

Abstract
A defining feature and necessary step of the retrovirus life cycle is the integration of the viral genome into the host genome. All retroviruses encode an integrase (IN) enzyme that catalyzes the covalent joining of viral to host DNA, which is known as strand transfer. Integration may be modeled in vitro with recombinant retroviral IN and DNA oligomers mimicking the ends of the viral genome. In order to more closely recapitulate the integration reaction that occurs in vivo, integration complexes are assembled from recombinant IN and synthetic oligomers by dialysis in a reduced salt concentration buffer. The integration complex, called an intasome, may be purified by size exclusion chromatography. In the case of prototype foamy virus (PFV), the intasome is a tetramer of IN and two DNA oligomers and is readily separated from monomeric IN and free oligomer DNA. The integration efficiency of PFV intasomes may be assayed under a variety of experimental conditions to better understand the dynamics and mechanics of retroviral integration.
Keywords: Immunology and Infection, Issue 133, Retrovirus, Prototype Foamy Virus, Intasome, Integrase, Protein Complex Assembly, Protein Complex Purification
Introduction
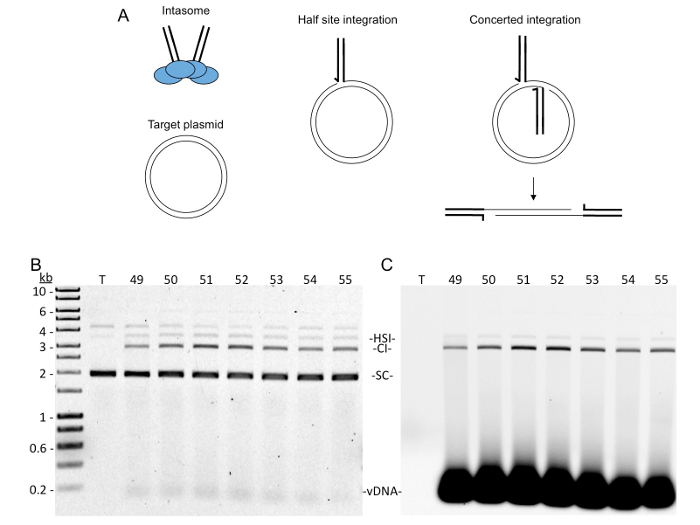
Integration of the viral genome into the host genome is a mandatory step in the life cycle of all retroviruses1. The viral enzyme integrase (IN) catalyzes the covalent joining of each end of the viral DNA genome to the host DNA. During a cellular infection, IN is part of a pre-integration complex that mediates integration. Recombinant IN complexed with double stranded DNA oligomers mimicking the viral DNA ends can also perform integration into a target DNA in vitro2. A common integration assay in vitro utilizes a supercoiled plasmid as the target DNA. Integration of both viral DNA oligomers (vDNA) to the plasmid results in a linear product and is termed concerted integration (Figure 2A). The integration assay in vitro may also yield products with only one vDNA covalently joined to the target plasmid resulting in a relaxed circle. This half-site integration product appears to be an artefact of the assay in vitro.
Recombinant IN and vDNA may perform integration in vitro, but they are not ideal reagents for the study of the dynamics or structure of integration complexes when monomeric IN would obscure relevant visualization. Purified integration complexes, or "intasomes," are required for dynamic single molecule analysis or structural studies. PFV IN and vDNAs may be assembled by dialysis from a relatively high salt concentration buffer to a lower salt concentration3,4. During dialysis, a precipitate forms. This precipitate is removed from dialysis and the salt concentration is increased. The higher salt concentration solubilizes the precipitate containing PFV intasomes. The intasomes are then purified by size exclusion chromatography (SEC). Recombinant prototype foamy virus (PFV) IN has been shown to exist as a monomer in solution at concentrations up to 225 µM5. SEC fractionation effectively separates the PFV intasomes (225.5 kDa), which includes a tetramer of PFV IN and two vDNAs, from monomeric PFV IN (44.4 kDa) and free vDNA (24.0 kDa). The PFV intasomes may be frozen and retain integration activity for at least six months of storage at -80 ˚C.
