

ABSTRACT
Pneumocystis species are opportunistic mammalian pathogens that cause severe pneumonia in immunocompromised individuals. These fungi are highly host specific and uncultivable in vitro. Human Pneumocystis infections present major challenges because of a limited therapeutic arsenal and the rise of drug resistance. To investigate the diversity and demographic history of natural populations of Pneumocystis infecting humans, rats, and mice, we performed whole-genome and large-scale multilocus sequencing of infected tissues collected in various geographic locations. Here, we detected reduced levels of recombination and variations in historical demography, which shape the global population structures. We report estimates of evolutionary rates, levels of genetic diversity, and population sizes. Molecular clock estimates indicate that Pneumocystis species diverged before their hosts, while the asynchronous timing of population declines suggests host shifts. Our results have uncovered complex patterns of genetic variation influenced by multiple factors that shaped the adaptation of Pneumocystis populations during their spread across mammals.
KEYWORDS: evolutionary biology, genetic diversity, genetic recombination, pneumonia, population structure
IMPORTANCE
Understanding how natural pathogen populations evolve and identifying the determinants of genetic variation are central issues in evolutionary biology. Pneumocystis, a fungal pathogen which infects mammals exclusively, provides opportunities to explore these issues. In humans, Pneumocystis can cause a life-threatening pneumonia in immunosuppressed individuals. In analysis of different Pneumocystis species infecting humans, rats, and mice, we found that there are high infection rates and that natural populations maintain a high level of genetic variation despite low levels of recombination. We found no evidence of population structuring by geography. Our comparisons of the times of divergence of these species to their respective hosts suggest that Pneumocystis may have undergone recent host shifts. The results demonstrate that Pneumocystis strains are widely disseminated geographically and provide a new understanding of the evolution of these pathogens.
INTRODUCTION
Pneumocystis species are opportunistic fungal pathogens that infect mammalian species. These organisms cause Pneumocystis pneumonia (PCP), which can be life-threatening in individuals with impaired immune systems, but they can also infect healthy individuals without any apparent clinical symptoms. Pneumocystis jirovecii, the species infecting humans, is a major cause of life-threating pneumonia in individuals with compromised immune systems, with mortality rates up to 30% (1). Organisms dwell almost exclusively in host lungs, and transmission to new hosts occurs through airborne infectious asci (84).
Pneumocystis species require a living host to survive at all stages of their life cycle. They cannot be maintained in continuous cultures, likely due to the lack of many metabolic and stress response pathways (2–5), and consequently can be obtained only from infected host tissues (<0.01% to 1% of extracted DNA).
The Pneumocystis life cycle alternates between metabolically active trophic forms and asci, which contain sexual spores (6). Haploid forms constitute 90% to 95% of the total population (7). The sexual phase takes place in mammals, which are the only known reservoirs of the organisms.
The genus belongs to the monophyletic clade of Taphrinomycotina (8), a group of early divergent fungi, which include the model organism Schizosaccharomyces pombe. Pneumocystis species harbor highly compact haploid genomes (7.4 to 8.3 Mb), with intron-rich genes. The genomic architecture is dynamic, with extensive chromosomal shuffling across species (5). Their evolution is marked by loss of genes encoding products that can be either recognized by host immune defenses (e.g., chitin and outer N-mannan chains [5]) or scavenged from the hosts (e.g., amino acids [2], cofactors [5], and sterols [9]).
Pneumocystis species are strictly host species specific as demonstrated by the consistent failures of experimental cross-species inoculations (10, 11) and have been detected in all mammalian species studied to date, suggesting an ancient long-term codivergence of these fungi with their hosts (12). These obligate and specific associations could have arisen through persistence of infections by an ancestor of Pneumocystis followed by parallel diversifications of the hosts with limited to no genetic exchanges between species contributing to the codiversification patterns. However, the validity of this scenario has not been tested using population genetic models and genomic data, and the molecular basis of host specificity is unknown.
Cophylogenetic studies have suggested that Pneumocystis species codiverge with their hosts (13), which implies a vertical mode of transmission. Although informative, these studies are based on a small number of highly conserved genes (e.g., large subunit of ribosomal DNA) and the congruence of the times of divergence has not been tested, which prevents a true understanding of the relationship between these organisms and their hosts. A survey of host-pathogen associations suggests that parasite cospeciation occurs predominantly after host shifts (14). Conflicting signals of cospeciation and host shifts have been inferred in Pneumocystis infecting rodents on the basis of two mitochondrial genes (15). However, the validity of the hypothesis of strict vertical transmission remains unchallenged.
Recent comparative genomics studies suggest that Pneumocystis species are homothallic (16), which removes the need for a partner and may greatly enhance sexual reproduction. Therefore, these fungi likely evolve as panmictic populations with no restrictions on mating. Homothallism in pathogenic fungi helps preserve a genomic structure that is well adapted for growth in the host, while still allowing organisms to undergo recombination and reshuffling of their genomes (17). The preference for inbreeding is expected to reduce diversity and the efficiency of spreading of a new allele in a population.
The population structure of Pneumocystis is not well understood and has been explored almost exclusively in P. jirovecii in the context of studying drug resistance and the epidemiology of PCP. While some studies have reported a strong local population structure mediated by the geographic distribution of susceptible individuals or clinical characteristics (8–20), other have suggested no or very limited global population structuring (21, 22). Methodological differences and the absence of whole-genome sequences make it hard to reconcile these findings. Most studies use a combination of multiple loci (typically <6), referred to as a MLG (multilocus genotype). These MLGs are often temporally stable (4 to 9 years) within the same geographic locations (23), which supports the idea of a lack of recombination among these MLGs. Others have not observed temporal clusters in MLGs and interpret this as evidence for recombination among MLGs (19). The few population structure studies in P. carinii suggest a lack of apparent structure and genetic diversity (24). Population structures of other Pneumocystis species are largely unknown.
The concept of mitotic recombination in Pneumocystis species is supported by the presence of all the genes required for homologous recombination (5, 25). Expression of subtelomeric antigenic genes is also likely mediated by telomeric recombination (26, 27). However, the frequency of recombination across the genomes is unknown. Coalescent methods are now widely used to infer population-scaled recombination rates directly from genome-wide single nucleotide polymorphism (SNP) data in a population (28). Unfortunately, key metrics such as mutation rates or population sizes are unknown for Pneumocystis.
Multiclonal infections of Pneumocystis are frequent (29), but their impact on transmission or clinical severity is unknown. Analysis of a small number of genomic regions of P. jirovecii has suggested that genetic diversity is low (30). However, genetic marker selection is of concern because most of these studies have used nonneutral loci, which can bias descriptions of population genetic structures. No equivalent analysis has been described in species infecting hosts other than humans, which calls into question the generality of these observations.
To investigate the diversity and demographic history of natural populations of Pneumocystis infecting humans, rats, and mice, we performed whole-genome and large-scale multilocus sequence typing (MLST) of 93 infected tissues collected in North America, Europe, and China. Here, we describe the genomic variation and population structures within each species.
RESULTS
Genomic variation and population structure.
We sequenced the whole genome of 52 strains of three distinct species (P. jirovecii, P. carinii, and P. murina) collected from North America and Denmark from 1983 to 2017 (see Table S1A in the supplemental material). P. jirovecii samples (n = 33) were collected from 26 patients. P. carinii samples (n = 8) were collected from rats in two animal facilities located in Bethesda (MD, USA) and Indianapolis (IN, USA). P. murina samples (n = 17) were collected from infected mice from two facilities located in Bethesda (MD, USA) and New Orleans (LA, USA). We combined these data with published whole-genome sequences from six P. jirovecii isolates collected in Lausanne, Switzerland (3, 27), one P. carinii isolate from Lausanne (3), and four P. carinii isolates from Cincinnati (OH, USA) and Bethesda (MD, USA) (5, 31) (Table S1B). Median coverage depths ranged from 0.1-fold to 1,387-fold, with 20% of the isolates having median coverage depths of greater than 5-fold. To identify genetic variants, we mapped reads to reference genomes (5). A total of 99% of the genomes could be mapped, and we applied stringent filtering to remove low (<5-fold)-coverage variants. We identified 57,764 SNPs in P. jirovecii, 56,771 in P. carinii, and 33,737 in P. murina (Table 1). Variants identified in genomic regions enriched with repeated subtelomeric gene families such as major surface glycoproteins (MSGs; ~6% of genomes) were excluded for analyses of infrapopulations.
TABLE 1 .
