

Conspectus
Assessment of protein structure and interaction is crucial for understanding protein structure/function relationships. Compared to high-resolution structural tools including X-ray crystallography, nuclear magnetic resonance (NMR) and cryo-EM, and traditional low-resolution methods such as circular dichroism, UV-Vis and florescence spectroscopy, mass spectrometry (MS)-based protein footprinting affords medium-to-high resolution (i.e., regional and residue-specific insights) by taking advantage of proteomics methods focused on the primary structure. The methodology relies on “painting” the reactive and solvent-exposed amino acid residues with chemical tags and using the pattern of modifications as footprints from analysis by bottom-up MS-based proteomics to deduce protein higher order structures. The outcome can refer to proteins in solution or even in cells and is complementary to those of X-ray crystallography and NMR. It particularly useful in mapping protein-ligand interfaces and conformational changes resulted from ligand binding, mutation and aggregation.
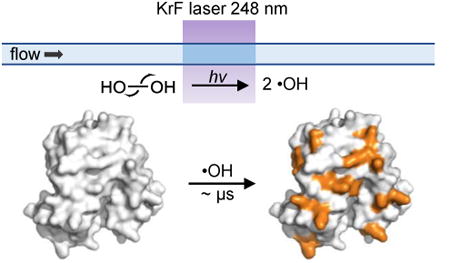
Fast photochemical oxidation of proteins (FPOP), in its original conception, is a type of hydroxyl-radical-based protein footprinting that utilizes a pulsed KrF laser (248 nm) to trigger hydrolysis of hydrogen peroxide to produce solution hydroxyl radicals, which subsequently modify the protein in-situ. The platform is expanding to adopt other reactive species including carbenes. The reactivity of the probe depends on the intrinsic reactivity of the radical with the residue side chain and the solvent accessibility of the residue as a function of the tertiary/quaternary structures. By introducing an appropriate scavenger to compete with hydroxyl radical self-quenching, the lifetime of the primary radicals is remarkably shortened to ∼ μs. Thus, the sampling timescale of FPOP is much faster than hydrogen-deuterium exchange and other covalent labeling methods relying on non-radical reactions.
The short footprinting timescale of FPOP offers two major advantages for protein structure elucidation:1) it allows the protein to be interrogated in its native or near-native state with minimum structural perturbation; 2) it exhibits high sensitivity toward alterations in protein higher order structures because its sampling time is short with respect to protein conformational changes and dynamic motion. In addition, the covalent and irreversible oxidation by the hydroxyl radical provides more flexibility in the downstream proteomics workflow and MS analysis, permitting high spatial resolution with residue-specific information.
Since its invention in 2005 by Hambly and Gross, FPOP has developed from proof-of-concept to a valuable biophysical tool for interrogating protein structure. In this account, we summarize the principles and experimental design of FPOP that enable its fast labeling, and describe the current and unique capabilities of the technique in protein higher order structure elucidation. Application examples include characterization of amyloid beta self-assembly, protein-ligand interactions with a special emphasis on epitope mapping for protein therapeutics (e.g., antibody, Fab and adnectin), protein folding detailed to residue-specific folding kinetics, and protein flexibility/dynamics. Additionally, the utility of FPOP-based oxidative footprinting should grow with our continuing developments of novel reagents (e.g., sulfate radical anion, carbene diradical and trifluoromethyl radical). These reactive reagents are compatible with the current FPOP platform and offer different reactivity and selectivity towards various types of amino acid residues, providing complementary insights into protein higher order structures for soluble proteins and ultimately for membrane-bound proteins.
Graphical abstract
Introduction
Mass spectrometry (MS)-based protein footprinting has become a compelling tool for proteins that are incompatible with or simply too demanding for traditional structural techniques of X-ray crystallography and NMR or not appropriate for CryoEM. As a highly practical technique that can be applied routinely, MS affords regional and residue-specific information in assessing structure and dynamics of proteins.
For perspective, we can view the commonly applied hydrogen-deuterium exchange (HDX) as a complementary and powerful means of protein footprinting. HDX interrogates protein higher order structures based on the stability, H-bonding, and structural protection of the protein backbone.1 The labile N-D bond after exchange, however, is prone to back exchange when the protein or its constituent peptides are placed in protic solvents for analysis, limiting the capacity of HDX in structural proteomics applications that require sophisticated sample handling (e.g., purification, proteolysis, buffer exchange) prior to MS analysis.
In contrast, hydroxyl-radical (·OH) footprinting, first introduced by Chance2-3 who used synchrotron radiation to ionize water and produce ·OH, yields irreversible modifications on the protein. Its elaboration as Fast Photochemical Oxidation of Proteins (FPOP) gives a method complementary to HDX. Hydroxyl radicals label the protein irreversibly, allowing application of rigorous downstream proteomics workflows to report accurately on solvent accessibility of the protein (Figure 1A). It accomplishes the labeling more rapidly and provides residue specificity, although with lower generality than HDX because labeling by ·OH requires the presence of reactive side chain(s) in a peptide segment.
Figure 1. A standard workflow of protein footprinting by FPOP.
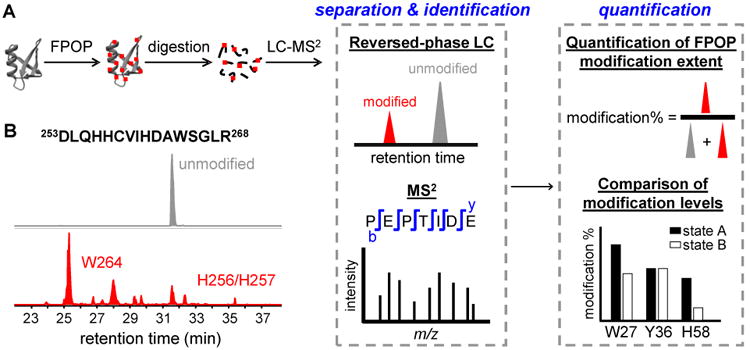
(A) Two different states, A and B, of a targeted protein (gray ribbon) are labeled by FPOP. The hydroxyl radical abstracts a hydrogen from the protein, and a series of follow-up reactions occur subsequently to produce stable covalent modifications (red dots) on the protein. The peptides produced in digestion are shown as black lines. Signals of unmodified and modified peptides observed in the HPLC chromatograms are shown in gray and red, respectively. MS2 is used to identify the modified residues. The extents of modification are quantified based on the signals of the unmodified and modified species and compared for proteins in different states. (B) Representative extracted-ion chromatograms showing the separation of the unmodified (grey) and FPOP-modified (red) peptides from the tryptic digests of interleukin-6 receptor (adapted with permission from ref. 8. Copyright 2017 American Chemical Society.). The peptide sequence and numbering are indicated above the chromatogram. The modified residues corresponding to each signal peak are labeled on the chromatogram.
