts. Scale bar: 30 μm. (n=3, *P < 0.05, Student’s t test) (D) Kaplan-Meier curves showing increased survival in GBM43 xenografts treated with SPC diet (n=7 for control and n=12 for SPC, **P < 0.01; Log-rank Mantel-Cox test) or Salovum (n=7 for control and n=5 for Salovum, *P < 0.05; Log-rank Mantel-Cox test) compared to control. SPC diet and Salovum also increased survival in EGFRvIII-expressing GBM6 xenografts (n=8 for control, n=13 for SPC, and n=10 for Salovum *P < 0.05, **P < 0.01; one-way ANOVA). (E) Immunocompetent GBM model based on established neurospheres from the subventricular zone of Ink4a/Arf null FVBn mice that were lentivirally transduced to express human EGFRvIII, mCherry, and LUC. Mouse GBM cells were allografted into adult FVBn mice and administered SPC diet on day 6. (F–G) SPC treatment resulted in AF induction in tumor cells (dotted line; tumor border) and induction of apoptosis (cleaved caspase 3). Scale bar: top panel 50 μm, bottom panel 20 μm. (n=3, **P < 0.01, Student’s t test). Kaplan-Meier (n=5 for control and n=5 for SPC, **P < 0.01; Log-rank Mantel-Cox test) curve showing increased survival following SPC treatment in GBM43 xenografts.
AF inhibits NKCC1 activity and osmotic adaption of GBM cells
We have previously demonstrated that the NKCC1 inhibitor bumetanide prevents restoration of cell volume by reducing transport of sodium, potassium, and chloride ions across the plasma membrane in GBM cultures (13). As a regulator of chloride permeability across neuronal membranes, AF protein or its derived octapeptide regulated permeability of chloride ions across neuronal membranes (45,46). To test whether AF induction similar to bumetanide regulates cell volume in GBM cells (13,47), we analyzed tumor cell volume following SPC treatment in GBM43 xenografts. Actin stainings showed that AF induction in tumor cells reduced cell growth (size) at terminal stages compared to control tumors (Fig. 4A and B). Measurements of actin (Phalloidin488) stainings from z-stacks with Imaris software provided an approximation for cell volumes (Fig. 4C and D). Tumor cells in SPC-treated mice displayed reduced relative values of total cell and nuclear volumes compared to controls (Fig. 4E and F). Using an antibody that recognizes residues 198-217 of human NKCC1, including the Thr203, Thr207, and Thr212 phosphorylation sites, we found reduced immunoreactivity in tumor cells of SPC treated GBM43 xenografts (Fig. 4G and H). To further evaluate the effects of AF on cell volume regulation of GBM cells in vitro, we loaded cultured GBM43 cells with cell-permeable calcein dye (calcein-AM), an established marker of intracellular cell volume (41). Treatment with AF peptide (5 μg/ml) or bumetanide (10 μM) inhibited regulatory volume increase of GBM43 cells grown under hypertonic conditions (Fig. 4I). In contrast to bumetanide-treated cells, tumor cells incubated with AF peptide failed to surpass the baseline when returned to isotonic conditions (Fig. 4J and K). AF peptide and bumetanide also prevented restoration of cell volume in GC22 (GBM) cultures (Supplementary Fig. S5A to C). We have shown that bumetanide regulates chloride permeability in GBM cells (13). Electroporation of GBM43 cells with the ratiometric chloride sensor ClopHensor (42) demonstrated that AF reduced chloride permeability when tumor cells were transferred to hypertonic conditions (Fig. 4L and M). Thus, AF’s ability to reduce NKCC1 activation in vivo and its paralleled effects with bumetanide on osmotic adaptation in vitro, provide a rationale for reduced cell survival in AF-treated GBM xenografts.
Fig. 4. AF prevents restoration of cell volume and chloride permeability under hyperosmolar conditions.