Genetic variation in three Pneumocystis species
| Species | Host | Annotation | No. of basesa |
Genome (%) |
No. of SNPs |
No. of insertions |
No. of deletions |
|---|---|---|---|---|---|---|---|
| P. jirovecii | Human | Genome | 8,396,240 | 100 | 57,764 | 269 | 548 |
| Exon | 5,364,089 | 63.8 | 53,466 | ||||
| Synonymous, frame conserved | 11,630 | ||||||
| Nonsynonymous, frameshift | 42,009 | ||||||
| Stop gain or loss | 3,012 | ||||||
| Start loss | 15 | ||||||
| 5′ or 3′ UTRb | 25,692 | 0.3 | 135 | ||||
| Intron | 911,414 | 10.8 | 1,139 | ||||
| Intergenicc | 2,049,052 | 24.4 | 1,409 | ||||
| P. carinii | Rat | Genome | 7,661,456 | 100 | 56,771 | 5,567 | 1,347 |
| Exon | 5,407,343 | 70.5 | 44,908 | ||||
| Synonymous, frame conserved | 9,265 | ||||||
| Nonsynonymous, frameshift | 35,768 | ||||||
| Stop gain or loss | 2,640 | ||||||
| Start gain or loss | 55 | ||||||
| 5′ or 3′ UTR | 292,852 | 3.8 | 1,687 | ||||
| Intron | 880,208 | 11.5 | 2,920 | ||||
| Intergenic | 1,379,590 | 18 | 3,395 | ||||
| P. murina | Mouse | Genome | 7,451,359 | 100 | 33,737 | 776 | 402 |
| Exon | 5,834,951 | 78.3 | 34,015 | ||||
| Synonymous, frame conserved | 6,340 | ||||||
| Nonsynonymous, frameshift | 25,725 | ||||||
| Stop gain or loss | 2,046 | ||||||
| Start gain or loss | 19 | ||||||
| 5′ or 3′ UTR | 330,022 | 4.4 | 1,291 | ||||
| Intron | 880,789 | 11.8 | 154 | ||||
| Intergenic | 1,411,549 | 18.9 | 1,208 |
Bases and annotations refer to the reference genomes (5).
UTR, untranslated region.
Intergenic regions include 500 bp upstream and 500 bp downstream of each gene and may overlap those of other genes.
(A) Clinical information and demographic data of the groups of individuals involved in the study (as determined using WGS). (B) Published data sets used in this study (WGS). (C) Clinical information and demographic data of the groups of individuals involved in the study (next-generation sequencing [NGS]-based amplicon multilocus sequence typing [MLST]). (D) Clinical information and demographic data of the groups of individuals involved in the acquisition of Pneumocystis jirovecii mitochondrion full-genome study data. Download TABLE S1, DOCX file, 0.03 MB (34.3KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Preliminary investigation of 17 P. murina isolates indicated the presence of clonal populations (see Fig. S1 in the supplemental material). As most of these isolates were from a limited number of animal facilities, they probably derived from isolated reproducing populations. A maximum likelihood phylogenetic tree based on pairwise SNP differences generated for P. jirovecii strains provides no evidence of geographic clustering (Fig. 1).
FIG 1 .

Phylogenomic analysis reveals a lack of global population structuring in Pneumocystis jirovecii. Each symbol represents a single P. jirovecii isolate. The color-coding scheme indicates the region (city or state and country) in which the isolate was obtained. There is no clustering by location but instead an intermixing of isolates from diverse geographic locations, consistent with a lack of geographic structure. The phylogenetic tree was estimated from genome-wide SNP differences of Pneumocystis jirovecii isolates sequenced in this project (832,669 segregating sites).
Initial analysis of Pneumocystis murina samples. The dendrogram was constructed from results of a hierarchical cluster analysis of 29,692 biallelic SNPs using z scores and an outlier threshold value of 5 to split the nodes. The population level of inbreeding was measured using coancestry coefficients, which are measures of relatedness between individuals in a population. Coancestry is expressed as the probability of identity by descent (IBD). Individual dissimilarities are calculated for each pair of individuals. The graph shows that most of the isolates were from clonal populations as defined by dissimilarity and coancestry coefficient values above 1 and below 0.5, respectively. The calculated kinship coefficient values, i.e., those corresponding to twins, a first sibling (FS), a half-sibling (HS), and a first cousin (FC), which are probabilistic measures of relatedness, are presented on the right axis. Download FIG S1, TIF file, 0.3 MB (278.1KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
The average level of pairwise diversity within 42 P. jirovecii strains was 1.3 × 10−2 substitutions per site, which is higher than previously reported for a comparison of two isolates (5), 5.4 × 10−3 substitutions/site for 14 P. carinii strains and 6.5 × 10−4 for 17 P. murina strains. These values are similar to the diversity levels observed within the yeasts Saccharomyces cerevisiae (5.7 × 10−3) (32) and Schizosaccharomyces pombe (3.0 × 10−3) (33). The reduced levels of polymorphisms in P. carinii and P. murina are characterized by high frequencies of rare alleles (negative Tajima’s D), increased high frequencies of derived alleles (negative Fay and Wu’s H), and stronger linkage disequilibrium (LD) in P. carinii (Fig. 2).
FIG 2 .

Comparison of population genetic statistics, nucleotide diversity (π), Tajima’s D, Fay and Wu’s H, and linkage disequilibrium (r2) in three Pneumocystis species. (A) Box plots of genome-wide nucleotide diversity. The data show elevated median levels of nucleotide diversity in P. jirovecii relative to P. carinii and P. murina populations. (B and C) Box plots of summaries of site frequency spectra (Tajima's D [B] and Fay and Wu’s H [C]) data show that these statistics are skewed toward low-frequency variants in P. carinii and, to a lesser extent, in P. murina but not in P. jirovecii (negative Tajima’s D values), which suggests that these species experienced different demographic events. (D) Box plots of squared correlations (r2) between pairs of SNPs indicate strong signals of linkage disequilibrium (LD) in P. jirovecii and P. carinii. Signs above the bars indicate significant differences between species determined by the Mann-Whitney U test (***, P value of <2.2 × 10−16). The boxes indicate medians and interquartile ranges, whiskers indicate 95% values, and additional points in each box plot represent outliers.
To describe the relatedness among strains in each species, we focused on SNPs located in the nuclear genomes. Pneumocystis species are predicted to be haploid and self-fertile (homothallic) (7, 16) and present substantial chromosomal shuffling among species (5). These traits are considered strong suppressors of recombination (34). Estimates of population structure can be biased by the presence of large inversions as these genomic regions are inherited without recombination (35). To explore the population structure, we selected sets of SNPs in P. jirovecii (n = 3,515), P. carinii (n = 6,696), and P. murina (n = 29,692) that were close to linkage equilibrium (pairwise r2, <0.5) and were randomly distributed across the genomes (Monte Carlo permutation test P = 0.0019; Fig. S2). These SNPs are more appropriate for population genetic models than SNPs in linkage disequilibrium, since their use assumes widespread recombination and no linkage between markers. In P. jirovecii, a principal-component analysis (PCA) of these SNPs indicated no evidence of clustering of strains by geography (Fig. S3). The mean Fst value (representing the proportion of between-population genetic variance for two populations) for comparisons between strains from the United States and Europe is 0.039, which indicates some population divergence. A similarly weak population structure was obtained with P. carinii strains, although nearly all samples were from diverse geographic regions of the United States (Fig. S3). Although no geographic clustering was possible for P. murina strains because of their common sampling origin, we found no evidence of clustering by collection dates (Fig. S3). Further phylogenetic and population genetic analysis (Hudson index) using P. jirovecii internal transcribed spacer 1 (ITS1) sequences extracted from our samples combined with published sequences from outbreaks in different places around the world revealed no evidence of clustering by geography (see Text S1 in the supplemental material; see also Fig. S4A and B at https://doi.org/10.5281/zenodo.1215631). This analysis was not conducted with P. carinii and P. murina because of the relatively low number of samples with distinct geographic origins.