FPOP was first developed by Hambly and Gross4 to utilize ·OH generated from photolysis of hydrogen peroxide (H2O2) to monitor solvent accessibility of the protein side chain. Compared to synchrotron radiolysis of water or use of the Fenton reaction, FPOP permits tuning the timescale of footprinting by introducing a scavenger to control the lifetime of the radical. This allows investigation of the protein structure in its native or near-native state without sampling altered conformations induced by modifications.
During the past decade, FPOP has been elaborated and applied to protein higher-order-structure characterization in both academic research and biotechnology development labs. Although most applications are occurring in biophysics, there is a significant need for new methods to support the discovery, development and quality control of protein therapeutics. In this review, we discuss the principles of FPOP and highlight selected applications including characterization of amyloid formation;5 of protein fast/slow folding;6-7 of protein-ligand interactions (emphasis on epitope mapping);8-10 and of protein dynamics and the identification of hidden conformations.11-12 These examples illustrate the power of FPOP to decipher the higher order structures of protein and protein complexes.
Fpop Fundamentals
FPOP setup
The FPOP apparatus uses a 248 nm KrF excimer laser to cleave H2O2 at low concentration (15 mM, 0.04%) (Figure 2). We chose 248 nm because the absorptions of water and most proteins are low at this wavelength. The laser beam is focused with convex lenses to give an exposure window of 2.0-3.0 mm wide on a 150 μM i.d. silica tubing (termed the FPOP capillary). The laser affords a high and converging flux of photons, maximizing the yield of ·OH in a small plug of an irradiated solution.
Figure 2. FPOP setup.
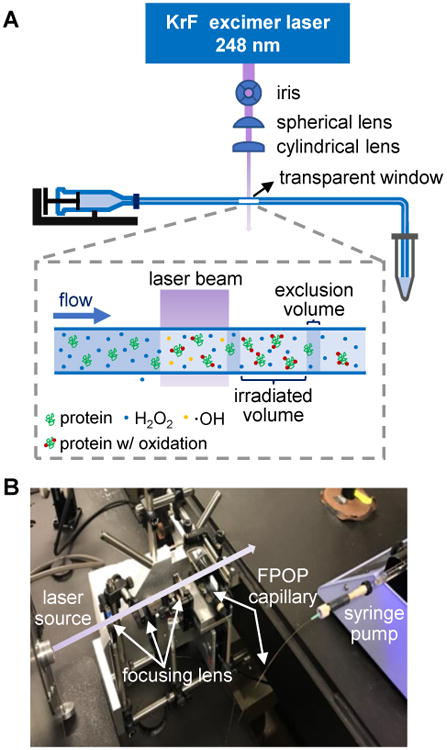
(A) Schematic of the FPOP platform. The laser beam (violet) generated from an excimer laser (blue square) is focused through an iris and then by two convex lenses. The FPOP capillary (blue line) made of silica tubing is connected to a syringe pump. The transparent window without polyimide coating indicates the location of laser irradiation (a blow-up of the transparent window is shown in the dashed box). A tube containing catalase and Met is placed at the end of the FPOP capillary to collect the FPOP-modified sample. (B) Photo of the FPOP apparatus. Primary components are labeled, and the violet arrow represents a visual pathway of the laser beam perpendicular to the FPOP capillary.
FPOP uses a flow system for protein footprinting, in which the capillary is placed perpendicular to the laser beam (Figure 2). Prior to FPOP, the protein is mixed with H2O2 and a radical scavenger (usually a free amino acid). The solution mixture is then immediately transported through the FPOP capillary by a syringe pump. When the solution passes the transparent window on the capillary, laser-triggered photolysis of H2O2 produces ·OH in nanoseconds. The radicals further undergo Haber-Weiss chain reactions in competition with self-quenching to re-form H2O2 (Scheme 1).
Scheme 1. Chain reactions involved in the generation of ·OH by FPOP.
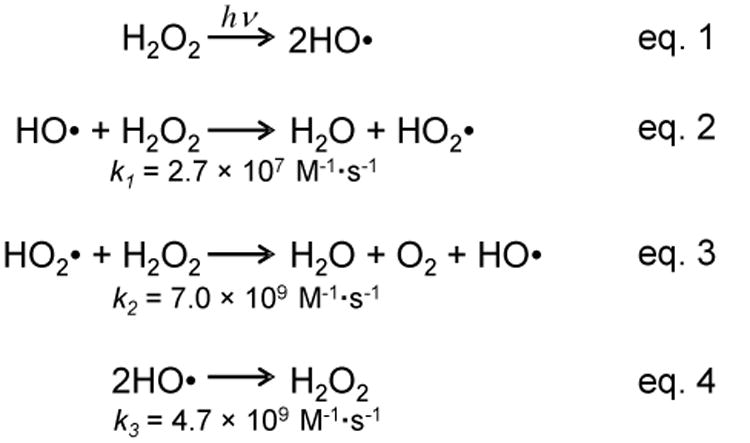
The rate constant is shown below each reaction.13
The hydroxyl radical induces protein oxidation by abstracting a hydrogen from the protein, which results in a protein radical that further undergoes a series of subsequent reactions to generate products with stable oxidation.2 Rate constants for reaction with individual amino acids range from 107 to 1010 M−1 s−1, serving as approximates of the intrinsic reactivities of protein side chains.2
An external pulse generator is incorporated to control frequency of the laser pulse, which together with a proper flow rate of the solution minimizes multiple laser irradiation to the same solution plug. To further avoid “double shooting”, a small volume is intentionally excluded from laser exposure to create a “barrier” between each exposed plug (termed an “exclusion volume”). The outlet of the FPOP capillary is placed into a sample-collection tube containing catalase and free methionine in buffer to remove leftover H2O2 and to prevent post-footprinting oxidation artifacts from any remaining reactive species. The control sample, used for correcting the background oxidation (< 5%, mainly on Met) of the protein induced by H2O2 or in post-FPOP sample handling is prepared the exact same way as the experimental sample, except the laser is omitted during the FPOP workflow.