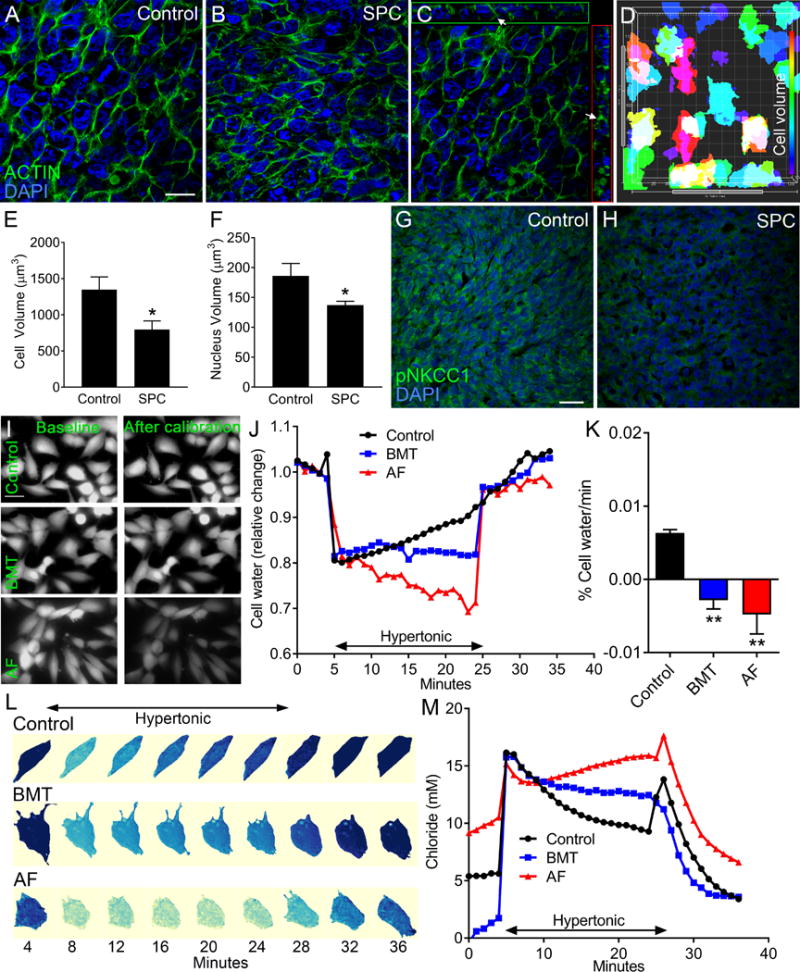
(A–F) Phalloidin (F-actin, green) and DAPI (nuclei, blue) stainings show significantly reduced cellular and nuclear volumes after SPC treatment of mice xenografted with GBM43 cells. (C) Orthogonal view show that Phalloidin (arrows) is expressed around DAPI+ nuclei. (D) Imaris software was used to demonstrate a correlation between approximate values for cell (Phalloidin, F-actin) and nuclear (DAPI) volumes in GBM cells. Scale bar: 20 μm. (n=3, *P < 0.05, Student’s t test). (G–H) SPC treatment reduced immunoreactivity of phosphorylated NKCC1 (pNKCC1) in GBM43 tumors. (I) AF peptide (5 μg/ml) and bumetanide (BMT, 10μM) reduced fluorescence of excited calcein-AM dye when GBM43 cells were transferred to hypertonic conditions (432 mOsm). Scale bar: 10 μm. (J) Cell shrinkage was induced with hypertonic exposure in both BMT and AF peptide treated cell with no regulatory volume increase response recorded within 20 minutes of hypertonic exposure. (K) Quantification of regulatory volume increase data. (n=3, *P < 0.05, one-way ANOVA) (L) Time-lapse recordings of intracellular chloride concentrations in GBM14 cells in the presence of BMT or AF peptide. (M) Quantifications following adjustment for pH changes showing changes in [Cl−] following incubation with BMT or AF peptide versus control (median values of 30, 39 and 39 time-lapse experiments for BMT, AF and control, respectively).
Compression-induced GBM cell proliferation is blocked by AF in a NKCC1-dependent manner
Total and phosphorylated NKCC1 were expressed on patient-derived GBM cells (Fig. 5A, Supplementary Fig. S6A). Immunoblotting confirmed that NKCC1 protein was expressed in human GBM tumorsphere cultures (Fig. 5B). Incubation of adherent human GBM cells with AF peptide had no effect on cell proliferation (Fig. 5C, Supplementary Fig. S6B). To mimic conditions during progression, human primary GBM cells were expanded as tumorspheres for four days and mixed in a hyaluronic acid-based 3D matrix followed by compression (30%) for 48 hours using a force bioreactor (Fig. 5D). Compression significantly expanded the average tumorsphere size across GBMs (GBM5, GBM14, GBM43) (Fig. 5E). Smaller spheres showed even further expansion in response to compression (Supplementary Fig. S7A to D). However, AF peptide blocked compression-induced expansion of GBM tumorspheres following static compression (30%) for 48 hours (Fig. 5F).
Fig. 5. NKCC1 is necessary for AF-mediated compression-induced proliferation in GBM tumorspheres.