Description of the methodology used for the analysis of genetic subdivisions, phylodynamics, within-individual populations, multilocus sequencing and analysis, evolutionary rates, and Pneumocystis macacae transcriptome sequencing. Download TEXT S1, DOCX file, 0.1 MB (103.1KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Random distribution of SNPs at linkage equilibrium. Polymorphic site (SNP) positions were analyzed using a Monte Carlo test implemented in the R package adegenet to determine if they are randomly distributed across chromosomes. The significance data are based on 500 replicates. Density plots of representative chromosomes, which clearly indicated a random distribution of SNPs (P value = 0.0019), are shown. The first chromosome (“chr1”) in each species corresponds to the following NCBI accession numbers: LFWA01000001 (P. jirovecii), LFVZ01000001 (P. carinii), and NW_006920852 (P. murina). Download FIG S2, TIF file, 1.1 MB (1.1MB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Principal-component analysis. Results of analysis of SNP sites at linkage equilibrium indicate a lack of population structure in three Pneumocystis species. (A and B) PCA of P. jirovecii isolates with (A) and without (B) outliers. The following codes are used: DEN (Denmark), CIN (Cincinnati, OH, USA), IND (Indianapolis, IN, USA), Bethesda (MD, USA), and SWZ (Switzerland). The P. murina samples for which whole-genome sequences could be obtained were all from Maryland (USA), and the color codes correspond to collection dates. There was no geographic clustering for P. jirovecii and P. carinii species and no apparent temporal clustering for P. murina populations. Download FIG S3, TIF file, 0.4 MB (452.2KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
(A) Haplotype network of Pneumocystis jirovecii in 13 patients based on the internal transcribed spacer 1 (ITS1) locus. Major haplotypes are m4_seq4, m3_seq2, and m19_seq1. (B) Geographic population structure of Pneumocystis jirovecii isolates based on internal transcribed spacer (ITS1) locus. An integer neighbor-joining network of 189 ITS1 sequences, including 48 sequences from this study and 141 previously reported in GenBank, is presented. For both panel A and panel B, each circle corresponds to a haplotype and its size is proportional to the total frequency. Circles are colored according to the isolate (A) or to geographic origin (B). Small circles represent single sequences, and line lengths represent pairwise nucleotide differences. Download FIG S4, PDF file, 0.4 MB (380.8KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Pneumocystis species show moderate clustering of strains as determined by local geography and the distribution of susceptible host populations. Investigation of P. jirovecii population structures has provided evidence of both weak global population structures and local geographic clusters (20, 22, 36). To assess the relationship in our samples, we applied unsupervised genetic clustering methods NGSAdmix (37) and fastStructure (38), which do not account for the geographic origins of the strains and rely on different algorithms (maximum likelihood and Bayesian) to detect population genetic partitioning patterns. No evidence of statistically significant clusters was detected in any of three species (see Fig. S5 at https://doi.org/10.5281/zenodo.1215631), which suggests a lack of population partitioning by geography and is consistent with a previous report (22).
Population structure of Pneumocystis determined by analysis using whole-genome sequence data. Results of analysis of genetic clustering on segregating sites identified in surveyed strains are presented. The estimated individual admixture was inferred using genotype likelihoods. Each isolate is represented by a single vertical bar, which is partitioned into K colored segments that represent the isolate’s estimated ancestry proportion in each of the K clusters. A single color represents an absence of admixture, whereas stacked colors show proportions of admixtures in each isolate. The color codes differ between the plots because the samples from each species were analyzed separately. Genetic clusters are ordered according to the geographic locations using the following codes: DEN (Denmark), CIN (Cincinnati, OH, USA), IND (Indianapolis, IN, USA), MD (Maryland, USA), and SWZ (Switzerland). Rat samples from CIN samples correspond to two distinct whole-genome sequence data sets corresponding to sequences previously published by Slaven and colleagues (31) and to the latest release of the genome in 2010 (http://pgp.cchmc.org). We assume that these data sets were obtained from at least two infected animals, while the possibility cannot be ruled out that they might represent multiple aliquots from the same animal. The IND sample was a generous gift from Chao-Hung Lee at Indiana University (USA). The P. murina samples for which whole-genome sequences could be obtained were all from Maryland (USA), so the genetic clusters are organized by collection dates instead of geographic locations. The population structure analysis does not show a clear separation of lineages in all three Pneumocystis species. Download FIG S5, TIF file, 0.5 MB (474.7KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Recombination landscape.
Meiotic recombination creates diversity influencing both natural population dynamics and selection. The genomic extent of linkage disequilibrium (LD), which represents the nonrandom association of alleles at physically distinct genomic loci, is expected to be reduced by recombination. LD is negatively correlated with recombination rates and thus provides information about the recombination frequency and population structure. We analyzed pairwise LD for segregating sites in P. jirovecii (n = 16,159), P. carinii (n = 22,904), and P. murina (n = 3,284). These data sets included exclusively biallelic SNPs, and variants located in multicopy major surface glycoproteins were excluded. The disequilibrium decayed to half of its maximum value at ~16 kb and fell below 0.2 after 100 kb in two species (P. jirovecii and P. carinii) (Fig. 3), which suggests low levels of recombination and rare outcrossing events. This remained true for P. jirovecii even when the analysis was restricted to the United States isolates (data not shown). We could not obtain reliable estimates of LD decay for P. murina because the r2 values could not be fitted to Hill’s decay function (39). In comparison, the LD fell to half of its maximum value at ~3 kb for S. cerevisiae (32) and fell below 0.2 at 67 kb for the selfing anther smut fungal pathogen Microbotryum (40).
FIG 3 .
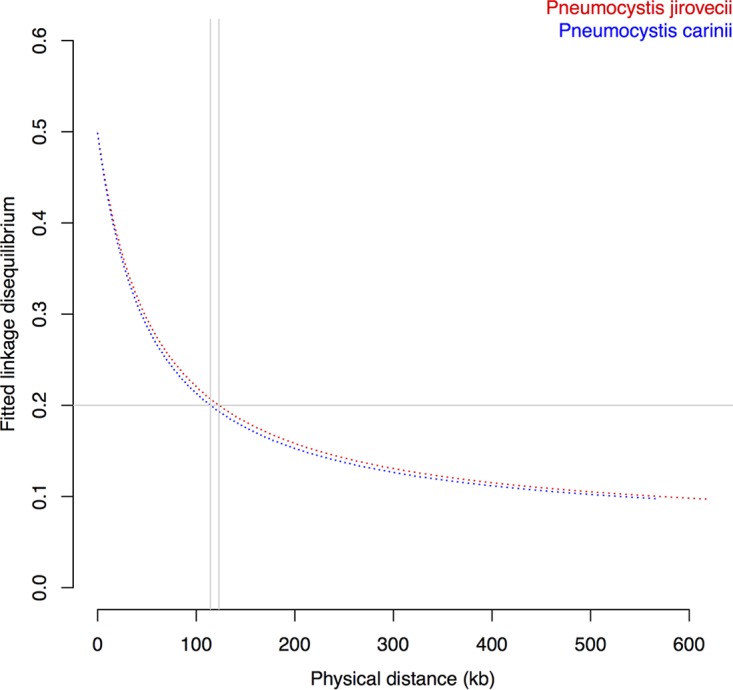
Decay of linkage disequilibrium (LD) as a function of distance. LD levels measured by the square of the correlation coefficient between two markers (r2) were calculated for all pairs of biallelic SNPs within a 1-kb genomic window and averaged. LD decay levels were below 0.2 at 123 and 114 kb for Pneumocystis jirovecii and P. carinii, respectively (shown as gray vertical lines). The low rates of LD decay suggest low rates of recombination in Pneumocystis, which would be at least 2-fold lower than Saccharomyces cerevisiae recombination rates (32).
For P. carinii, we estimated a mean population scale recombination rate of ρ = 8.4 × 10−3 event per base pair per generation, assuming an effective population size (Ne) of 7,500 individuals (Ne values are derived from BEAST analyses as described in Text S1) and a generation length of 4.5 days (41) (~80 mitotic events per year). In P. jirovecii, we obtained a mean ρ value of 3.1 × 10−4, assuming a Ne level of 46,250 individuals before population decline and a generation time of 4.5 days. Of note, the generation length for P. jirovecii is not known. The reported generation times for other Pneumocystis species range from 1.7 to 10.5 days (41).
Within-individual infection complexity.
Pneumocystis jirovecii pneumonia cases are often caused by multiclonal infections (29). In viruses, multiclonal infections increase the likelihood of recombination among individuals of one species that occur in the same host individual (infrapopulation) (42). To investigate the genetic complexity of our isolates, we used genome-wide read sequence alignments to estimate the multiplicity of infections (MOI) of each isolate. The MOI here refers to the number of different parasite genotypes coinfecting a single individual. Only samples with a genome-wide sequencing coverage level of >5-fold were selected, and genomic regions, including those corresponding to multicopy surface glycoprotein families, were excluded. Overall, 63% of all isolates had genetically mixed infections, with a median of 2 populations per sample (see Text S1; see also Fig. S6 at https://doi.org/10.5281/zenodo.1215631).
Scatterplot of genome-wide multiplicity of infection (MOI) estimates from whole-genome sequencing data excluding multicopy gene families across Pneumocystis isolates. The MOI value refers to the number of different genotypes coinfecting a single host, and each dot represents an individual isolate. P. jirovecii isolates (n = 7) are shown in green, P. carinii isolates (n = 9) in orange, and P. murina isolates (n = 23) in blue. Multiplicity of infection (MOI) values are plotted against the fraction of the genome covered per sample (x axis). Download FIG S6, TIF file, 0.3 MB (294.9KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Because many samples were not amenable to direct genome sequencing due to their low Pneumocystis DNA content, we extracted panels of polymorphic sites from whole-genome-sequencing (WGS) data (2,886 SNPs in 114 loci in P. jirovecii, 1,874 SNPs in 93 loci in P. carinii, and 817 SNPs in 35 loci in P. murina). We used PCR to capture these SNPs in 22, 8, and 13 samples for each species, respectively, and sequenced the PCR products by Illumina sequencing (Table S1C and Text S1). SNP allele frequencies were used to infer the number of strains present in each sample.