If there is a concern that significant oxidation will occur when mixing the protein with H2O2, then H2O2 can be supplied prior to laser irradiation by using a T-shaped micromixer. In this setup, one syringe pump delivers the protein in buffer, and another delivers H2O2 and the scavenger for T-mixing prior to laser irradiation. This setup improves reproducibility by minimizing the exposure of the protein to H2O2 and hence any non-FPOP oxidation by the peroxide.14
Timescale of the FPOP probe
The radical lifetime in FPOP is tuned through varying the type and/or concentration of the scavenger.4, 15 In the absence of a scavenger, the radical lifetime in FPOP is limited by the recombination reaction and can extend to approximately 100 μs. Allowing a protein to react for this time may lead to unfolding, exposing buried residues to solvent exposure. Addition of a scavenger (e.g., 20 mM Gln) to the protein solution effectively shortens the lifetime of the primary hydroxyl radical to ∼ 1 μs, limiting the modification to reactive and solvent-exposed residues in the native conformation. Gau et al.16 studied the population distribution of the FPOP oxidation (0, +16, +32 Da etc.) for three oxidation-sensitive proteins (β-lactoglobulin, apo calmodulin and lysozyme), and proposed that a fit of the distribution to a Poisson signals conformational homogeneity of the protein during FPOP labeling. That is, protein samples with FPOP properly conducted by including a radical scavenger and suitable quenching of peroxide (catalase and Met) undergo footprinting of a single conformational state without perturbing the native conformation during labeling. These results underscore the indispensable roles of the flow system, the radical scavenger, and the prompt removal of peroxide in FPOP.
Any radical reaction leads to production of a secondary or higher order radicals, which have lifetimes in the millisec range.17 If these longer-lived radicals modify the protein, the time of labeling is longer than ∼1 μs that we claimed.4 Unfortunately, there is little direct evidence that reactions of secondary radicals like ·OOH modify the protein. Some indirect demonstration of the rapid labeling, however, comes from the application of FPOP in probing protein fast folding (see “Protein folding” section)7. The clear time-dependence seen in that study suggests that the radical reactions are not continuing onto the millisecond time range.
Protein modification depends not only on lifetime but also the number of radicals produced by the laser. Using d0/d5 Phe as an isotope-encoded dosimeter, for 15 mM H2O2, we found that the initial concentration of ·OH is 0.95 mM.15 Because the amount of the radical scavenger is in a large excess with respect to the solvent-exposed, reactive sites in the protein (e.g., 20 mM free Gln vs. 10 μM protein), the dominating chemistry that controls the lifetime of ·OH in FPOP is the reaction with the scavenger. Therefore, moderate variations in the protein concentration or the presence of a ligand will not significantly affect the overall oxidative yield on the protein or the relative yields on various side chains.18 From our experience, this remains valid for compounds with a size up to that of an antibody (∼ 150 kDa).19 Thus, observed FPOP variations between the protein in different conformational states (e.g., apo and holo) correlate only with their different solvent accessibilities.
Post-FPOP proteomics workflow
The FPOP-modified protein is usually analyzed by a bottom-up proteomics strategy to obtain sub-regional and residue-specific structural information (i.e., the footprint) (Figure 1). Owing to the covalent and irreversible modification by FPOP, many improvements made for analysis and determination of the primary structure (analytical proteomics) can be applied to facilitate the FPOP approach for higher order structure characterization. For example, the irreversible modification (unlike in HDX) offers high flexibility in proteolysis. Digestion with specific (e.g., trypsin, Asp-N), semi-specific (e.g., chymotrypsin) and nonspecific (e.g., pepsin) proteases can be performed in tandem or separately to generate overlapping peptide fragments that give comprehensive coverage of the protein sequence. Unlike identifying primary structure in proteomics, footprinting requires high coverage of the protein sequence, but this is manageable because usually a single, known protein or a simple mixture of proteins is used.
The resultant peptides are analyzed by liquid chromatography and tandem mass spectrometry (LC-MS2) to identify the modification sites and to quantify the levels. Oxidation by FPOP makes the peptide less hydrophobic, permitting separation of the modified peptide from its unmodified form by reversed-phase LC (see Figure 1B for an example). Generally, the modified peptides elute earlier than the unmodified ones, which allows a clear identification of the modified peptide on the basis of its accurate mass and product-ion spectrum (MS2). We quantify the peptide modification level based on the primary modification species (i.e., products of OH substitution (+15.9949 Da)). To calculate the modification percentage for a specific peptide, signal intensities of the unmodified peptide (Iu) and its modified species (ΣIox) are taken as peak areas from extracted ion chromatograms (XICs). The modification fraction for a certain peptide is calculated from the following equation: Fraction-modified =ΣIox/(Iu + ΣIox). This algorithm is straightforward and sufficiently sensitive to report the solvent accessibility change.5
Applications of Fpop
To show that the method is reliable and versatile, we now review four topics that show the utility of FPOP in biophysics and structural proteomics.