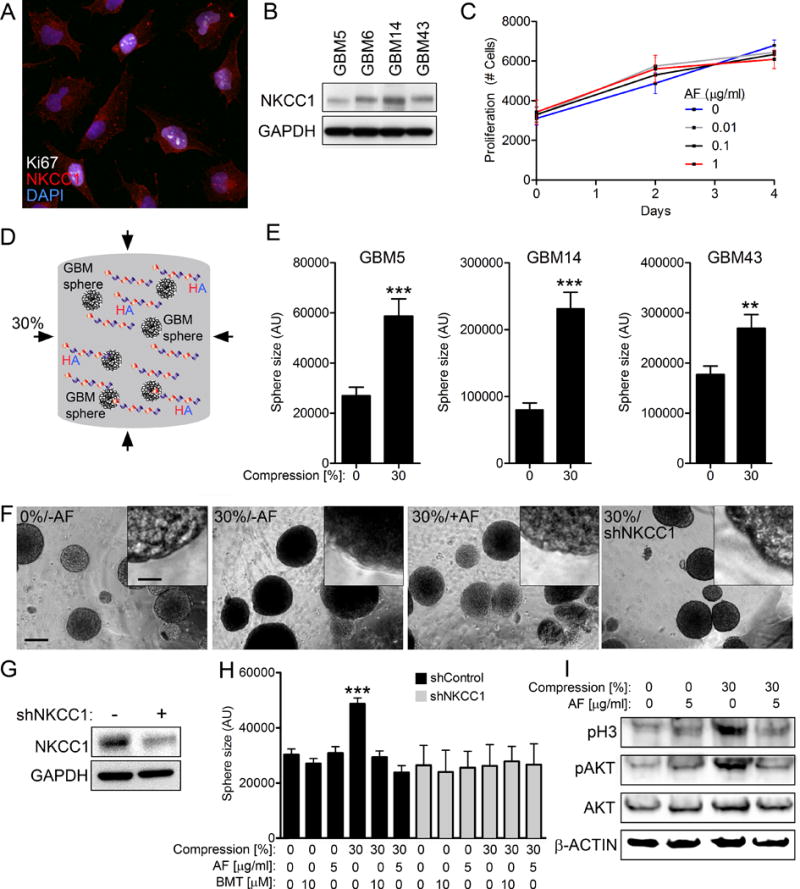
(A) NKCC1 is expressed in human GMB43 cells at the cell membrane and in the cytoplasm. Scale bar: 10μm. (B) NKCC1 protein is expressed in four human GMB tumorsphere cultures. (C) AF treatment has no proliferative effects on GBM43 cells grown under atmospheric pressure. (D) Human GBM tumorspheres were grown under static compression (30%) for 48 hours in HA-gels. (E) Tumorsphere size was measured following 48 h compression (30%) for human GBM tumorspheres grown in 3D HA-gels. (n=3, **P < 0.01, ***P < 0.001, Student’s t test). (F) AF and NKCC1 knockdown reversed compression-induced expansion of tumorsphere size. Scale bar: 100 μm (insert 7 μm). (G) Knockdown of NKCC1 by shRNA in GBM43 cells. (H) shRNA-mediated knockdown of NKCC1 and the NKCC1 inhibitor bumetanide (BMT) restored compression-induced expansion of GBM tumorspheres. No further effects of AF or BMT were found in shNKCC1 cells. (I) AF peptide reverses the protein levels of pH3 and pAKT in cells subjected to static pressure (30%) for 48 hours. (n=3, ***P < 0.001, one-way ANOVA).
Similarly, shRNA-mediated knockdown of NKCC1 or the FDA-approved NKCC1 inhibitor bumetanide restored compression-induced expansion of GBM tumorspheres (Fig. 5F–H). The average tumorsphere size was not further reduced when shRNA NKCC1 cells were incubated with AF peptide or bumetanide, consistent with NKCC1 as a target of both agents (Fig. 5H). The normalizing effect of AF peptide on compression-induced tumorsphere size was attributed to restored cell proliferation since levels of phosphorylated mitotic proteins histone H3 (pH3) and AKT were normalized (Fig. 5I). Transcriptional profiling of patient-derived GBMs (GBM5, GBM14, GBM43, GBM34) showed that compression significantly altered the expression profile of >3,000 genes (Supplementary Fig. 7E and F). Expression profiles of compressed cells incubated with AF peptide were not distinguishable from cells under atmospheric conditions. Compression preferentially regulated genes involved in translational control (SNORA60, SNORA75, MIR21), stress response (ATG5, FOSB, GADD45B), and members of the solute carrier family (SLCA5P2, SLC1A4, SLC9A6) (Table S1). Thus, our data show that NKCC1 function is necessary for compression-induced proliferation and transcriptional changes in GBM cells, a phenotype that was completely reversed by AF peptide.