Recombinant PFV intasomes may also be modified to include IN amino acid substitutions or truncation mutations or vDNAs labeled with fluorophores or biotin4,6. The purified PFV intasomes readily perform integration into a supercoiled plasmid target DNA in vitro. Bulk biochemical integration assays with intasomes may test the effects of IN mutations, IN inhibitors, or other chemical additives. Biotinylated intasomes can be used to probe affinity with nucleic acids or proteins. PFV intasomes are functional at ambient temperature allowing for single molecule microscopy analysis by magnetic tweezers to measure the time between joining of the two vDNA ends or total internal reflection fluorescence to visualize the intasome search on target DNA6. In addition, PFV intasomes were the first to be structurally characterized significantly impacting the field of retroviral integration3.
Protocol
1. Annealing of vDNA
Combine 1X TEN Buffer (10X TEN stock: 100 mM Tris-HCl, pH 8.0, 1 M NaCl, 10 mM EDTA), 10 µM Oligo 1 (5' ATTGTCATGGAATTTTGTATATTGAGTGGCGCCCGAACAG 3', 100 µM stock), and 10 µM Oligo 2 (5' CTGTTCGGGCGCCACTCAATATACAAAATTCCATGACA 3', 100 µM stock) in a final volume of 1.5 mL. Aliquot 25 µL into sixty 0.2-mL polymerase chain reaction (PCR) tubes. NOTE: Depending on the application, oligomers may be modified with fluorophores or tags at the 5'-end of Oligo 2 or as an internal amino-T at base 13 from the 5'-end of Oligo 1. Modified oligomers should be purified by high-performance liquid chromatography (HPLC) before annealing. We have found that 5'-end Cy3 or Cy5 fluorophore labeling of Oligo 2 reduces the intasome assembly efficiency 10-fold. Internal fluorophore labeling does not reduce assembly efficiency.
Anneal using a thermocycler with the following times and temperatures: 1 cycle at 94.0 ˚C 3 min, 99 cycles at (94.0 ˚C 1 min, 93.6 ˚C 1 min) decreasing both temperatures by 0.8 ˚C per cycle (the last cycle is 14.8 ˚C 1 min, 14.4 ˚C 1 min), and store at 4.0 ˚C.
Combine the annealing reactions from all sixty tubes. Load 500 µL to two 0.5-mL 3 kDa molecular weight cutoff (MWCO) ultracentrifugal filter concentration units. Concentrate the annealed vDNA by centrifugation in a microcentrifuge at 14,000 x g for 10 min at room temperature (RT). NOTE: The retentate volume should be ~50 µL in each concentration unit.
Discard the flow through. Add 250 µL of the remaining annealed vDNA to each filter unit. Repeat the spin and discard the flow through. Buffer exchange into TE buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA) by washing three times with 450 µL TE.
Invert the filter unit and spin at 1,000 x g for 2 min at RT. The final retentate volume for each filter unit should be ~50 µL. Combine the retentates and transfer to a 1.5 mL screw cap tube.
Measure the 260-nm ultraviolet (UV) absorbance of the vDNA. Use this value to calculate the final molar DNA concentration. The extinction coefficient (ε260) of this vDNA is 622,803 cm-1 M-1 and its molecular weight is 23,972 Da. Store at -20 ˚C.
2. Intasome Assembly
If possible, perform the intasome assembly in a small room with a variable thermostat. Adjust the thermostat to approximately 18-22 ˚C. NOTE: In our experience, intasome assembly at 4 ˚C is inefficient.
Prepare 1 L of dialysis buffer (20 mM Bis-tris propane, pH 7.5, 200 mM NaCl, 2 mM dithiothreitol (DTT), 25 µM ZnCl2) in a 2 L beaker with a stir bar on a stir plate. Equilibrate the buffer in the 18-22 ˚C room for at least 6 h.