Amyloid protein aggregation
Aggregation of amyloid beta (Aβ) is one of the primary pathogenic events leading to Alzheimer's disease. Characterizing the products of Aβ aggregation, however, remains challenging owing to the vast heterogeneity and transient nature of the intermediate aggregates. We recently used FPOP to monitor amyloid formation and solvent accessibility of Aβ1-42, the 42- amino acid form of Aβ, at global, peptide, and amino-acid levels (Figure 3).5 FPOP footprinting immediately captures the solvent accessibilities of Aβ1-42 intermediates, filling in the gap between solution NMR for the early state oligomers and solids NMR and cryo-EM for the final fibrils. The readout from FPOP is clear, because the irreversible modification by FPOP should not be affected by Aβ aggregation and/or conformational change occurring post-footprinting.
Figure 3. FPOP and kinetic modeling characterize the time-dependent aggregation of Aβ1-42 at protein, peptide and amino-acid residue levels.
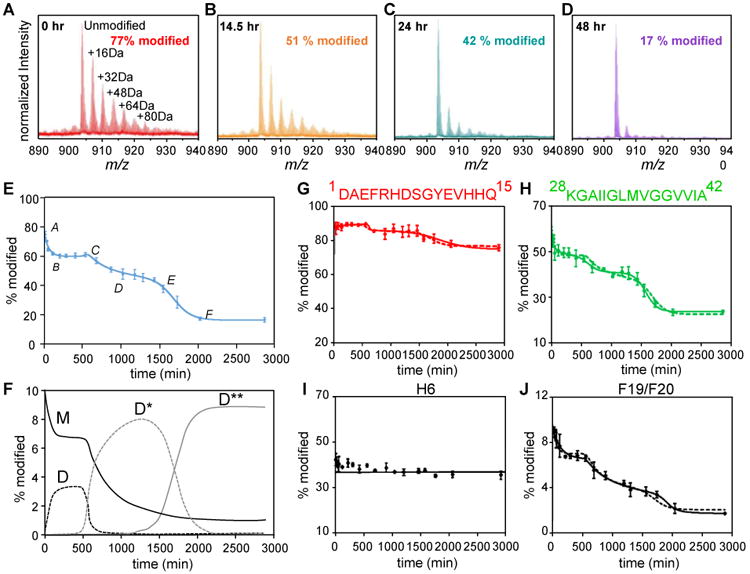
(A-D) Mass spectra showing extents of FPOP modification for intact Aβ1-42 (5+ charge) as a function of the incubation time. The determined modification percentage is shown in each panel. (E) Characterization of Aβ1-42 aggregation on the global (full-polypeptide) level by a kinetic simulation. Points represent experimental data, and the solid curve is a model fit based on two auto-catalytic reactions. (F) Concentrations (in monomeric equivalents) change of representative Aβ1-42 species (M-monomer, D-paranuclei, D*-protofibrils and D**-fibrils) from kinetic simulation. (G-H) Aggregation curves of N-terminal region 1-15 and C-terminal region 28-42. (I-J) Aggregation curves of representative Aβ1-42 residues H6 and F19/F20. In G-J, the solid and dashed curves are model fits independent of or constrained by the global rates, respectively. Adapted with permission from ref. 5. Copyright 2016 American Chemical Society.
We modified Aβ1-42 aggregates in various oligomeric states by FPOP. Occurring are extensive modifications for Aβ1-42 monomer owing to its intrinsically disordered structure (Figure 3A). As Aβ1-42 assembles and reorganizes into higher order oligomers, the FPOP modification of Aβ1-42 deceases owing to formation of secondary/tertiary structures (Figure 3B and C). At the end of aggregation, the modification extent decreases to a modest level (Figure 3D), indicating the formation of compact amyloid fibrils with a core structure that resists modification. A fit of a plot of FPOP modifications as a function of incubation time, adopting the conventional nucleation-condensation mechanism (Finke-Watzky), affords a sigmoidal curve that shows sequential formation of the major species of Aβ1-42 during aggregation (Figure 3E). We also characterized the interconversion of those representative Aβ species by simulating their concentration change as a function of the incubation time (Figure 3F).
FPOP also provides a high-resolution view of aggregation at regional and amino-acid levels for some residues. We characterized the modification of three Aβ1-42 sub regions, including the N-terminal peptide 1-15, central domain 16-27, and C-terminal peptide 28-42 from digestion of the full-length Aβ1-42. FPOP modification for the N-terminal peptide remains high (∼ 80%) with little variation as Aβ1-42 aggregates, indicating that the N-terminus remains structurally flexible and solvent accessible, with little participation in Aβ1-42 aggregation (Figure 3G). This finding is further supported by NMR results showing that the N-terminus of Aβ1-42 is disordered in a variety of oligomers and in the mature fibril. By contrast, the critical roles of the central domain and hydrophobic C-terminus in promoting self-association is highlighted by a dramatic decrease in FPOP modification from monomers to fibrils (Figure 3H). Furthermore, measuring the modification at the amino-acid level allows characterization of the aggregation tendencies for Aβ1-42 at a residue level. For example, H6 shows relatively constant FPOP modification, during aggregation, indicating little change in its solvent-accessibility and conformation (Figure 3I). On the other hand, F19/F20 are residues primarily responsible for the modification change observed for the central domain (Figure 3J), and we propose that interactions of F19/F20 contribute a driving force for Aβ1-42 aggregation by serving as the hydrophobic nucleation interface.
We envision that this FPOP platform can reveal how interactions of Aβ with other molecules affect site-specific aggregation. Application to drug candidates and natural products that promote/inhibit oligomerization should be particularly fruitful.
Protein folding
Protein folding and stability is crucial in understanding the biological effects of mutations and the pathogenesis of diseases associated with aggregation of misfolded proteins. Protein folding is transient and rapid (usually sub-millisecond), challenging structural characterization of kinetic intermediates formed during folding. Previously, spectroscopic methods, including fluorescence emission, circular dichroism, and electron-spin resonance, have provided insights into the underlying mechanism of protein folding. Approaches having high spatial resolution and sensitivity, however, are still sparse. Because the labeling speed of FPOP is faster than most protein folding, it should be effective in probing folding by measuring protein solvent-accessibility changes accompanying the conformational change. Meanwhile, the high spatial resolution of FPOP allows direct assessment of the folding down to amino-acid residues in some cases.