A combination of AF and temozolomide treatment blocks progression and regrowth in GBM xenografts
Temozolomide is an integral part of standard care in GBM patients (1). We have previously demonstrated that bumetanide-mediated NKCC1 inhibition accelerated temozolomide-induced apoptosis and reduced temozolomide-induced migration in human GBM cultures (13,47). Temozolomide alone, or in combination with SPC diet effectively reduced bioluminescence levels in GBM43 xenografts (Fig. 6A and B). However, temozolomide-treated GBM43 xenografts underwent regrowth and 70% succumbed to the disease displayed 30-120 days post-injection (Fig. 6C to E). In contrast, mice administered temozolomide in combination with SPC showed cessation of tumor growth and 100% overall survival at 120 days, compared to either monotherapy. These data demonstrate that AF therapy, similar to NKCC1 inhibition in vitro, accelerates temozolomide-induced apoptosis of GBM cells, preventing tumor regrowth in xenografted mice.
Fig. 6. Combined AF and temozolomide treatment blocks regrowth in GBM xenografts.
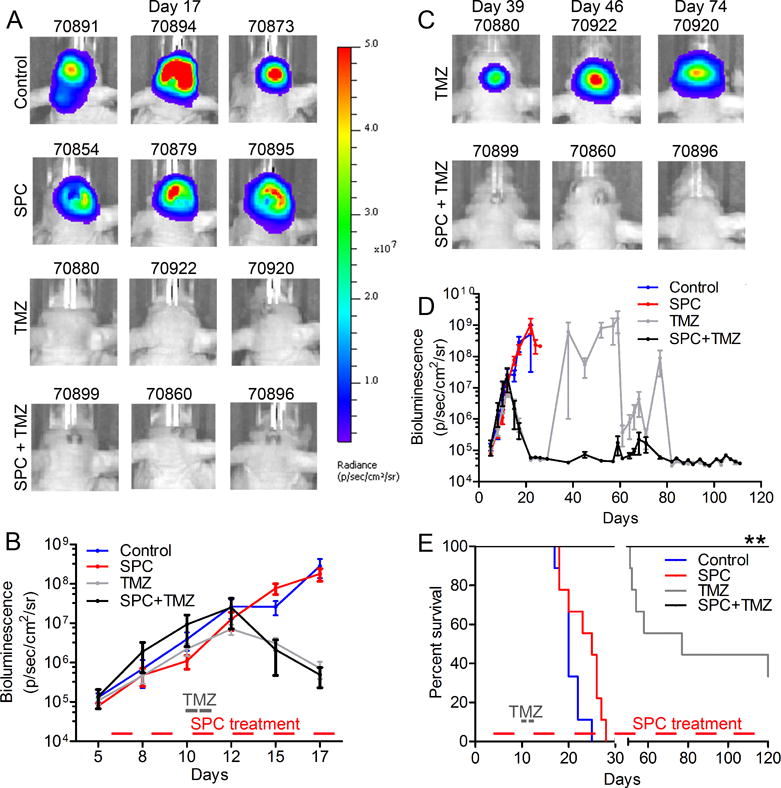
(A) Bioluminescence data of mice treated with control diet, SPC, TMZ (5 mg/kg, 2 consecutive days) alone, or in combination. (B) TMZ alone or in combination with SPC diet effectively reduced tumor growth at early stages of tumor development. (C–D) Bioluminescence showing regrowth of tumors following TMZ treatment, but not after a combination of TMZ and SPC diet. (E) Kaplan-Meier (n=9 for control, n=8 for SPC, n=9 for TMZ and n=7 for SPC + TMZ, **P < 0.01; Log-rank Mantel-Cox test) curve displays regrowth after ~50 days post-TMZ treatment. Tumors in mice treated with a combination of SPC and TMZ never displayed regrowth.
Discussion
High IFP drives compression and osmotic swelling in solid tumors. In patient-derived GBM xenografts, we used telemetry-based methodology to show that IFP levels parallel tumor growth during progression. Here, we show that AF therapy reduced IFP and increased drug uptake in GBM xenografts. Collectively, AF’s effects on osmotic adaptation in vitro, its ability to reduce NKCC1 activation in vivo, and its inability to block compression-induced proliferation in NKCC1 knockdown cells, suggest that intact NKCC1 function is necessary for AF-mediated anti-tumor activity in GBM. These results suggest that AF therapy represents an IFP-reducing approach that should be tested in clinical trials for GBM patients.