To assemble the intasomes, combine 50 mM Bis-tris propane, pH 7.5, 500 mM NaCl, 120 µM PFV IN, and 50 µM vDNA in a total volume of 150 µL. NOTE: The PFV IN should be free of contaminating nuclease activity7,8. If the concentration of PFV IN is too dilute to accommodate 120 µM IN in 150 µL volume, then it is possible to increase the final volume to 200 µL. The critical factors of this step are to maintain the molar ratio of IN to vDNA (2.4:1) and to have enough complex to visualize during chromatography.
Clean a ~10-cm long piece of 10-mm 6-8 kDa MWCO dialysis tubing and clips with double distilled water (ddH2O), or the highest purity water available. Clip one end of the tubing. NOTE: Dialysis tubing should be handled with care to prevent any possible contamination from the lab space.
Make 15 mL of dialysis tubing rinse buffer (50 mM Bis-tris propane, pH 7.5, 500 mM NaCl). Rinse the inside of the dialysis tubing three times with 1-2 mL rinse buffer. Remove as much of the rinse buffer as possible with a pipette and thin gel loading tip.
If necessary, use a clean razor blade or scalpel to cut the dialysis tubing so that a thin gel loading tip may reach the end of the tubing.
Transfer the intasome assembly from step 2.3 to the dialysis tubing with a pipette and a thin gel loading tip. Clip the open end of the dialysis tubing, minimizing the amount of air introduced within the tubing. Place the tubing into the dialysis buffer.
Dialyze overnight (16-20 h) with gentle stirring so that the tubing rotates but is not in a vortex. Ensure that the dialysis tubing is submerged and mobile. After approximately one hour, observe a visible precipitate inside the tubing. NOTE: With Cy3 or Cy5 fluorescently labeled vDNA, the precipitate is more obvious. This is true for both end labeled and internally labeled fluorescent vDNA.
3. Intasome Solubilization
Prepare two wide bore pipette tips by removing 2-4 mm from the end of a 200 µL tip with a new razor or scalpel blade on a clean surface. NOTE: The tips will be used in step 3.3 and 3.5.
Remove the dialysis tubing from the dialysis buffer. In order to prevent dilution of the intasome sample with dialysis buffer that may remain at the end of the tubing near the clip, use a micropipette with a small pipette tip to remove any excess dialysis buffer. Remove the clip from one end of the dialysis tubing.
Use a wide bore pipette tip from step 3.1 to transfer the sample including the precipitate inside the dialysis tubing to a 1.5-mL tube on ice. Note the total volume of the recovered material; the total volume is typically ~140 µL.
The sample with precipitate is at 200 mM NaCl; increase the salt to a final concentration of 320 mM NaCl by adding the appropriate volume of a stock 5 M NaCl solution. For example, for a sample of 150 µL, add 3.9 µL 5 M NaCl and 1.1 µL ddH2O for a final volume of 155 µL. Transfer ice bucket with sample into a 4 ˚C cold room.
Use a wide bore pipette tip from step 3.1 to resuspend and solubilize the precipitate by pipetting. Repeat every 20 min for at least 1 h. Observe that most of the precipitate should go into solution. NOTE: Pipetting to resuspend the precipitate can be stopped when it is apparent that the precipitate is no longer noticeably reduced between time points. When vDNA is not labeled or is internally labeled, the precipitate is reduced by ~90% based on visual inspection. In the case of Cy5 end labeled vDNA, the precipitate is reduced by only ~20%; most of the precipitate with fluorophore end labeled vDNA will not solubilize. The amount of precipitate that is effectively solubilized is variable and should be empirically determined.
4. Intasome Purification
Prepare 250 mL of size exclusion chromatography (SEC) running buffer (20 mM Bis-tris propane, pH 7.5, 320 mM NaCl, 10% glycerol). Sterile filter the buffer with a 0.2-µm filter unit and store at 4 ˚C. NOTE: All purification steps are performed in a 4 ˚C cold room.