To explore this opportunity, we designed a two laser-based platform that combines a temperature jump (one laser) and FPOP (second laser) to study protein fast folding (Figure 4A).7 This platform employed a Raman-shifted Nd:YAG laser (1900 nm) to generate a nanosecond temperature jump (∼ 20 °C) in the buffer solution and initiate protein (barstar) refolding, followed by a folding time-dependent FPOP footprinting triggered by the KrF excimer laser. Control of the heating and the FPOP laser, and hence the folding times is achieved by two delay circuits, which are used to adjust the time between firing the two lasers.
Figure 4. Folding of barstar characterized by a T-jump FPOP.
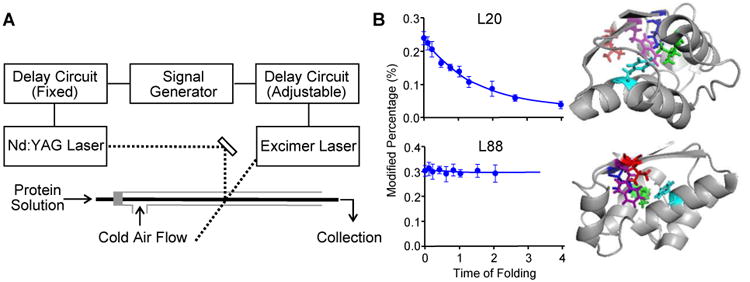
(A) Schematic representation of the two-laser FPOP platform. As in Figure 2A, the transparent window is located where the two laser beams (dash lines) are incorporated. (B) Left: FPOP modification percentage of two representative residues as a function of the protein (barstar) folding time. Solid lines in the plots are obtained from kinetic fitting. Right: Five critical residues identified by FPOP mapped on native barstar structure. Two views are provided to show the side chains of the amino acid L88 (red), F74 (cyan), I5 (blue), L20 (green), and W53 (purple). Adapted with permission from ref. 7. Copyright 2012 American Chemical Society.
The test protein, barstar, a small globulin protein with a single domain structure composed of four α-helixes and three β-sheets, is a well-characterized model for protein folding. We probed its folding from a fraction of a ms to 2 ms, a time domain spanning its early folding and found residues with distinct kinetic features in terms of their solvent accessibility (Figure 4B). Modifications of H17, L20 and L24, all located in helix1, show an exponential decrease in the first 2 ms of folding (a control is represented by L88 in Figure 4B). Their modification at 2 ms time is attenuated owing to its folding. These residues showing dramatically decreased FPOP modification are consistent with folding via hydrophobic interactions in the first 2 ms. Our findings agree with the spectroscopic results20 but bring spatial resolution, demonstrating that the hydrophobic collapse on helix1 is the initial step in barstar folding.
FPOP can also be used to probe protein slow folding. An example is study of the refolding of a viral fusion protein, parainfluenza virus 5 F (PIV5 F).6 We triggered the irreversible refolding of PIV5 F by heat, and captured five conformations on the basis of their FPOP footprints: PIV5 F in the pre-fusion state (before heating), three intermediate states (45, 55, and 65 °C), and the post-fusion state. The experiment required the FPOP capillary contiguous to the laser window to be placed in a heated chamber that maintains the temperature of the protein solution. The modification of the PIV5 F decreases upon transition from the pre-fusion to the post-fusion state, indicating that the structure of PIV5 F in the pre-fusion state is more open and solvent-exposed.
The modifications of the PIV5 F peptides afford structural information on regions becoming solvent-exposed or protected along the refolding pathway. This allows us to propose a model describing the dynamic refolding of PIV5 F and to compare the differences in FPOP modification of the pre-fusion and post-fusion PIV5 F with changes in the solvent accessible surface area (SASA) from X-ray structures. There are greater changes in solvent accessibility for nine regions, as reported by FPOP but not predicted by SASA. This indicates an underestimation of the pre-fusion PIV5 F's SASA calculated from the static crystal structure and points to a higher structural flexibility in solution. Interestingly, most of those regions contain the epitopes of PIV5 F for the prefusion-specific neutralizing antibodies, suggesting the importance of those regions for PIV5 F biological function.
Epitope mapping
Understanding antibody action relies on identifying the binding sites of the antibody on the target antigen. Methods for this, called epitope mapping, include X-ray crystallography, NMR, site-directed mutagenesis, and MS-based approaches. Those techniques offer complementary information on the epitope and on binding-induced conformational changes. To illustrate the capability of FPOP for epitope mapping, our lab has collaborated with Bristol-Myers Squibb and Genentech to characterize epitopes for a number of targets upon binding of protein therapeutics.
Early in the development, we applied FPOP to investigate the epitope of human epidermal growth factor receptor (EGFR) for adnectin binding at both the peptide and amino-acid residue levels.10 The FPOP-determined binding interface involves various amino-acid and peptide regions near the N-terminus of EGFR. Our data correlate well with the previously determined epitopes from the crystal structure. Additionally, successful characterization of epitopes in thrombin19 and vascular endothelial growth factor 18 serves as other good examples of FPOP's ability to identify both the epitope and regions with remote conformational changes.
We later combined HDX-MS, site-directed mutagenesis, and FPOP to study the epitope of interleukin-23 (IL-23) upon binding of an anti-IL-23 antibody (Figure 5).9 Using HDX-MS, we found five peptide regions on IL-23 showing reduced backbone amide solvent accessibility upon antibody binding. Five different peptides of IL-23 are identified by FPOP, among which three regions are identified by HDX as well. Additional FPOP analysis at residue level allows us to assign potential critical-binding residues. More recently, we implemented FPOP, HDX, and carboxyl group footprinting to map interactively the epitope of IL-6 receptor (IL-6R) for two adnectins with distinct affinities (Kd, Adnectin1 ∼ 6.2 pM vs Kd, Adnectin2 ∼ 46 nM).8 Besides identifying the epitope as a flexible loop that connects two β-strands in the cytokine-binding domain, our results reveal that two loops, located beyond the conserved epitope undergo reduced dynamic motion upon adnectin1 binding. Those local effects on the IL-6R structure are attenuated or not observed in the case of adnectin2 binding. Apparently, binding of adnectin1, the stronger ligand, stabilizes IL-6R by reducing the flexibility of those dynamic regions.