Our data suggest that AF regulates osmotic adaptation by inhibiting NKCC1 function, resulting in reduced survival and increased chemosensitivity of GBM cells. The activity of NKCC1 is stimulated by low chloride ion levels during swelling in astrocytes (48). NKCC1 promotes proliferation via decreasing intracellular chloride concentrations, driving efflux through chloride ion channels and triggering membrane depolarization and voltage-sensitive calcium influx (49). The neurotransmitter GABA also regulates osmotic swelling in neural stem cells through type A receptor GABAAR signaling (50). NKCC1 knockdown or bumetanide treatment inhibits GABAAR-induced depolarization and reduced proliferation of neural progenitors (51). Interestingly, low levels of AF peptide reduced GABA diffusion and chloride ion conductance across cell membranes (29,45). It is therefore plausible that AF and bumetanide treatment regulate osmotic adaptation and proliferation by converging on GABAARs in human GBMs.
Anti-angiogenic therapies effectively reduce IFP levels in solid tumors but can also contribute to invasive recurrent tumors. We found that SPC treatment induced endogenous AF protein in tumor cells but not in the surrounding tumor vasculature (Fig. 1F). Previous studies showed that NKCC1 knockdown or bumetanide-treatment disrupted migration of GBM cells (11,47). Interactions between activated NKCC1 and ezrin, acting as a linker between membrane proteins and the actin cytoskeleton, were critical to promote invasion of GBM cells (11). Thus, osmotic regulators that inhibit NKCC1 function on tumor cells rather than tumor vasculature may achieve the benefits of anti-angiogenic therapies without also contributing invasive resistance.
Activated by growth factor signaling pathways genetically altered in a majority of GBMs and transforming when overexpressed (52), NKCC1 is emerging as a contributor to GBM aggressiveness. The expression of NKCC1 along with kinases including upstream with-no-K (Lysine) kinase family members, oxidative stress response kinase (OSR1), and ste-20-related, proline-alanine-rich kinase (SPAK) all increase with glioma grade (11,12). Administration of oral SPC takes advantage of an endogenous response, leading to induction of AF protein in regions of active fluid secretion and osmotic imbalance. Based on our findings and previous work, blood brain barrier permeable small molecule inhibitors that target NKCC1 or its upstream kinases represent a promising strategy to block osmotic adaptation of tumor cells, to improve outcome in GBM patients.
The interplay between extracellular matrix mechanics and fluid pressure in solid tumors remains unclear. Here, we found that AF-mediated reduction in IFP was paralleled by reduced swelling of GBM cells. More work is needed to determine to what extent osmotic pressure in tumor cells contributes to elevated IFP and poor drug uptake in solid cancers. Although our studies suggest that endogenous AF induction and exogenous AF administration inhibit NKCC1 activation, the systemic action on NKCC1 function and other potential genes need further clarification. Preceding clinical trials, additional studies are needed to determine the optimal treatment regimen to achieve AF-mediated NKCC1 inhibition in GBMs.
Here, we report that AF-mediated inhibition of osmotic adaptation of GBM cells reduced IFP levels and tumor growth. These findings imply that osmotic adaptation is critical for survival of GBM cells. As a result, strategies that regulate osmotic adaptation emerge as tractable approaches to improve outcome in cancer patients.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank patients for giving their consent and the staff at the UCSF Neurosurgery Tissue Core for providing biopsies. We also thank Stefan Lange at Sahlgrenska University Hospital in Sweden for providing information regarding AF function.
Financial support: Supported by U54CA163155 (A.I.P., W.A.W., V.M.W.), R21NS088114 (A.I.P.), R01NS091620 (W.A.W.), 1U01CA217864 (W.A.W), R01NS081117 (J.J.P), R01NS075995 (D.S.), Children’s Tumor Foundation (W.A.W.), the Samuel Waxman Cancer Research (W.A.W.), the National Brain Tumor Society (A.I.P.), the Loglio Collaborative (M.S.B, A.I.P., W.A.W.), the TDC Foundation (A.I.P.), and the Guggenhime Endowment Fund (A.I.P.), the Swedish Brain Foundation (S.I.), the Swedish Childhood Cancer Foundation (S.I. and O.R.L.) and the Foundation BLANCEFLOR Boncompagni Ludovisi (S.I. and H.S.), the Swedish Research Council (H.S.), the European Molecular Biology Organization (H.S.), and the American Brain Tumor Association Basic Research Fellowship in memory of Joel A. Gingras, Jr (H.S).
Footnotes
Conflicting interests: The authors have no financial or competing interest. Dr. Persson is a co-author on patent WO2005030246 A1 entitled “Novel use of antisecretory factor”.