We first utilized the internal transcribed spacer 1 (ITS1) locus in P. jirovecii for MLST studies because this is the only locus for which sequences are available from multiple outbreaks around the world. The ITS1 locus is in a single copy per genome in Pneumocystis species (43), has an evolutionary rate similar to that of other fungi (44), and has been extensively used for epidemiological studies. In analyzing 48 ITS1 sequences from 13 patients, we detected three major haplotypes present in most samples (Fig. S4A). Haplotypes clustered more frequently with sequences from different individuals instead of clustering exclusively with those from the same individual, suggesting recurrent introductions of organisms or introductions of multiple populations at one time. To compare these results to other data sets, we merged these 48 sequences with 141 published ITS1 sequences (Fig. S4B). Analysis of nucleotide differences revealed a low level of genetic diversity (π = 0.0052; Tajima’s D, −2.62) and no recombination at this locus (P = 0.35). Our 48 ITS1 sequences fell into two categories: 81% were nearly identical to published sequences, whereas 19% were novel sequences shared by different patients in this study. We found that some strains clustered independently of geography, which, together with the high frequency of multiple infections, suggests high transmission efficiency within host populations.
Neighborhood connectivity analysis of 56 networks (with each network corresponding to a distinct locus in addition to ITS1) that included 292 sequences confirmed that infections by multiple genetically distinct populations are more frequent than clonal expansions (78.4% versus 21.6%). Similar findings were obtained with P. carinii (26 networks, 140 haplotypes in 12 rats, 68.5% with multiclonal infections) and P. murina (28 networks, 223 haplotypes in 13 mice, 92.3% with multiclonal infections). Recombination was detected in ~3% of loci (pairwise homoplasy index test P < 0.005). The overrepresentation of multiple introductions over clonal expansions strongly suggests that these haplotypes are widespread in the environment.
Estimation of the evolutionary rates.
Pneumocystis species are globally distributed, but their evolutionary rate is unknown. To accurately estimate evolutionary rates, we sequenced 10 full-length P. jirovecii mitochondrial genomes and used five that were previously reported (3, 45). The samples were collected from 1986 to 2013 from different geographic locations in the United States, Europe, and China (Table S1D). Fitting root-to-tip regression data indicated a diffuse positive relationship between the sampling dates (years) and the expected number of nucleotide substitutions (r2 = 0.41), demonstrating the presence of a temporal signal suitable for phylogenetic molecular clock analysis (Fig. 4) (a detailed protocol is presented in Text S1). Mantel tests as implemented in Murray’s R scripts (46) showed no evidence for confounding factors of temporal and genetic structures (P = 0.23). Bayes factors favor relaxed over strict clocks (log BF = 5.4), which indicate variable genetic rates over the mitogenomes. We used the BEAST framework (47) to account for variations in population size and relaxation in molecular clock data. We found an estimated rate of evolution of 5.9 × 10−5 substitutions per site per year.
FIG 4 .

Estimation of evolutionary rates using P. jirovecii mitogenome data. (A) Midpoint rooted maximum likelihood phylogenetic tree of 15 full-length mitochondrial genomes, with bootstrap values shown on the branches. The tree tips indicate the collection date (year) and the sample identifier; the color code indicates the country of origin for the sample. (B) Root-to-tip distances (numbers of substitutions per site × 10−3) significantly correlate with the collection dates, indicating the presence of temporal signals. Calculations of regression of distances against the dates were performed using Murray’s R scripts (46). Statistical significance data are based on 1,000 random permutations.
Changes in past population sizes.
We evaluated different population modes and applied the extended Bayesian skyline model implemented in BEAST. To infer the demographic history, different coalescent models of effective population size (Ne) as well as molecular clocks were tested using only samples for which collection dates were available (Text S1). Loci encoding MSG sequences were excluded. Our data sets include 36 P. jirovecii isolates collected from 1983 to 2017 (65 nonrecombinant nuclear gene loci and 15 full-length mitogenomes) and 11 P. carinii isolates collected from 1994 to 2010 (34 nonrecombinant nuclear gene loci). P. murina isolates were not investigated because the sampling was limited to a single animal facility. We evaluated two parametric models (constant size and exponential growth) and one nonparametric model (extended Bayesian skyline plot [EBSP]) using our estimated evolutionary rate of 5.9 × 10−5 substitutions/site per year. Initial analysis supported the extended Bayesian skyline model against the null hypothesis of constant size and exponential models (log BF = 3.69). We used the EBSP model and strict clocks to reconstruct historical demographics. In both P. jirovecii and P. carinii, the EBSPs rejected a constant size model because the 95% highest posterior density (HPD) excluded the value 0. Population size change tests for P. jirovecii indicated significant signals of reduction of effective population size, which started about 400,000 years ago (Fig. 5). A decline in population size was observed in P. carinii populations, with a severe bottleneck around 16,000 years ago.
FIG 5 .

Extended Bayesian skyline plot (EBSP) results for populations of two Pneumocystis species. The gray areas represent the upper and lower 95% highest posterior density (HPD) data, and the black dashed line represents the median estimate of 95% highest-posterior-density bounds. (A) Each graph represents the results of an EBSP analysis for Pneumocystis jirovecii (A1) and for P. jirovecii (A2), with the timeline restricted to 500,000 years before now. The purple dot indicates the approximate time of population decline. (B) Pneumocystis carinii (B1) and P. carinii (B2). The timeline was restricted to 20,000 years before now. The y axis shows the effective population size (Ne) per generation time (~4.5 days).
Dating the species divergence.
To place these events in the context of speciation, we estimated the timing of species divergence using a time-calibrated phylogeny. To improve the resolution of the tree, we first sequenced the transcriptome of the Pneumocystis species infecting rhesus macaques, which we have designated in the manuscript as P. macacae (formerly referred to as Pneumocystis carinii f. sp. macacae [NCBI taxon identifier 112250]) (see Text S1 in the supplemental material). We built two nuclear gene data sets. Data set 1 contains exclusively Pneumocystis and related Taphrinomycotina fungi (43 orthologs), and data set 2 contains 10 genes conserved in Pneumocystis and their respective hosts despite an estimated divergence time of ~1.2 billion years (48). The sole purpose of data set 2 was to infer the divergence times of Pneumocystis species and their hosts in the same analysis so that the species estimates would be comparable. The low number of genes in data set 1 was due to the high fragmentation of the low-coverage P. macacae transcriptome assembly. We used fossil-calibrated nodes as priors instead of evolutionary rates because of differences in the rates of heterogeneity across sites among distantly related species.
The divergence times for the two data sets were similar, although some discrepancies were observed (see Fig. S7A and B at https://doi.org/10.5281/zenodo.1215631). The well-supported maximum clade credibility coalescent tree (posterior probability of 1) indicated a median value of 123 million years ago (Mya) (95% confidence interval [CI], 104.4 to 145.5) for an initial emergence of Pneumocystis (Fig. 6), which is consistent with previous estimates (49). Our dated phylogenetic trees, based on conserved genes in both Pneumocystis and their hosts, indicated that P. jirovecii and P. macacae diverged ~65 Mya ago (95% CI, 51.4 to 72.5) and humans and macaques ~15 Mya ago (95% CI, 13 to 17), which suggested that P. jirovecii and P. macacae may have diverged before their respective hosts. P. jirovecii populations experienced a decline (0.4 Mya; Fig. 5), which might indicate a host shift. Our estimated divergence times also indicated that P. carinii and P. murina species diverged before their hosts (42 Mya [95% CI, 35.8 to 49.8] versus mouse-rat separation at 25 to 32 Mya ago). The presence of a distinctive bottleneck at 16,000 years might again indicate a host shift (Fig. 5).
FIG 6 .

Overview of the timing and evolution of Pneumocystis and their mammalian hosts. (A) Phylogeny and coalescent divergence time estimates of Pneumocystis and their mammalian hosts were determined using nuclear genic alignments. The divergence time estimates (in millions of years) are displayed at the nodes. (B) Time line of events in Pneumocystis evolution relative to the mammal evolutionary history. For all Pneumocystis species, the time of divergence precedes the time of divergence of their respective hosts, which is inconsistent with the hypothesis that Pneumocystis coevolved with its hosts.