Figure 5. Epitope regions of IL-23 identified by FPOP, HDX-MS, and alanine shave mutagenesis.
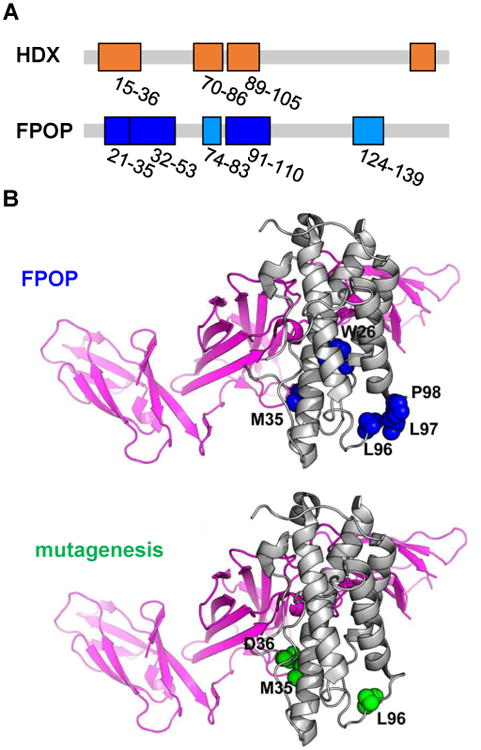
(A) Critical peptides discovered by HDX (top) and FPOP (bottom). These regions are mapped on the linear sequence of IL-23 (in orange for HDX results and blue for FPOP). (B) Critical residues identified by FPOP (top) and alanine-shave mutagenesis (bottom) mapped on the IL-23 crystal structure. Amino acids identified by FPOP are in blue, and by mutagenesis in green. Gray regions suggest no significant difference in the conformation of IL-23 upon binding with Fab. Adapted with permission from ref. 9. Copyright 2017 American Chemical Society.
Outcomes from the above studies highlight the capability of FPOP as a sensitive tool for epitope mapping, motivating its further applications in protein discovery and development either as a stand-alone method or in conjunction with orthogonal structural approaches (e.g., X-ray, SAXS, EM) and other MS-based methods (e.g., HDX-MS, ion mobility). In addition, the combined use of MS-based methods provides critical information on therapeutic targets in the absence of an X-ray structure, has high throughput, and may be a major advance for the discovery and development of biopharmaceuticals.
Protein dynamics and hidden conformations
FPOP can report variations in protein dynamics and flexibility because its sampling time is short with respect to local dynamics of the protein. For example, a study of the oligomerization interface of apolipoprotein E3 (ApoE3) reveals that the C-terminal helix of ApoE3 and an unstructured hinge region proceeding the helix undergo decreases in solvent accessibilities in the tetramer compared to a monomeric mutant.12 The C-terminal helix of ApoE3 has long been hypothesized to be the primary interface for self-association leading to tetramer formation. Evidence for the change in the mobility of the hinge region associated with oligomerization cannot be seen by HDX and carboxyl group footprinting on ApoE3, done subsequently. Thus, the preceding hinge region is stabilized owing to formation of the oligomerization interface by the C-terminus of ApoE3. The inability to see this by HDX and carboxyl-group footprinting stems from their relatively long labeling time compared to that of FPOP.
FPOP also effectively reveals the difference in local dynamics between wildtype (WT) TEM-1 and its cefotaximase variant. TEM β-lactamase is an enzyme responsible for the antibiotic resistance in pathogenic Gram-negative bacteria. The cefotaximase variant of TEM, favored by a E104K/G238S mutant, hydrolyzes cefotaxime 1,400-fold more efficiently than does WT TEM-1.11 Although the function and specificity of various TEM variants can vary dramatically, no apparent conformational changes are found for those variants (Figure 6A and B). To uncover the structural difference between the cefotaximase variant and WT TEM-1, our collaborators, using Markov state models-based simulation, proposed that the decreased flexibility of the Ω-loop in the cefotaximase variant is responsible for its high enzymatic efficiency by stabilizing the binding-competent state (Figure 6C). To test, we turned to FPOP and found significantly decreased FPOP modification on the Ω-loop itself and the region preceding the Ω-loop in the cefotaximase variant (Figure 6D). In addition, the decreased solvent accessibility pertains to the C-terminus, suggesting that the E104K/G238S substitution not only reduces the flexibility in the Ω-loop, but that of the C-terminus remotely via long-range interaction. In this case, FPOP provides a test for the theory from molecular dynamics simulation and demonstrates its capability in probing protein solution dynamics that are inaccessible by static crystal structures.
Figure 6. Modeling and FPOP reveal restricted motion in the Ω-loop in cefotaximase variant.