Equilibrate a cross-linked agarose SEC column (diameter = 10 mm; length = 300 mm; bed volume = 24 mL; sample volume = 25 - 500 µL; maximum pressure = 1.5 MPa; exclusion limit = 4 x 107 Da; separates molecular weights between 5 and 5,000 kDa, see the Table of Materials) with SEC running buffer at a flow rate of 0.4 mL/min. NOTE: This purification may be adapted to different size exclusion columns if they are able to effectively separate 300 kDa from 44 kDa, such as Superose 12 10/300 GL or Superose 6 Increase. Superose 12 10/300 GL has lower resolution, leading to more overlap of peaks. Conversely, Superose 6 Increase has higher resolution and offers better separation.
Centrifuge the intasome sample in a microfuge at 14,000 x g for 10 min at 4 ˚C to pellet any remaining precipitate. Carefully remove the supernatant. Load the supernatant to a 200 µL injection loop (tubing that can hold ~200 μL). Apply the sample to the SEC column. NOTE: Smaller load volumes allow greater resolution by SEC.
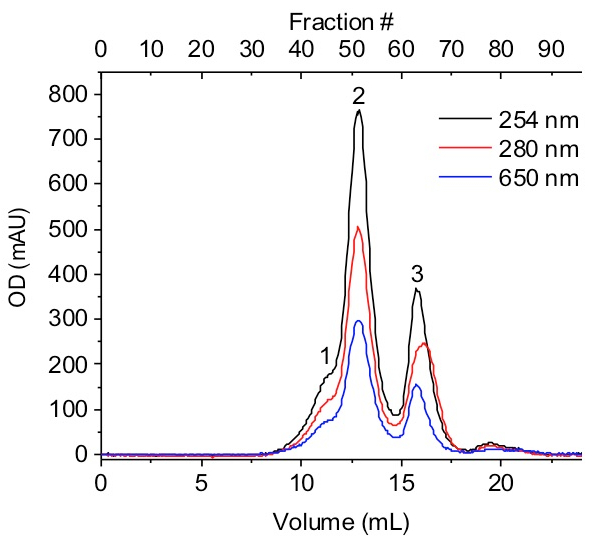
Elute with 25 mL SEC running buffer and collect 95 fractions of 270 µL. Observe that the SEC chromatogram displays three A280/A260 peaks (Figure 1). NOTE: The first should be an aggregate (~9.0 - 11.5 mL elution), followed by the PFV tetramer intasome peak (~12.0 - 14.75 mL elution) and PFV IN monomer with free vDNA peak (~15.0 - 18.0 mL).
5. Integration Strand Transfer Assay, Fraction Selection, and Storage
Combine 2 µL of each intasome peak fraction and 50 ng 3 kb supercoiled plasmid DNA (stock concentration is 50 ng/µL) in reaction buffer (10 mM Bis-tris propane, pH 7.5, 110 mM NaCl, 5 mM MgSO4, 4 µM ZnCl2, 10 mM DTT) in a final reaction volume of 15 µL. Incubate at 37 ˚C for 5 min.
Include a negative control with no added PFV intasome. Stop the reaction with 0.75 µL proteinase K (20 mg/mL stock solution) and 0.75 µL SDS (10% stock solution). Incubate at 55 ˚C for 1 h. Store samples as needed at -20 ˚C for later analyses. NOTE: This assay is a qualitative assessment of integration activity. The intasome molar concentration of each fraction is not determined prior to this assay. Fractions included in the integration strand transfer assay can be stored on ice (including overnight storage) until integration activity is confirmed and selected fractions are aliquoted. Here we use pMP2, a pUC19 derivative9. We have also successfully used the 5.4 kb plasmid pcDNA 3.1. Any plasmid that may be resolved to relaxed circle, linear, and supercoiled forms may be used as a target for integration.
Prepare a 120 mL 1% weight per volume (w/v) agarose gel in 1X TAE buffer (40 mM Tris-acetate, 1 mM EDTA) with 0.1 μg/mL ethidium bromide (EtBr, 10 mg/mL stock solution)10. Melt the agarose solution and pour it into a 15 cm x 10 cm gel casting tray. Insert a 15-well comb with 5-mm wide, 0.75-mm thick wells. NOTE: Narrower wells yield better band resolution compared to 1.5-mm thick wells.