(A) Time-calibrated phylogeny of Pneumocystis species and their mammalian hosts. A maximum clade credibility tree of Pneumocystis species was obtained from 10 orthologous nuclear genes using BEAST2. The 95% highest-posterior-density (HPD) intervals (i.e., confidence intervals) are presented at the nodes, and the blue bands are proportional to confidence intervals. The scale bar at the bottom depicts time in millions of years back in time toward the left. The following codes are used: MACA (Macaque), PCAR (P. carinii), PMUR (P. murina), PJIR (P. jirovecii), and PMK (P. macacae). (B) Time-calibrated phylogeny of Pneumocystis species. A maximum clade credibility tree of Pneumocystis species was obtained from 43 orthologous nuclear genes using BEAST2. The 95% highest-posterior-density (HPD) intervals (i.e., confidence intervals) are presented at the nodes, and the blue bands are proportional to confidence intervals. The scale bar at the bottom depicts time in millions of years back in time toward the left. Download FIG S7, PDF file, 0.3 MB (300.6KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
DISCUSSION
The genetic diversity of a species is strongly correlated with life history traits such as recombination rates, mating systems, gene densities, generation times, and mutation rates (50). Our results suggest that recombination levels are low for Pneumocystis, which would be consistent with the presence of recombination reducing factors such as selfing and asexuality (34) and, in theory, should lead to reduced genetic diversity. However, we found that genetic diversity in P. jirovecii and P. carinii is similar to that of free-living yeasts, despite the fact that Pneumocystis species appear to reside almost exclusively in the host lungs, which is where diversity must develop, rather than in the external environment. One possible explanation is that outcrossing events (mating between nonrelatives) occur but might be limited to inbred subpopulations. This would seem to be supported by the fact that we detected ~3% of intralocus recombination among MLST data from multiple patients. Another possibility is that there is a selective advantage in maintaining diversity (standing genetic variation [51]), for example, to avoid detection by the host immune system. Alternatively, high rates of coinfection with genetically different parasite populations could theoretically increase genetic diversity via competitive interactions among them (52), which would be consistent with our findings that most Pneumocystis infections are multiclonal. We tend to favor the last possibility, although a combination of multiple scenarios is possible. The transmission and population structures of Pneumocystis are likely influenced by the constant movement of human populations around the globe, which has potentially contributed to mixing of genetically distinct Pneumocystis populations following the advent of widespread global travel. In this context, immunocompetent individuals are powerful agents of dissemination.
Low levels of recombination and strong population structure represent a challenge for most population genetic models. Gold standard summary statistics such as Tajima’s D depend on recombination rates (53). We applied the recommended methodology in this situation by combining different neutrality tests (54) and by separately analyzing regions with sufficient levels of recombination.
We did not attempt to detect positive selection (i.e., selective sweeps) because current methods to detect such events are sensitive to strong LD and assume widespread recombination (55). Demographic variations (e.g., bottlenecks and founder effects) also impair the detection of positive selection due to their confounding nature. A reliable long-term culturing method would ideally be available to investigate the genome-wide impact of selection in these populations.
We found diffuse positive relationships between collection dates and genetic distances. Several factors may explain this distortion in the regression. First, root-to-tip regression assumes that all branches evolve at a constant rate (strict clock) and that there is no population structure (56). We found that relaxed molecular clocks are more appropriate to our mitogenome data. A weak geographic structure may exist even if it is not statistically significant (Mantel test P = 0.23). Regression correlations were improved by analyzing P. jirovecii mitogenomes from North America and Europe separately, although the P values were not significant (data not shown). This initial finding prompted us to consider an alternative strategy incorporating variations in evolution rates across branches as well as population size. The use of an extremely short sampling period (57) or oversampling of closely related sequences (imbalanced trees) (58) could also lead to biased estimates. These factors should not significantly impact our results because our samples were collected over 27 years from different continents and contained a significant amount of genetic divergence. Nonetheless, our estimation of evolutionary rates presents some caveats: (i) the calculated rates are based on P. jirovecii mitogenomes, but they might be different in other species; (ii) the P. jirovecii samples were from individuals and were probably collected following an antifungal treatment, which could affect the evolutionary rates. For instance, positively selected mutations in dihydrofolate synthase have been associated with sulfa/sulfone prophylaxis (59).
Our estimates of species divergence strongly suggest that Pneumocystis species diverge before their hosts. This means, for example, that P. jirovecii, which is uniquely able to infect humans, was probably infecting another (possibly extinct) species before the divergence of human and macaque lineages. This suggests that Pneumocystis species do not strictly cospeciate along with their hosts and that host shifts likely occurred at some point in their evolution. The P. jirovecii populations seem to have been constant for ~15 Mya, possibly since their separation from P. macacae, and suddenly to have declined at ~0.4 Mya, which roughly coincides with the emergence of Homo sapiens species 0.4 to 0.7 Mya ago (60). Given our relatively small size sampling (<50 individuals), it is possible that our Bayesian demographic reconstructions lack power to capture recent population expansions, as shown by simulations (61). We speculate that this sudden population decline could indicate a host shift. The continuous population declines of Pneumocystis might indicate that these species are heading toward extinction as a consequence of the activity of mechanisms such as Muller’s ratchet (62). The strict host species specificity of Pneumocystis is consistent with this scenario because extreme host specialization of parasites often results in extinction (63).
Pneumocystis organisms exhibit diversity in karyotype patterns among species (64, 65). Unfortunately, the karyotype variability cannot be addressed in the context of the current study since karyotypes are not available for the samples that were used in this study. For karyotyping, fresh samples need to be processed in a very short period of time in a manner that minimizes degradation of DNA, and we utilized frozen samples that in a number of cases had been stored for many years and thus could not be used for karyotyping.
P. wakefieldiae is another species that infects rats and which is almost always found associated with P. carinii. None of the rat samples that we used contained P. wakefieldiae (verified at the sequence level); therefore, we cannot comment on the evolutionary profile of P. wakefieldiae or on its potential interaction with P. carinii.
The lack of a culture system and the difficulty in obtaining sufficiently pure Pneumocystis DNA are major obstacles in the research field. Bronchoalveolar lavage (BAL) fluid samples, which represent the most accessible source for P. jirovecii DNA, are not suitable for WGS without enrichment. The use of whole-genome amplification methods is not ideal but is necessary to obtain sufficient sequence data. By scanning the sequencing depth in our samples, we found no evidence of preferential amplification or reduction of particular sequences. We minimized these effects by applying stringent bioinformatics filtering to produce a comprehensive catalog of polymorphisms that reasonably capture genetic variation patterns.
Conclusion.
Our results demonstrate the feasibility of comparative population genome analyses of uncultivable organisms directly obtained from host tissues even with low levels of infection. Our results have uncovered complex patterns of genetic variation influenced by multiple factors that ultimately shape the adaptation of Pneumocystis populations during their spread across mammalian hosts. These results provide a dynamic view of the evolution of Pneumocystis populations and improve our understanding of its transmission.
MATERIALS AND METHODS
Sampling.
Animal and human subject experimentation guidelines of the National Institutes of Health were followed in the conduct of these studies. P. jirovecii samples were obtained from infected autopsy lung tissues or BAL fluid pellets; some samples had been utilized in previous studies (3, 5). P. murina-infected lung samples were obtained from CD40 ligand knockout mice (66), and P. carinii-infected lung samples were obtained from immunosuppressed Sprague-Dawley male rats. Rhesus macaque lung samples were obtained from a simian immunodeficiency virus (SIV)-infected animal with PCP.
Nucleic acid extraction and sequencing.
Genomic DNA was extracted using a MasterPure yeast DNA purification kit (Epicentre). Total RNA was extracted using an RNeasy minikit (Qiagen).
Samples with low Pneumocystis DNA content (mostly BAL fluid samples) were enriched by the use of a NEBNext microbiome DNA enrichment kit (New England Biolabs, Inc.) that selectively removes CpG-methylated host DNA and preserves the microbially diverse populations. Enriched samples were purified by ethanol purification. Five microliters of each DNA was amplified in a 50-vol reaction using an Illustra GenomiPhi DNA V3 DNA amplification kit (GE Healthcare, United Kingdom). Purified samples were bar-coded, pooled, and sequenced using an Illumina HiSeq platform with a Nextera library preparation kit. P. jirovecii full-length mitogenome DNA was extracted, sequenced as described previously (45), and annotated using MFannot (https://github.com/BFL-lab/Mfannot).
Read alignment, SNP calling, and filtering.
DNA reads were aligned to high-quality reference genomes of P. jirovecii, P. carinii, and P. murina (5) using BWA-MEM v.0.7 (67). Alignment files (BAM) were filtered using Picard v.2.1.1 (http://broadinstitute.github.io/picard/) with the MarkDuplicates option. Individual variant calls were performed using Genome Analysis Toolkit (GATK) v3.5-0 with the HaplotypeCaller module (68) and following recommended best practices (69). The “VariantFiltration” module was applied using the following criteria to remove false positives: -window 35 -cluster 3 -filterName FS -filter “FS > 30.0” -filterName QD -filter “QD < 2.0.” Multisample variant call format (VCF) files were generated using FermiKit (70). SNPs were annotated using snpEff (71). Polymorphic site positions were analyzed using R package adegenet v.2.0.1 (72).