References
- 1.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Alberti E, Hartmann A, Schutz HJ, Schreckenberger F. The effect of large doses of dexamethasone on the cerebrospinal fluid pressure in patients with supratentorial tumors. J Neurol. 1978;217(3):173–81. doi: 10.1007/BF00312958. [DOI] [PubMed] [Google Scholar]
- 3.Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure - an obstacle in cancer therapy. Nat Rev Cancer. 2004;4(10):806–13. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- 4.Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell. 2012;22(1):21–35. doi: 10.1016/j.ccr.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Habela CW, Ernest NJ, Swindall AF, Sontheimer H. Chloride accumulation drives volume dynamics underlying cell proliferation and migration. J Neurophysiol. 2009;101(2):750–7. doi: 10.1152/jn.90840.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watkins S, Sontheimer H. Hydrodynamic cellular volume changes enable glioma cell invasion. J Neurosci. 2011;31(47):17250–9. doi: 10.1523/JNEUROSCI.3938-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ernest NJ, Habela CW, Sontheimer H. Cytoplasmic condensation is both necessary and sufficient to induce apoptotic cell death. J Cell Sci. 2008;121(Pt 3):290–7. doi: 10.1242/jcs.017343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jayakumar AR, Norenberg MD. The Na-K-Cl Co-transporter in astrocyte swelling. Metab Brain Dis. 2010;25(1):31–8. doi: 10.1007/s11011-010-9180-3. [DOI] [PubMed] [Google Scholar]
- 9.Thrane AS, Rappold PM, Fujita T, Torres A, Bekar LK, Takano T, et al. Critical role of aquaporin-4 (AQP4) in astrocytic Ca2+ signaling events elicited by cerebral edema. Proc Natl Acad Sci U S A. 2011;108(2):846–51. doi: 10.1073/pnas.1015217108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ding T, Zhou Y, Sun K, Jiang W, Li W, Liu X, et al. Knockdown a water channel protein, aquaporin-4, induced glioblastoma cell apoptosis. PLoS One. 2013;8(8):e66751. doi: 10.1371/journal.pone.0066751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garzon-Muvdi T, Schiapparelli P, ap Rhys C, Guerrero-Cazares H, Smith C, Kim DH, et al. Regulation of brain tumor dispersal by NKCC1 through a novel role in focal adhesion regulation. PLoS Biol. 2012;10(5):e1001320. doi: 10.1371/journal.pbio.1001320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haas BR, Cuddapah VA, Watkins S, Rohn KJ, Dy TE, Sontheimer H. With-No-Lysine Kinase 3 (WNK3) stimulates glioma invasion by regulating cell volume. Am J Physiol Cell Physiol. 2011;301(5):C1150–60. doi: 10.1152/ajpcell.00203.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Algharabil J, Kintner DB, Wang Q, Begum G, Clark PA, Yang SS, et al. Inhibition of Na(+)-K(+)-2Cl(-) cotransporter isoform 1 accelerates temozolomide-mediated apoptosis in glioblastoma cancer cells. Cell Physiol Biochem. 2012;30(1):33–48. doi: 10.1159/000339047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flamenbaum W, Friedman R. Pharmacology, therapeutic efficacy, and adverse effects of bumetanide, a new “loop” diuretic. Pharmacotherapy. 1982;2(4):213–22. doi: 10.1002/j.1875-9114.1982.tb03188.x. [DOI] [PubMed] [Google Scholar]
- 15.Tollner K, Brandt C, Topfer M, Brunhofer G, Erker T, Gabriel M, et al. A novel prodrug-based strategy to increase effects of bumetanide in epilepsy. Ann Neurol. 2014;75(4):550–62. doi: 10.1002/ana.24124. [DOI] [PubMed] [Google Scholar]
- 16.Lonnroth I, Lange S. Purification and characterization of the antisecretory factor: a protein in the central nervous system and in the gut which inhibits intestinal hypersecretion induced by cholera toxin. Biochim Biophys Acta. 1986;883(1):138–44. doi: 10.1016/0304-4165(86)90144-3. [DOI] [PubMed] [Google Scholar]
- 17.Hanner P, Rask-Andersen H, Lange S, Jennische E. Antisecretory factor-inducing therapy improves the clinical outcome in patients with Meniere’s disease. Acta Otolaryngol. 2010;130(2):223–7. doi: 10.3109/00016480903022842. [DOI] [PubMed] [Google Scholar]
- 18.Johansson E, Lange S, Lonnroth I. Identification of an active site in the antisecretory factor protein. Biochim Biophys Acta. 1997;1362(2–3):177–82. doi: 10.1016/s0925-4439(97)00066-5. [DOI] [PubMed] [Google Scholar]