Allow the gel to solidify at ambient temperature. Immerse the gel in 1X TAE with 0.1 μg/mL EtBr.
Add 3 µL 50% glycerol to each integration reaction from step 5.2. Load the entire reaction volume to the gel. Load 1 µL of 10 kb DNA size marker (150 ng/µL stock concentration) to lanes on either side of the samples; in other words, the DNA size marker should flank the sample lanes.
In an outer lane, load 6 µL Orange G dye (0.25% Orange G, 30% glycerol). Run the gel at constant voltage, 10 V/cm (100 V) at ambient temperature for 1 h, or until the Orange G dye front reaches the end of the gel.
Immediately image the agarose gel with a fluorescent scanner set to detect EtBr (532 nm excitation, 610 nm emission filter). If fluorophore labeled vDNAs were used, also image with appropriate fluorophore settings. NOTE: For example, to detect Cy5 labeled DNA, image using 633 nm excitation and 670 nm emission filters. Concerted integration products (CI, linear DNA) should run true to size at ~3 kb, unreacted supercoiled plasmid (SC) should run faster (~2 kb), and half-site integration products (HSI, relaxed circle DNA) should run slower (~3.5 kb). Unreacted vDNA should run true to size at ~40 bp.
Quantify the bands in each lane with imaging software7. Calculate the fraction of DNA in each lane that is CI, HSI, or SC; vDNA is not included in this calculation. NOTE: Integration efficiency, or the fraction of SC converted to a linear product, was calculated by dividing the pixel volume of concerted integration products (CI) by the total pixel volume of three bands (HSI+CI+SC). If intasomes are fluorophore labeled, the HSI and CI products may also be quantitated by fluorescence.
Select fractions that have the most concerted integration activity. NOTE: In our experience, the peak concerted integration activity coincides with the protein peak identified as tetrameric PFV intasomes. Measure the A280 of each of these fractions and calculate the final intasome concentration. The ε280 for a tetramer of wild type PFV IN with two vDNAs described here is 908,339 cm-1M-1 and the molecular weight is 225.5 kDa. The concentrations should follow the same pattern as the SEC chromatogram peak and in the strand transfer assay.
Distribute each fraction in 5 µL aliquots and snap freeze in liquid nitrogen. Store aliquots at -80 ˚C. Fractions selected for storage are typically between 200 - 600 nM.
Representative Results
PFV intasomes are assembled from recombinant IN and vDNA. After assembly, intasomes are purified by SEC (Figure 1). Integration activity of each fraction is assayed with a supercoiled DNA target and agarose gel electrophoresis (Figure 2). This gel is imaged with a fluorescent scanner set to detect EtBr (and fluorophore, if fluorophore-labeled vDNA is used). Using image analysis software, band pixel volumes can be used to calculate the integration efficiency (Figure 3). Fractions with the highest integration efficiency are snap frozen for later use. Frozen fractions retain integration activity (Figure 4), allowing for long term storage of assembled intasomes. Any label molecule and its position within the vDNA may affect intasome assembly efficiency (Figure 5).