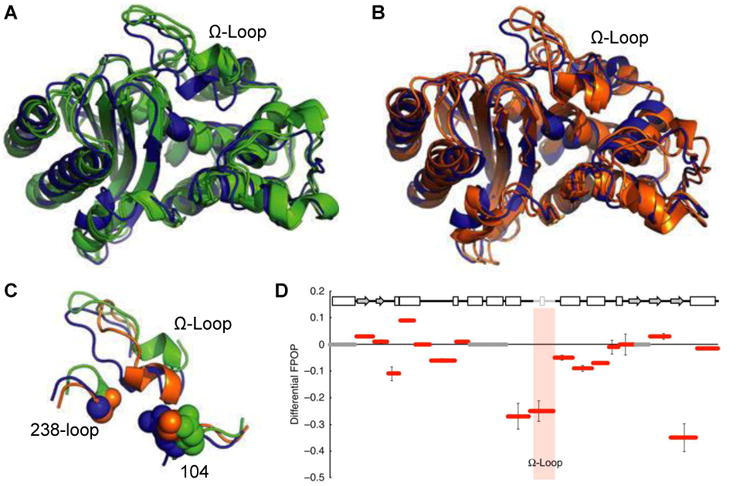
A crystal structure of TEM-1 (blue) is overlaid with the two most populated structures taken from modeling of (A) the non-cefotaximase states (green, favored by wild type) and (B) cefotaximase states (orange, favored by the E104K/G238S). (C) Large structural rearrangements in the Ω-loop distinguish low-energy non-cefotaximase states from cefotaximase states. (D) FPOP data reveal experimentally the restricted motion in cefotaximase variant predicted from modeling. Hydroxyl labelling of TEM variants are shown as the difference in percent labelled between cefotaximase variant and wild type. The Ω-loop (164–179) is shaded grey in the sequence and pink in the graph. Adapted with permission from ref. 11. Copyright 2016 Nature Publishing Group.
Conclusions and Outlook
FPOP coupled with MS has emerged as a powerful approach to study protein higher order structures. The robustness of the platform has been further enhanced by recent advances in methodology, allowing the approach to provide improved quantitative information and broader coverage in footprinting for various types of amino acid residue.
One example is incorporation of a reporter peptide into the protein sample to resolve discrepancies in the radical dosage. In this scheme, the protein and the reporter peptide are modified by FPOP simultaneously under the same condition. Thus, the modification extent of the reporter peptide correlates with the radical lifetime in the sample. By varying the scavenger concentration (i.e., the radical lifetime), the reporter peptide approach permits a normalized, time-dependent measurement of the modification on the protein.21 In addition, the FPOP platform has been extended by Jones to footprint live cells,22 allowing investigation of protein conformation and interactions in the native cellular environment.
The versatility of FPOP to accommodate new reagents in addition to the ·OH is illustrated by the developments of the sulfate radial anion (SO4-·)23 and iodine radical (I·)24 as alternative footprinting reagents. We also adapted the carbene radical, a laser-based footprinting reagent developed by Schriemer25 using a Nd-YAG laser at 355 nm, into the FPOP platform to modify Asp/Glu.26 More recently, Cheng et al. developed trifluoromethyl radical (·CF3) as a novel reagent that can modify 18 of 20 amino acids residues, including Gly, Ala, Ser, Thr, Asp and Glu, which have low reactivities with ·OH.27
The ability to accommodate new reagents on the FPOP platform allows construction of a “library&-rdquo; of reagents that offer different chemical reactivities and specificities to footprint proteins and distinguish structural changes that occur from apo to holo, bound to unbound, and wild type to mutant. As these improvements grow, FPOP-based oxidative footprinting approaches will play an increasingly important role by serving as constrains of the macromolecule structure, much like chemical shifts do in NMR. The ultimate goal is to use chemistry and MS-based proteomics to determine coarse-grained structure of proteins and their assemblies. This will require high coverage of the protein residues in footprinting and motivates continued development and application of FPOP.
Acknowledgments
This work was supported by the National Institutes of Health P41GM103422.
Biographies
Ke Sherry Li received her B.S. in Chemistry from Xiamen University in 2012. She is currently working towards a Ph.D. under the supervision of Michael Gross. Her research interest has focused on development and application of mass spectrometry-based approaches for protein characterization.
Liuqing Shi received her B.S. in Pharmaceutical Sciences from Tianjin University in 2011 and Ph.D. in Analytical Chemistry from Indiana University in 2015 with Prof. David Clemmer. She is currently a postdoctoral researcher in the Gross laboratory.
Michael L. Gross is a chemist who has worked independently in mass spectrometry since 1968. He began his career at the University of Nebraska-Lincoln, where he was distinguished professor of chemistry and director of an NSF Center for Mass Spectrometry until 1994. He then moved to Washington University as professor of chemistry and principal investigator of the WU NIH NIGMS Mass Spectrometry Research Resource. His primary research interests have evolved to structural proteomics and MS-based biophysics.
Footnotes
Notes: The authors declare no competing financial interest.
References
- 1.Engen JR. Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal Chem. 2009;81:7870–7875. doi: 10.1021/ac901154s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu G, Chance MR. Hydroxyl Radical-Mediated Modification of Proteins as Probes for Structural Proteomics. Chem Rev. 2007;107:3514–3543. doi: 10.1021/cr0682047. [DOI] [PubMed] [Google Scholar]
- 3.Maleknia SD, Brenowitz M, Chance MR. Millisecond radiolytic modification of peptides by synchrotron X-rays identified by mass spectrometry. Anal Chem. 1999;71:3965–73. doi: 10.1021/ac990500e. [DOI] [PubMed] [Google Scholar]