- 19.Lange S, Lonnroth I. The antisecretory factor: synthesis, anatomical and cellular distribution, and biological action in experimental and clinical studies. Int Rev Cytol. 2001;210:39–75. doi: 10.1016/s0074-7696(01)10003-3. [DOI] [PubMed] [Google Scholar]
- 20.Ulgheri C, Paganini B, Rossi F. Antisecretory factor as a potential health-promoting molecule in man and animals. Nutr Res Rev. 2010;23(2):300–13. doi: 10.1017/S0954422410000193. [DOI] [PubMed] [Google Scholar]
- 21.Eriksson A, Shafazand M, Jennische E, Lonnroth I, Lange S. Antisecretory factor-induced regression of Crohn’s disease in a weak responder to conventional pharmacological treatment. Inflamm Bowel Dis. 2003;9(6):398–400. doi: 10.1097/00054725-200311000-00013. [DOI] [PubMed] [Google Scholar]
- 22.Johansson E, Al-Olama M, Hansson HA, Lange S, Jennische E. Diet-induced antisecretory factor prevents intracranial hypertension in a dosage-dependent manner. Br J Nutr. 2013;109(12):2247–52. doi: 10.1017/S0007114512004552. [DOI] [PubMed] [Google Scholar]
- 23.Al-Olama M, Wallgren A, Andersson B, Gatzinsky K, Hultborn R, Karlsson-Parra A, et al. The peptide AF-16 decreases high interstitial fluid pressure in solid tumors. Acta Oncol. 2011;50(7):1098–104. doi: 10.3109/0284186X.2011.562240. [DOI] [PubMed] [Google Scholar]
- 24.Laurenius A, Wangberg B, Lange S, Jennische E, Lundgren BK, Bosaeus I. Antisecretory factor counteracts secretory diarrhoea of endocrine origin. Clin Nutr. 2003;22(6):549–52. doi: 10.1016/s0261-5614(03)00057-8. [DOI] [PubMed] [Google Scholar]
- 25.Eriksson A, Shafazand M, Jennische E, Lange S. Effect of antisecretory factor in ulcerative colitis on histological and laborative outcome: a short period clinical trial. Scand J Gastroenterol. 2003;38(10):1045–9. doi: 10.1080/00365520310005064. [DOI] [PubMed] [Google Scholar]
- 26.Jennische E, Bergstrom T, Johansson M, Nystrom K, Tarkowski A, Hansson HA, et al. The peptide AF-16 abolishes sickness and death at experimental encephalitis by reducing increase of intracranial pressure. Brain Res. 2008;1227:189–97. doi: 10.1016/j.brainres.2008.05.083. [DOI] [PubMed] [Google Scholar]
- 27.Lange S, Lonnroth I, Skadhauge E. Effects of the antisecretory factor in pigs. Pflugers Arch. 1987;409(3):328–32. doi: 10.1007/BF00583485. [DOI] [PubMed] [Google Scholar]
- 28.Lonnroth I, Lange S, Jennische E, Johansson E, Jonson I, Torres J. Cholera toxin protects against action by Clostridium difficile toxin A. The role of antisecretory factor in intestinal secretion and inflammation in rat Apmis. 2003;111(10):969–77. doi: 10.1034/j.1600-0463.2003.1111009.x. [DOI] [PubMed] [Google Scholar]
- 29.Kim M, Wasling P, Xiao MY, Jennische E, Lange S, Hanse E. Antisecretory factor modulates GABAergic transmission in the rat hippocampus. Regul Pept. 2005;129(1–3):109–18. doi: 10.1016/j.regpep.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 30.Dowlatshahi Pour M, Jennische E, Lange S, Ewing AG, Malmberg P. Food-induced changes of lipids in rat neuronal tissue visualized by ToF-SIMS imaging. Sci Rep. 2016;6:32797. doi: 10.1038/srep32797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johansson E, Jonson I, Bosaeus M, Jennische E. Identification of flotillin-1 as an interacting protein for antisecretory factor. Regul Pept. 2008;146(1–3):303–9. doi: 10.1016/j.regpep.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 32.Boucher Y, Jain RK. Microvascular pressure is the principal driving force for interstitial hypertension in solid tumors: implications for vascular collapse. Cancer Res. 1992;52(18):5110–4. [PubMed] [Google Scholar]
- 33.Wiig H, Tveit E, Hultborn R, Reed RK, Weiss L. Interstitial fluid pressure in DMBA-induced rat mammary tumours. Scand J Clin Lab Invest. 1982;42(2):159–64. [PubMed] [Google Scholar]