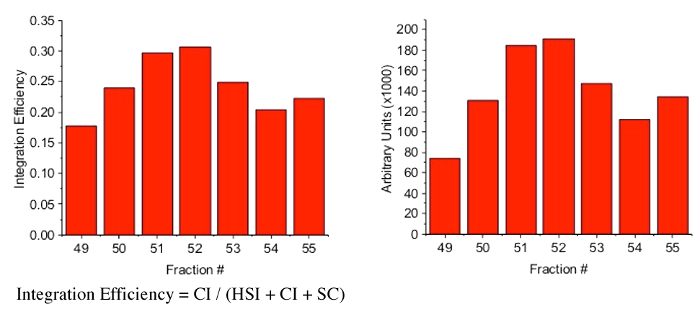
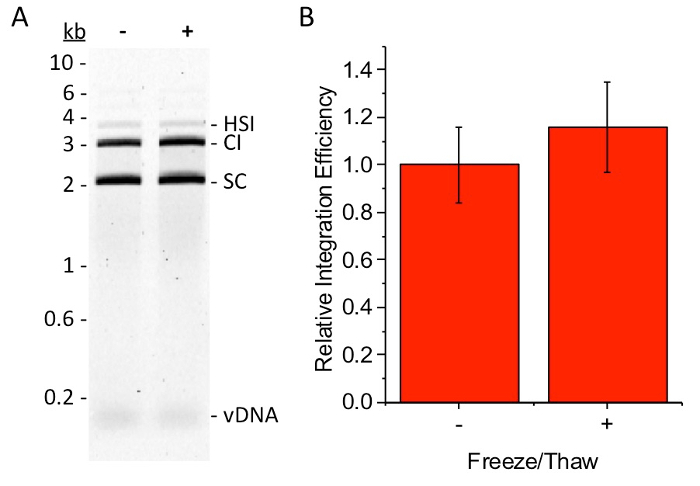
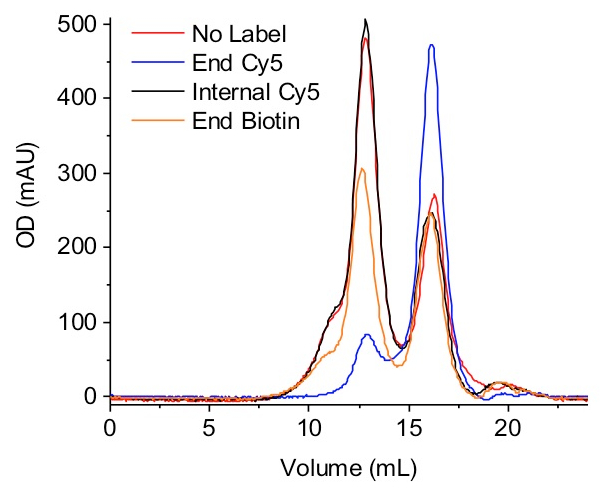
Figure 1: Size exclusion chromatogram. A PFV intasome assembly was separated with an agarose SEC column. This column allows for the separation of the aggregate (1), PFV intasome consisting of a tetramer of PFV IN and two vDNA (2), and monomeric PFV IN and vDNA (3). This example included vDNA with an internal Cy5 fluorophore label detected by 650 nm excitation. The y-axis denotes optical density (OD) in milli-absorbance units (mAU). Please click here to view a larger version of this figure.
Figure 2: Integration assay of SEC fractions. (A) Schematic of integration strand transfer reaction. Left, cartoon of a PFV intasome including a tetramer of IN (blue circles) and two vDNA (thick black lines), and a target plasmid (thin black lines). The supercoils (SC) of the target plasmid are not drawn. Center, cartoon of a half-site integration (HSI) product when one vDNA has been covalently joined to the target plasmid. The joining reaction introduces a nick and relaxes the supercoils. Right, cartoon of a concerted integration (CI) product where two vDNAs have been joined to the target DNA. This integration product results in a linear DNA with vDNAs at the ends. (B) Supercoiled plasmid DNA was added to each SEC fraction #49-55. Following incubation, the integration reactions were deproteinated and separated by 1% agarose gel electrophoresis containing EtBr. Negative control was plasmid DNA with no protein (T). DNA size markers are shown in kb. Supercoiled plasmid (SC), concerted integration products (CI), half-site integration products (HSI), and vDNA are indicated. (C) The agarose gel was scanned for the presence of Cy5. Only the vDNA, CI, and HSI products are visible. Please click here to view a larger version of this figure.