- 4.Hambly DM, Gross ML. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J Am Soc Mass Spectrom. 2005;16:2057–63. doi: 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 5.Li KS, Rempel DL, Gross ML. Conformational-Sensitive Fast Photochemical Oxidation of Proteins and Mass Spectrometry Characterize Amyloid Beta 1–42 Aggregation. J Am Chem Soc. 2016;138:12090–12098. doi: 10.1021/jacs.6b07543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poor TA, Jones LM, Sood A, Leser GP, Plasencia MD, Rempel DL, Jardetzky TS, Woods RJ, Gross ML, Lamb RA. Probing the paramyxovirus fusion (F) protein-refolding event from pre- to postfusion by oxidative footprinting. Proceedings of the National Academy of Sciences. 2014;111:E2596–E2605. doi: 10.1073/pnas.1408983111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, Rempel DL, Gau BC, Gross ML. Fast photochemical oxidation of proteins and mass spectrometry follow submillisecond protein folding at the amino-acid level. J Am Chem Soc. 2012;134:18724–31. doi: 10.1021/ja307606f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li KS, Chen G, Mo J, Huang RYC, Deyanova EG, Beno BR, O'Neil SR, Tymiak AA, Gross ML. Orthogonal Mass Spectrometry-Based Footprinting for Epitope Mapping and Structural Characterization: The IL-6 Receptor upon Binding of Protein Therapeutics. Anal Chem. 2017;89:7742–7749. doi: 10.1021/acs.analchem.7b01748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, Wei H, Krystek SR, Bond D, Brender TM, Cohen D, Feiner J, Hamacher N, Harshman J, Huang RYC, Julien SH, Lin Z, Moore K, Mueller L, Noriega C, Sejwal P, Sheppard P, Stevens B, Chen G, Tymiak AA, Gross ML, Schneeweis LA. Mapping the Energetic Epitope of an Antibody/Interleukin-23 Interaction with Hydrogen/Deuterium Exchange, Fast Photochemical Oxidation of Proteins Mass Spectrometry, and Alanine Shave Mutagenesis. Anal Chem. 2017;89:2250–2258. doi: 10.1021/acs.analchem.6b03058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan Y, Chen G, Wei H, Huang RYC, Mo J, Rempel DL, Tymiak AA, Gross ML. Fast Photochemical Oxidation of Proteins (FPOP) Maps the Epitope of EGFR Binding to Adnectin. J Am Soc Mass Spectrom. 2014;25:2084–2092. doi: 10.1007/s13361-014-0993-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hart KM, Ho CM, Dutta S, Gross ML, Bowman GR. Modelling proteins' hidden conformations to predict antibiotic resistance. Nat Commun. 2016;7:12965. doi: 10.1038/ncomms12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gau B, Garai K, Frieden C, Gross ML. Mass Spectrometry-Based Protein Footprinting Characterizes the Structures of Oligomeric Apolipoprotein E2, E3, and E4. Biochemistry. 2011;50:8117–8126. doi: 10.1021/bi200911c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elliot AJ, Chalk River Nuclear L, System C. System Chemistry & Corrosion Branch. Chalk River Laboratories; Chalk River, Ont: 1994. Corrosion Rate constants and G-values for the simulation of the radiolysis of light water over the range 0-300°. [Google Scholar]
- 14.Zhang Y, Rempel DL, Zhang H, Gross ML. An Improved Fast Photochemical Oxidation of Proteins (FPOP) Platform for Protein Therapeutics. J Am Soc Mass Spectrom. 2015;26:526–529. doi: 10.1007/s13361-014-1055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Niu B, Zhang H, Giblin D, Rempel DL, Gross ML. Dosimetry Determines the Initial OH Radical Concentration in Fast Photochemical Oxidation of Proteins (FPOP) J Am Soc Mass Spectrom. 2015;26:843–846. doi: 10.1007/s13361-015-1087-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gau BC, Sharp JS, Rempel DL, Gross ML. Fast Photochemical Oxidation of Protein Footprints Faster than Protein Unfolding. Anal Chem. 2009;81:6563–6571. doi: 10.1021/ac901054w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vahidi S, Konermann L. Probing the Time Scale of FPOP (Fast Photochemical Oxidation of Proteins): Radical Reactions Extend Over Tens of Milliseconds. J Am Soc Mass Spectrom. 2016;27:1156–1164. doi: 10.1007/s13361-016-1389-x. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Wecksler AT, Molina P, Deperalta G, Gross ML. Mapping the Binding Interface of VEGF and a Monoclonal Antibody Fab-1 Fragment with Fast Photochemical Oxidation of Proteins (FPOP) and Mass Spectrometry. J Am Soc Mass Spectrom. 2017;28:850–858. doi: 10.1007/s13361-017-1601-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jones LM, B Sperry J, A Carroll J, Gross ML. Fast Photochemical Oxidation of Proteins for Epitope Mapping. Anal Chem. 2011;83:7657–7661. doi: 10.1021/ac2007366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agashe VR, Shastry MC, Udgaonkar JB. Initial hydrophobic collapse in the folding of barstar. Nature. 1995;377:754–7. doi: 10.1038/377754a0. [DOI] [PubMed] [Google Scholar]
- 21.Niu B, Mackness BC, Rempel DL, Zhang H, Cui W, Matthews CR, Zitzewitz JA, Gross ML. Incorporation of a Reporter Peptide in FPOP Compensates for Adventitious Scavengers and Permits Time-Dependent Measurements. J Am Soc Mass Spectrom. 2017;28:389–392. doi: 10.1007/s13361-016-1552-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Espino JA, Mali VS, Jones LM. In Cell Footprinting Coupled with Mass Spectrometry for the Structural Analysis of Proteins in Live Cells. Anal Chem. 2015;87:7971–7978. doi: 10.1021/acs.analchem.5b01888. [DOI] [PubMed] [Google Scholar]
- 23.Gau BC, Chen H, Zhang Y, Gross ML. Sulfate Radical Anion as a New Reagent for Fast Photochemical Oxidation of Proteins. Anal Chem. 2010;82:7821–7827. doi: 10.1021/ac101760y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J, Cui W, Giblin D, Gross ML. New Protein Footprinting: Fast Photochemical Iodination Combined with Top-down and Bottom-up Mass Spectrometry. J Am Soc Mass Spectrom. 2012;23:1306–1318. doi: 10.1007/s13361-012-0403-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jumper CC, Schriemer DC. Mass Spectrometry of Laser-Initiated Carbene Reactions for Protein Topographic Analysis. Anal Chem. 2011;83:2913–2920. doi: 10.1021/ac102655f. [DOI] [PubMed] [Google Scholar]
- 26.Zhang B, Rempel DL, Gross ML. Protein Footprinting by Carbenes on a Fast Photochemical Oxidation of Proteins (FPOP) Platform. J Am Soc Mass Spectrom. 2016;27:552–555. doi: 10.1007/s13361-015-1313-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng M, Zhang B, Cui W, Gross ML. Laser-Initiated Radical Trifluoromethylation of Peptides and Proteins: Application to Mass-Spectrometry-Based Protein Footprinting. Angew Chem Int Ed. 2017;56:14007–14010. doi: 10.1002/anie.201706697. [DOI] [PMC free article] [PubMed] [Google Scholar]