- 34.Lindberg OR, McKinney A, Engler JR, Koshkakaryan G, Gong H, Robinson AE, et al. GBM heterogeneity as a function of variable epidermal growth factor receptor variant III activity. Oncotarget. 2016;7(48):79101–16. doi: 10.18632/oncotarget.12600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, et al. Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell. 2012;21(4):488–503. doi: 10.1016/j.ccr.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishidate M, Yamamoto K, Masuda C, Aikawa H, Hayashi M, Kawanishi T, et al. MALDI mass spectrometry imaging of erlotinib administered in combination with bevacizumab in xenograft mice bearing B901L, EGFR-mutated NSCLC cells. Sci Rep. 2017;7(1):16763. doi: 10.1038/s41598-017-17211-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li N, Maly DJ, Chanthery YH, Sirkis DW, Nakamura JL, Berger MS, et al. Radiotherapy followed by aurora kinase inhibition targets tumor-propagating cells in human glioblastoma. Mol Cancer Ther. 2015;14(2):419–28. doi: 10.1158/1535-7163.MCT-14-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carvalho BS, Irizarry RA. A framework for oligonucleotide microarray preprocessing. Bioinformatics. 2010;26(19):2363–7. doi: 10.1093/bioinformatics/btq431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98(9):5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448–9. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 41.Lenart B, Kintner DB, Shull GE, Sun D. Na-K-Cl cotransporter-mediated intracellular Na+ accumulation affects Ca2+ signaling in astrocytes in an in vitro ischemic model. J Neurosci. 2004;24(43):9585–97. doi: 10.1523/JNEUROSCI.2569-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arosio D, Ricci F, Marchetti L, Gualdani R, Albertazzi L, Beltram F. Simultaneous intracellular chloride and pH measurements using a GFP-based sensor. Nat Methods. 2010;7(7):516–8. doi: 10.1038/nmeth.1471. [DOI] [PubMed] [Google Scholar]
- 43.Hashizume R, Ozawa T, Gryaznov SM, Bollen AW, Lamborn KR, Frey WH, 2nd, et al. New therapeutic approach for brain tumors: Intranasal delivery of telomerase inhibitor GRN163. Neuro Oncol. 2008;10(2):112–20. doi: 10.1215/15228517-2007-052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lujan HL, Janbaih H, Feng HZ, Jin JP, DiCarlo SE. Ventricular function during exercise in mice and rats. Am J Physiol Regul Integr Comp Physiol. 2012;302(1):R68–74. doi: 10.1152/ajpregu.00340.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lange S, Lonnroth I, Palm A, Hyden H. The effect of antisecretory factor on the permeability of nerve cell membrane to chloride ion. Pflugers Arch. 1987;410(6):648–51. doi: 10.1007/BF00581326. [DOI] [PubMed] [Google Scholar]
- 46.Rapallino MV, Cupello A, Lange S, Lonnroth I. Antisecretory factor peptide derivatives specifically inhibit [3H]-gamma-amino-butyric acid/36Cl- out–>in permeation across the isolated rabbit Deiters’ neuronal membrane. Acta Physiol Scand. 2003;179(4):367–71. doi: 10.1111/j.1365-201X.2003.01173.x. [DOI] [PubMed] [Google Scholar]
- 47.Zhu W, Begum G, Pointer K, Clark PA, Yang SS, Lin SH, et al. WNK1-OSR1 kinase-mediated phospho-activation of Na+-K+-2Cl- cotransporter facilitates glioma migration. Mol Cancer. 2014;13:31. doi: 10.1186/1476-4598-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su G, Kintner DB, Flagella M, Shull GE, Sun D. Astrocytes from Na(+)-K(+)-Cl(−) cotransporter-null mice exhibit absence of swelling and decrease in EAA release. Am J Physiol Cell Physiol. 2002;282(5):C1147–60. doi: 10.1152/ajpcell.00538.2001. [DOI] [PubMed] [Google Scholar]
- 49.Nardou R, Ferrari DC, Ben-Ari Y. Mechanisms and effects of seizures in the immature brain. Semin Fetal Neonatal Med. 2013;18(4):175–84. doi: 10.1016/j.siny.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 50.Cesetti T, Ciccolini F, Li Y. GABA Not Only a Neurotransmitter: Osmotic Regulation by GABA(A)R Signaling. Front Cell Neurosci. 2012;6:3. doi: 10.3389/fncel.2012.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Young SZ, Taylor MM, Wu S, Ikeda-Matsuo Y, Kubera C, Bordey A. NKCC1 knockdown decreases neuron production through GABA(A)-regulated neural progenitor proliferation and delays dendrite development. J Neurosci. 2012;32(39):13630–8. doi: 10.1523/JNEUROSCI.2864-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Panet R, Marcus M, Atlan H. Overexpression of the Na(+)/K(+)/Cl(−) cotransporter gene induces cell proliferation and phenotypic transformation in mouse fibroblasts. J Cell Physiol. 2000;182(1):109–18. doi: 10.1002/(SICI)1097-4652(200001)182:1<109::AID-JCP12>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.