Figure 3: Quantitation of integration assay. The agarose gel from Figure 2 was scanned and quantified for the presence of both (A) EtBr and (B) Cy5. (A) For the EtBr calculation, HSI, CI, and SC band pixel volumes were obtained using gel analysis software. Integration efficiency was calculated by dividing the pixel volume of concerted integration products (CI) by the total pixel volume of all three bands (HSI+CI+SC). For example, the pixel values in the EtBr channel for the bands of fraction #49 are: 110565.2 SC, 25152.56 CI, and 6313.04 HSI. The total DNA pixel value for fraction #49 is the sum of these values, 142030.8. The integration efficiency is the CI pixel volume of 25152.56 divided by the total DNA value 142030.8 equaling 0.18. (B) Cy5 CI band pixel volumes are graphed as arbitrary units. Fractions with peak integration activity are individually aliquoted and frozen. In this example, fractions #50-53 were selected. Aliquots are snap frozen with liquid nitrogen and stored at -80 ˚C for future use.
Figure 4: Effect of freezing on intasome activity. Half of one SEC fraction was flash frozen with liquid nitrogen, stored at -80 ˚C for 1 h, and then slowly thawed on ice. The remaining half of the SEC fraction was kept on ice in a cold room while the first half was frozen and thawed. Intasomes were tested for activity without (-) and with (+) freeze/thawing. Integration efficiency was measured as described above. (A) EtBr stained agarose gel of integration reaction products. Supercoiled plasmid (SC), concerted integration products (CI), half-site integration products (HSI), and unreacted vDNA are indicated. DNA size markers are shown in kb. (B) Quantitation of the integration efficiency from the EtBr image. Calculations are described in Figure 3 legend. There is no loss of integration activity following freezing and thawing. The average integration efficiency is shown for 5 independent experiments with 2 intasome preparations. Error bars indicate the standard deviation. Paired t-test analyses yielded a two-tailed P = 0.011, suggesting that the freeze/thaw may have slightly enhanced integration activity. Please click here to view a larger version of this figure.
Figure 5: Label molecule and position impacts intasome assembly. PFV intasome assemblies included vDNAs that were unlabeled (red), end labeled on Oligo 2 with Cy5 (blue), internally labeled on Oligo 1 with Cy5 (black), or end labeled on Oligo 2 with biotin (orange). All assemblies were separated with an agarose SEC column. The y-axis denotes OD in mAU. Chromatograms are representative of at least 2 independent assemblies of each intasome type. End labeling reduces the yield of PFV intasomes with Cy5 10-fold and biotin 1.8-fold. End Cy5 and biotin assemblies have noticeably more precipitate remaining after high salt resolubilization (step 3.5), resulting in apparent loss of material on the size exclusion column. Internal labeling of the vDNA with the Cy5 fluorophore leads to an equal yield of intasomes as unlabeled vDNA. Please click here to view a larger version of this figure.
Discussion
Retroviral INs form a multimeric complex with viral genomic DNA to perform integration and continue the viral life cycle. The number of IN monomers per intasome may be tetramers, octamers, or possibly higher order multimers11,12,13,14. PFV intasomes are a tetramer of recombinant IN with double-stranded DNA oligomers that mimic viral genomic DNA ends3. These intasomes have been used in structural and dynamic studies3,5,13,14.
The assembly of PFV intasomes occurs during dialysis from a relatively high salt concentration buffer to a lower salt concentration. This results in the formation of a precipitate which includes the PFV intasomes. The intasomes are solubilized by increasing the salt concentration. PFV IN has been shown to be monomeric at concentrations up to 225 µM5. The complexes are then isolated from monomeric IN and free vDNA by SEC. We have modified the purification of PFV intasomes to include glycerol in the SEC buffer6. The addition of glycerol allows the PFV intasomes to be frozen for future use without loss of activity (Figure 4).
There are several key features of this protocol. Although protein purification is often performed at low temperatures, we have found empirically that PFV intasome assembly is inefficient at 4 ˚C. Instead, the assembly dialysis should occur in the range of 18-22 ˚C. It is also important to use recombinant PFV IN that is free of contaminating bacterial nuclease7,8. Finally, the ratio of IN to vDNA is a key factor of the assembly. If the starting IN protein concentration is lower than recommended here, the vDNA concentration must be proportionally decreased. While it is possible to assemble PFV intasomes at lower concentrations, the complexes must be at a high enough concentration for detection during SEC separation.
