Figure 1. Within-host diversity of IAV populations.
(A) Boxplots (median, 25th and 75th percentiles, whiskers extend to most extreme point within median ±1.5 x IQR) of the number of viral genomes per microliter transport media stratified by day post symptom onset. Notches represent the approximate 95% confidence interval of the median. (B) Boxplots (median, 25th and 75th percentiles, whiskers extend to most extreme point within median ±1.5 x IQR) of the number of iSNV in 249 high quality samples stratified by day post symptom onset. (C) The number of iSNV in each isolate stratified by vaccination status. The red lines indicate the median. (D) Location of all identified iSNV in the influenza A genome. Mutations are colored nonsynonymous (blue) and synonymous (gold) relative to that sample’s consensus sequence. Mutations are considered nonsynonymous if they are nonsynonymous in any known influenza open reading frame. Triangles signify mutations that were found in more than one individual in a given season.

Figure 1—figure supplement 1. Sequence coverage for all samples.

Figure 1—figure supplement 2. Approximate maximum likelihood trees of the concatenated coding sequences for high quality H1N1 samples.

Figure 1—figure supplement 3. Approximate maximum likelihood trees of the concatenated coding sequences for high quality H3N2 samples.

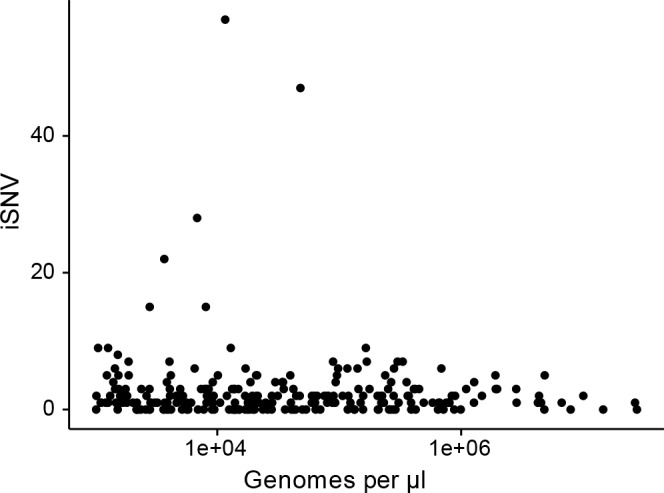
Figure 1—figure supplement 4. The effect of titer on the number of iSNV identified.