Population structure and recombination rates.
We used filtered variants (VCF) to visualize relationships between isolates using principal-component analysis (PCA) implemented in SNPRelate v.1.10.2 (73). Summary statistics were computed using VCFtools (74). Neutrality tests were performed using ANGSD (75). Genetic clustering was performed using NGSAdmix (37) and fastStructure (38).
The linkage disequilibrium was calculated as r2, which indicates the square of the correlation coefficient between two SNPs, using VCFtools and excluding singletons and multiallelic SNPs. The values corresponding to the mean LD for each nonoverlapping 1-kb window within the genomes were averaged. The estimate decay of LD with distance was determined by fitting the observed r2 values to the decay function (39) in R. The population-scaled recombination rates (ρ = 2 Ner [where Ne represents the effective population size and r is the recombination rate]) were calculated using whole-genome scaffolds and excluding small chromosomes enriched with MSGs using the INTERVAL program in LDhat v.2.2 (76). Data sets for use with the INTERVAL program were extracted from VCF files using VCFtools. We excluded multiallelic sites and sites with minor-frequency allele values of <0.1 or with a proportion of missing data of >0.5. INTERVAL’s Markov chain Monte Carlo scheme was run for 1e6 iterations, with a block penalty of 5, a sampling step performed every 5,000 iterations, and a burn-in phase of 100. Likelihood look-up tables were generated from precomputed tables using LDhat’s lkgen program (https://github.com/auton1/LDhat) and adjusted for Pneumocystis total nucleotide diversity estimated from 10-kb nonoverlapping windows. Results were summarized using Stat Interface LDhat with a 10% burn-in value.
Species divergence dating.
Nuclear gene sequences in the following categories were downloaded from NCBI (last accessed January 2018): fungi (P. jirovecii, P. carinii, P. murina, Schizosaccharomyces pombe, and Taphrina deformans) and mammals (Homo sapiens, Macaca mulatta, Rattus norvegicus, and Mus musculus). One-to-one orthology was inferred using a reciprocal best BLASTN hit with an E value of 10−10 as the threshold (77). Two data sets were constructed. The first data set included only fungi, and the second data set included Pneumocystis species and their mammalian hosts. Data set 1 included 43 single-copy orthologs, and data set 2 included 10 genes. Multiple alignments were generated using MUSCLE (78), manually inspected for inconsistencies in Jalview (79), and concatenated. The best-fitting substitution model was the GTR+G model as estimated by jModel v.2.1.7 (80) using the corrected Akaike information criterion. Divergence times were estimated using BEAST2 v.2.4.6 (47). For data set 1, we applied an internal calibration point corresponding to the separation of S. pombe and T. deformans (median, 467 MYA) (81) as a constraint following a lognormal prior distribution. For data set 2, we applied calibration points to the human-macaque node (median divergence, 28 MYA) (82) and to the mouse-rat node (16 MYA) (83). BEAST inputs were prepared using BEAUTi with a strict clock model, and a calibrated Yule model was used to estimate the divergence times and the credibility intervals. Trees were summarized using TreeAnnotator (http://beast.bio.ed.ac.uk/treeannotator) and visualized using FigTree v.1.4.2 (http://tree.bio.ed.ac.uk/software/figtree) to obtain the means and 95% higher posterior densities (HPD).
Data availability.
All sequence data have been linked to NCBI Umbrella project PRJNA385300. The mitochondrial genomes have been deposited into GenBank under accession codes MH010437 to MH010446. Data sets used for molecular clocks and phylodynamic analyses are available at https://doi.org/10.5281/zenodo.1215631.
ACKNOWLEDGMENTS
We thank the Broad Institute and Leidos genomics platforms for Illumina and 454 sequencing. We also thank Jung-ho Youn for performing MISEQ Illumina sequencing. We thank Jannik Helweg-Larsen, Müller Nicolas, Andreas Sing, and Laurence Huang for providing P. jirovecii samples for mitogenome sequencing. We thank Chao-Hung Lee for providing several P. carinii samples. This study used the Office of Cyber Infrastructure and Computational Biology (OCICB) high-performance computing (HPC) cluster at the National Institute of Allergy and Infectious Diseases (NIAID), Bethesda, MD.
This project was funded in whole or in part with federal funds from the Intramural Research Program of the United States National Institutes of Health Clinical Center; the National Institute of Allergy and Infectious Diseases; the National Cancer Institute; the National Institutes of Health under contract HHSN261200800001E; and the National Human Genome Research Institute (grant U54HG003067 to the Broad Institute). P.M.H. and M.P. hold grant number 310030_165825 of the Swiss National Science Foundation.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Citation Cissé OH, Ma L, Wei Huang D, Khil PP, Dekker JP, Kutty G, Bishop L, Liu Y, Deng X, Hauser PM, Pagni M, Hirsch V, Lempicki RA, Stajich JE, Cuomo CA, Kovacs JA. 2018. Comparative population genomics analysis of the mammalian fungal pathogen Pneumocystis. mBio 9:e00381-18. https://doi.org/10.1128/mBio.00381-18.
REFERENCES
- 1.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, White TC. 2012. Hidden killers: human fungal infections. Sci Transl Med 4:165rv13. doi: 10.1126/scitranslmed.3004404. [DOI] [PubMed] [Google Scholar]
- 2.Hauser PM, Burdet FX, Cissé OH, Keller L, Taffé P, Sanglard D, Pagni M. 2010. Comparative genomics suggests that the fungal pathogen pneumocystis is an obligate parasite scavenging amino acids from its host’s lungs. PLoS One 5:e15152. doi: 10.1371/journal.pone.0015152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cissé OH, Pagni M, Hauser PM. 2012. De novo assembly of the Pneumocystis jirovecii genome from a single bronchoalveolar lavage fluid specimen from a patient. MBio 4:e00428-12. doi: 10.1128/mBio.00428-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cissé OH, Pagni M, Hauser PM. 2014. Comparative genomics suggests that the human pathogenic fungus Pneumocystis jirovecii acquired obligate biotrophy through gene loss. Genome Biol Evol 6:1938–1948. doi: 10.1093/gbe/evu155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma L, Chen Z, Huang DW, Kutty G, Ishihara M, Wang H, Abouelleil A, Bishop L, Davey E, Deng R, Deng X, Fan L, Fantoni G, Fitzgerald M, Gogineni E, Goldberg JM, Handley G, Hu X, Huber C, Jiao X, Jones K, Levin JZ, Liu Y, Macdonald P, Melnikov A, Raley C, Sassi M, Sherman BT, Song X, Sykes S, Tran B, Walsh L, Xia Y, Yang J, Young S, Zeng Q, Zheng X, Stephens R, Nusbaum C, Birren BW, Azadi P, Lempicki RA, Cuomo CA, Kovacs JA. 2016. Genome analysis of three Pneumocystis species reveals adaptation mechanisms to life exclusively in mammalian hosts. Nat Commun 7:10740. doi: 10.1038/ncomms10740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de W, Benchimol M. 2005. Basic biology of Pneumocystis carinii: a mini review. Mem Inst Oswaldo Cruz 100:903–908. doi: 10.1590/S0074-02762005000800013. [DOI] [PubMed] [Google Scholar]
- 7.Wyder MA, Rasch EM, Kaneshiro ES. 1998. Quantitation of absolute Pneumocystis carinii nuclear DNA content. Trophic and cystic forms isolated from infected rat lungs are haploid organisms. J Eukaryot Microbiol 45:233–239. doi: 10.1111/j.1550-7408.1998.tb04531.x. [DOI] [PubMed] [Google Scholar]
- 8.Lutzoni F, Kauff F, Cox CJ, McLaughlin D, Celio G, Dentinger B, Padamsee M, Hibbett D, James TY, Baloch E, Grube M, Reeb V, Hofstetter V, Schoch C, Arnold AE, Miadlikowska J, Spatafora J, Johnson D, Hambleton S, Crockett M, Shoemaker R, Sung GH, Lücking R, Lumbsch T, O’Donnell K, Binder M, Diederich P, Ertz D, Gueidan C, Hansen K, Harris RC, Hosaka K, Lim YW, Matheny B, Nishida H, Pfister D, Rogers J, Rossman A, Schmitt I, Sipman H, Stone J, Sugiyama J, Yahr R, Vilgalys R. 2004. Assembling the fungal tree of life: progress, classification, and evolution of subcellular traits. Am J Bot 91:1446–1480. doi: 10.3732/ajb.91.10.1446. [DOI] [PubMed] [Google Scholar]