This method may be used for PFV intasomes with unlabeled or labeled vDNA. We have found that the vDNA may be efficiently labeled with a fluorophore or biotin6. Other small molecule labels may also be possible. In the case of Cy5 fluorophore, we find that end labeled vDNA reduces the yield of assembled intasomes with the peak concentration reduced 10-fold. However, internally Cy5 labeled vDNA has an equivalent yield to unlabeled vDNA. This assembly method may also be used with point mutations or truncation mutations of PFV IN4. Since PFV IN is inhibited by clinically relevant IN strand transfer inhibitor (INSTI) drugs targeting HIV-1 IN, it is possible to use these PFV intasomes to study the mechanism of INSTIs3. In addition, some INSTI resistant mutations of HIV-1 IN occur at conserved residues in PFV IN, allowing studies of drug resistance with engineered PFV intasomes.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by NIH AI099854 and AI126742 to KEY.
References
- Coffin JM, Hughes SH, Varmus HE. Retroviruses. Cold Spring Harbor Laboratory Press; 1997. [PubMed] [Google Scholar]
- Valkov E, et al. Functional and structural characterization of the integrase from the prototype foamy virus. Nucleic Acids Res. 2009;37(1):243–255. doi: 10.1093/nar/gkn938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature. 2010;464(7286):232–236. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Lin S, Craigie R. Outer domains of integrase within retroviral intasomes are dispensible for catalysis of DNA integration. Protein Sci. 2016;25(2):472–478. doi: 10.1002/pro.2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta K, et al. Solution conformations of prototype foamy virus integrase and its stable synaptic complex with U5 viral DNA. Structure. 2012;20(11):1918–1928. doi: 10.1016/j.str.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones ND, et al. Retroviral intasomes search for a target DNA by 1D diffusion which rarely results in integration. Nat Commun. 2016;7:11409. doi: 10.1038/ncomms11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez MA, Mackler RM, Altman MP, Yoder KE. Detection and removal of nuclease contamination during purification of recombinant prototype foamy virus integrase. J Vis Exp. 2017. [DOI] [PMC free article] [PubMed]
- Lopez MA, Mackler RM, Yoder KE. Removal of nuclease contamination during purification of recombinant prototype foamy virus integrase. J Virol Methods. 2016;235:134–138. doi: 10.1016/j.jviromet.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier MG, Bussiek M, Langowski J, Widom J. Spontaneous access to DNA target sites in folded chromatin fibers. J Mol Biol. 2008;379(4):772–786. doi: 10.1016/j.jmb.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PY, Costumbrado J, Hsu CY, Kim YH. Agarose gel electrophoresis for the separation of DNA fragments. J Vis Exp. 2012. [DOI] [PMC free article] [PubMed]
- Ballandras-Colas A, et al. Cryo-EM reveals a novel octameric integrase structure for betaretroviral intasome function. Nature. 2016;530(7590):358–361. doi: 10.1038/nature16955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballandras-Colas A, et al. A supramolecular assembly mediates lentiviral DNA integration. Science. 2017;355(6320):93–95. doi: 10.1126/science.aah7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passos DO, et al. Cryo-EM structures and atomic model of the HIV-1 strand transfer complex intasome. Science. 2017;355(6320):89–92. doi: 10.1126/science.aah5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, et al. Crystal structure of the Rous sarcoma virus intasome. Nature. 2016;530(7590):362–366. doi: 10.1038/nature16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maertens GN, Hare S, Cherepanov P. The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature. 2010;468(7321):326–329. doi: 10.1038/nature09517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskell DP, et al. Structural basis for retroviral integration into nucleosomes. Nature. 2015;523(7560):366–369. doi: 10.1038/nature14495. [DOI] [PMC free article] [PubMed] [Google Scholar]