- 9.Worsham DN, Basselin M, Smulian AG, Beach DH, Kaneshiro ES. 2003. Evidence for cholesterol scavenging by Pneumocystis and potential modifications of host-synthesized sterols by the P. carinii SAM:SMT. J Eukaryot Microbiol 50:678–679. doi: 10.1111/j.1550-7408.2003.tb00683.x. [DOI] [PubMed] [Google Scholar]
- 10.Durand-Joly I, Aliouat el M, Recourt C, Guyot K, François N, Wauquier M, Camus D, Dei-Cas E. 2002. Pneumocystis carinii f. sp. hominis is not infectious for SCID mice. J Clin Microbiol 40:1862–1865. doi: 10.1128/JCM.40.5.1862-1865.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gigliotti F, Harmsen AG, Haidaris CG, Haidaris PJ. 1993. Pneumocystis carinii is not universally transmissible between mammalian species. Infect Immun 61:2886–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aliouat-Denis CM, Chabé M, Demanche C, el Aliouat el M, Viscogliosi E, Guillot J, Delhaes L, Dei-Cas E. 2008. Pneumocystis species, co-evolution and pathogenic power. Infect Genet Evol 8:708–726. doi: 10.1016/j.meegid.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 13.Hugot JP, Demanche C, Barriel V, Dei-Cas E, Guillot J. 2003. Phylogenetic systematics and evolution of primate-derived Pneumocystis based on mitochondrial or nuclear DNA sequence comparison. Syst Biol 52:735–744. doi: 10.1080/10635150390250893. [DOI] [PubMed] [Google Scholar]
- 14.de Vienne DM, Refrégier G, López-Villavicencio M, Tellier A, Hood ME, Giraud T. 2013. Cospeciation vs host-shift speciation: methods for testing, evidence from natural associations and relation to coevolution. New Phytol 198:347–385. doi: 10.1111/nph.12150. [DOI] [PubMed] [Google Scholar]
- 15.Latinne A, Bezé F, Delhaes L, Pottier M, Gantois N, Nguyen J, Blasdell K, Dei-Cas E, Morand S, Chabé M. 2017. Genetic diversity and evolution of Pneumocystis fungi infecting wild Southeast Asian murid rodents. Parasitology 9:1–16. doi: 10.1017/S0031182017001883. [DOI] [PubMed] [Google Scholar]
- 16.Almeida JMGCF, Cissé OH, Fonseca Á, Pagni M, Hauser PM. 2015. Comparative genomics suggests primary homothallism of Pneumocystis species. MBio 6:e02250-14. doi: 10.1128/mBio.02250-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nielsen K, Heitman J. 2007. Sex and virulence of human pathogenic fungi. Adv Genet 57:143–173. doi: 10.1016/S0065-2660(06)57004-X. [DOI] [PubMed] [Google Scholar]
- 18.Esteves F, de Sousa B, Calderón EJ, Huang L, Badura R, Maltez F, Bassat Q, de Armas Y, Antunes F, Matos O. 2016. Multicentre study highlighting clinical relevance of new high-throughput methodologies in molecular epidemiology of Pneumocystis jirovecii pneumonia. Clin Microbiol Infect 22:566.e9–566.e19. doi: 10.1016/j.cmi.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 19.Esteves F, Gaspar J, Tavares A, Moser I, Antunes F, Mansinho K, Matos O. 2010. Population structure of Pneumocystis jirovecii isolated from immunodeficiency virus-positive patients. Infect Genet Evol 10:192–199. doi: 10.1016/j.meegid.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 20.Alanio A, Gits-Muselli M, Guigue N, Desnos-Ollivier M, Calderon EJ, Di Cave D, Dupont D, Hamprecht A, Hauser PM, Helweg-Larsen J, Kicia M, Lagrou K, Lengerova M, Matos O, Melchers WJG, Morio F, Nevez G, Totet A, White LP, Bretagne S. 2017. Diversity of Pneumocystis jirovecii across Europe: a multicentre observational study. EBioMedicine 22:155–163. doi: 10.1016/j.ebiom.2017.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monroy-Vaca EX, de Armas Y, Illnait-Zaragozí MT, Diaz R, Toraño G, Vega D, Alvarez-Lam I, Calderón EJ, Stensvold CR. 2014. Genetic diversity of Pneumocystis jirovecii in colonized Cuban infants and toddlers. Infect Genet Evol 22:60–66. doi: 10.1016/j.meegid.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 22.Parobek CM, Jiang LY, Patel JC, Alvarez-Martínez MJ, Miro JM, Worodria W, Andama A, Fong S, Huang L, Meshnick SR, Taylor SM, Juliano JJ. 2014. Multilocus microsatellite genotyping array for investigation of genetic epidemiology of Pneumocystis jirovecii. J Clin Microbiol 52:1391–1399. doi: 10.1128/JCM.02531-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller RF, Lindley AR, Ambrose HE, Malin AS, Wakefield AE. 2003. Genotypes of Pneumocystis jiroveci isolates obtained in Harare, Zimbabwe, and London, United Kingdom. Antimicrob Agents Chemother 47:3979–3981. doi: 10.1128/AAC.47.12.3979-3981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palmer RJ, Settnes OP, Lodal J, Wakefield AE. 2000. Population structure of rat-derived Pneumocystis carinii in Danish wild rats. Appl Environ Microbiol 66:4954–4961. doi: 10.1128/AEM.66.11.4954-4961.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kutty G, Achaz G, Maldarelli F, Varma A, Shroff R, Becker S, Fantoni G, Kovacs JA. 2010. Characterization of the meiosis-specific recombinase Dmc1 of pneumocystis. J Infect Dis 202:1920–1929. doi: 10.1086/657414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keely SP, Stringer JR. 2009. Complexity of the MSG gene family of Pneumocystis carinii. BMC Genomics 10:367. doi: 10.1186/1471-2164-10-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmid-Siegert E, Richard S, Luraschi A, Mühlethaler K, Pagni M, Hauser PM. 2017. Mechanisms of surface antigenic variation in the human pathogenic fungus Pneumocystis jirovecii. MBio 8:e01470-17. doi: 10.1128/mBio.01470-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stumpf MP, McVean GA. 2003. Estimating recombination rates from population-genetic data. Nat Rev Genet 4:959–968. doi: 10.1038/nrg1227. [DOI] [PubMed] [Google Scholar]
- 29.Alanio A, Gits-Muselli M, Mercier-Delarue S, Dromer F, Bretagne S. 2016. Diversity of Pneumocystis jirovecii during infection revealed by ultra-deep pyrosequencing. Front Microbiol 7:733. doi: 10.3389/fmicb.2016.00733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jarboui MA, Mseddi F, Sellami H, Sellami A, Makni F, Ayadi A. 2013. Genetic diversity of Pneumocystis jirovecii strains based on sequence variation of different DNA region. Med Mycol 51:561–567. doi: 10.3109/13693786.2012.744879. [DOI] [PubMed] [Google Scholar]
- 31.Slaven BE, Meller J, Porollo A, Sesterhenn T, Smulian AG, Cushion MT. 2006. Draft assembly and annotation of the Pneumocystis carinii genome. J Eukaryot Microbiol 53:S89–S91. doi: 10.1111/j.1550-7408.2006.00184.x. [DOI] [PubMed] [Google Scholar]
- 32.Schacherer J, Shapiro JA, Ruderfer DM, Kruglyak L. 2009. Comprehensive polymorphism survey elucidates population structure of Saccharomyces cerevisiae. Nature 458:342–345. doi: 10.1038/nature07670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeffares DC, Rallis C, Rieux A, Speed D, Převorovský M, Mourier T, Marsellach FX, Iqbal Z, Lau W, Cheng TM, Pracana R, Mülleder M, Lawson JL, Chessel A, Bala S, Hellenthal G, O’Fallon B, Keane T, Simpson JT, Bischof L, Tomiczek B, Bitton DA, Sideri T, Codlin S, Hellberg JE, van Trigt L, Jeffery L, Li JJ, Atkinson S, Thodberg M, Febrer M, McLay K, Drou N, Brown W, Hayles J, Carazo Salas RE, Ralser M, Maniatis N, Balding DJ, Balloux F, Durbin R, Bähler J. 2015. The genomic and phenotypic diversity of Schizosaccharomyces pombe. Nat Genet 47:235–241. doi: 10.1038/ng.3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taylor JW, Hann-Soden C, Branco S, Sylvain I, Ellison CE. 2015. Clonal reproduction in fungi. Proc Natl Acad Sci U S A 112:8901–8908. doi: 10.1073/pnas.1503159112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seich Al Basatena NK, Hoggart CJ, Coin LJ, O’Reilly PF. 2013. The effect of genomic inversions on estimation of population genetic parameters from SNP data. Genetics 193:243–253. doi: 10.1534/genetics.112.145599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matos O, Esteves F. 2010. Pneumocystis jirovecii multilocus gene sequencing: findings and implications. Future Microbiol 5:1257–1267. doi: 10.2217/fmb.10.75. [DOI] [PubMed] [Google Scholar]
- 37.Skotte L, Korneliussen TS, Albrechtsen A. 2013. Estimating individual admixture proportions from next generation sequencing data. Genetics 195:693–702. doi: 10.1534/genetics.113.154138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raj A, Stephens M, Pritchard JK. 2014. fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197:573–589. doi: 10.1534/genetics.114.164350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hill WG, Robertson A. 1968. Linkage disequilibrium in finite populations. Theor Appl Genet 38:226–231. doi: 10.1007/BF01245622. [DOI] [PubMed] [Google Scholar]
- 40.Badouin H, Gladieux P, Gouzy J, Siguenza S, Aguileta G, Snirc A, Le Prieur S, Jeziorski C, Branca A, Giraud T. 2017. Widespread selective sweeps throughout the genome of model plant pathogenic fungi and identification of effector candidates. Mol Ecol 26:2041–2062. doi: 10.1111/mec.13976. [DOI] [PubMed] [Google Scholar]
- 41.Aliouat el-M, Dujardin L, Martinez A, Duriez T, Ricard I, Dei-Cas E. 1999. Pneumocystis carinii growth kinetics in culture systems and in hosts: involvement of each life cycle parasite stage. J Eukaryot Microbiol 46:S116–S117. [PubMed] [Google Scholar]
- 42.Poon AF, Kosakovsky Pond SL, Bennett P, Richman DD, Leigh Brown AJ, Frost SD. 2007. Adaptation to human populations is revealed by within-host polymorphisms in HIV-1 and hepatitis C virus. PLoS Pathog 3:e45. doi: 10.1371/journal.ppat.0030045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang X, Bartlett MS, Smith JW, Lu JJ, Lee CH. 1998. Determination of copy number of rRNA genes in Pneumocystis carinii f. sp. hominis. J Clin Microbiol 36:2491–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fischer JM, Keely SP, Stringer JR. 2006. Evolutionary rate of ribosomal DNA in Pneumocystis species is normal despite the extraordinarily low copy-number of rDNA genes. J Eukaryot Microbiol 53:S156–S158. doi: 10.1111/j.1550-7408.2006.00213.x. [DOI] [PubMed] [Google Scholar]
- 45.Ma L, Huang DW, Cuomo CA, Sykes S, Fantoni G, Das B, Sherman BT, Yang J, Huber C, Xia Y, Davey E, Kutty G, Bishop L, Sassi M, Lempicki RA, Kovacs JA. 2013. Sequencing and characterization of the complete mitochondrial genomes of three Pneumocystis species provide new insights into divergence between human and rodent Pneumocystis. FASEB J 27:1962–1972. doi: 10.1096/fj.12-224444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murray GG, Wang F, Harrison EM, Paterson GK, Mather AE, Harris SR, Holmes MA, Rambaut A, Welch JJ. 2016. The effect of genetic structure on molecular dating and tests for temporal signal. Methods Ecol Evol 7:80–89. doi: 10.1111/2041-210X.12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu CH, Xie D, Suchard MA, Rambaut A, Drummond AJ. 2014. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol 10:e1003537. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parfrey LW, Lahr DJ, Knoll AH, Katz LA. 2011. Estimating the timing of early eukaryotic diversification with multigene molecular clocks. Proc Natl Acad Sci U S A 108:13624–13629. doi: 10.1073/pnas.1110633108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keely SP, Fischer JM, Stringer JR. 2003. Evolution and speciation of Pneumocystis. J Eukaryot Microbiol 50:624–626. doi: 10.1111/j.1550-7408.2003.tb00655.x. [DOI] [PubMed] [Google Scholar]
- 50.Ellegren H, Galtier N. 2016. Determinants of genetic diversity. Nat Rev Genet 17:422–433. doi: 10.1038/nrg.2016.58. [DOI] [PubMed] [Google Scholar]
- 51.Barrett RD, Schluter D. 2008. Adaptation from standing genetic variation. Trends Ecol Evol 23:38–44. doi: 10.1016/j.tree.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 52.Bashey F. 2015. Within-host competitive interactions as a mechanism for the maintenance of parasite diversity. Philos Trans R Soc Lond B Biol Sci 370:20140301. doi: 10.1098/rstb.2014.0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thornton K. 2005. Recombination and the properties of Tajima’s D in the context of approximate-likelihood calculation. Genetics 171:2143–2148. doi: 10.1534/genetics.105.043786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramírez-Soriano A, Ramos-Onsins SE, Rozas J, Calafell F, Navarro A. 2008. Statistical power analysis of neutrality tests under demographic expansions, contractions and bottlenecks with recombination. Genetics 179:555–567. doi: 10.1534/genetics.107.083006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pavlidis P, Alachiotis N. 2017. A survey of methods and tools to detect recent and strong positive selection. J Biol Res (Thessalon) 24:7. doi: 10.1186/s40709-017-0064-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rambaut A, Lam TT, Max Carvalho L, Pybus OG. 2016. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol 2:vew007. doi: 10.1093/ve/vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Navascués M, Emerson BC. 2009. Elevated substitution rate estimates from ancient DNA: model violation and bias of Bayesian methods. Mol Ecol 18:4390–4397. doi: 10.1111/j.1365-294X.2009.04333.x. [DOI] [PubMed] [Google Scholar]
- 58.Duchêne D, Duchêne S, Ho SY. 2015. Tree imbalance causes a bias in phylogenetic estimation of evolutionary timescales using heterochronous sequences. Mol Ecol Resour 15:785–794. doi: 10.1111/1755-0998.12352. [DOI] [PubMed] [Google Scholar]
- 59.Ma L, Borio L, Masur H, Kovacs JA. 1999. Pneumocystis carinii dihydropteroate synthase but not dihydrofolate reductase gene mutations correlate with prior trimethoprim-sulfamethoxazole or dapsone use. J Infect Dis 180:1969–1978. doi: 10.1086/315148. [DOI] [PubMed] [Google Scholar]
- 60.Endicott P, Ho SY, Stringer C. 2010. Using genetic evidence to evaluate four palaeoanthropological hypotheses for the timing of Neanderthal and modern human origins. J Hum Evol 59:87–95. doi: 10.1016/j.jhevol.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 61.Grant WS. 2015. Problems and cautions with sequence mismatch analysis and Bayesian skyline plots to infer historical demography. J Hered 106:333–346. doi: 10.1093/jhered/esv020. [DOI] [PubMed] [Google Scholar]
- 62.Muller HJ. 1963. The need for recombination to prevent genetic deterioration. Genetics 48:903. [Google Scholar]
- 63.Poulin R. 2011, Evolutionary ecology of parasites. Princeton University Press, Princeton, NY. [Google Scholar]
- 64.Hong ST, Steele PE, Cushion MT, Walzer PD, Stringer SL, Stringer JR. 1990. Pneumocystis carinii karyotypes. J Clin Microbiol 28:1785–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cushion MT, Kaselis M, Stringer SL, Stringer JR. 1993. Genetic stability and diversity 2883 of Pneumocystis carinii infecting rat colonies. Infect Immun 61:4801–4813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bishop LR, Helman D, Kovacs JA. 2012. Discordant antibody and cellular responses to Pneumocystis major surface glycoprotein variants in mice. BMC Immunol 13:39. doi: 10.1186/1471-2172-13-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, Banks E, Garimella KV, Altshuler D, Gabriel S, DePristo MA. 2013. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li H. 2015. FermiKit: assembly-based variant calling for Illumina resequencing data. Bioinformatics 31:3694–3696. doi: 10.1093/bioinformatics/btv440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jombart T, Ahmed I. 2011. Adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27:3070–3071. doi: 10.1093/bioinformatics/btr521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS. 2012. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28:3326–3328. doi: 10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R; 1000 Genomes Project Analysis Group . 2011. The variant call format and VCFtools. Bioinformatics 27:2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Korneliussen TS, Albrechtsen A, Nielsen R. 2014. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15:356. doi: 10.1186/s12859-014-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Auton A, McVean G. 2007. Recombination rate estimation in the presence of hotspots. Genome Res 17:1219–1227. doi: 10.1101/gr.6386707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. 2009. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Darriba D, Taboada GL, Doallo R, Posada D. 2012. JModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772. doi: 10.1038/nmeth.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Beimforde C, Feldberg K, Nylinder S, Rikkinen J, Tuovila H, Dörfelt H, Gube M, Jackson DJ, Reitner J, Seyfullah LJ, Schmidt AR. 2014. Estimating the Phanerozoic history of the Ascomycota lineages: combining fossil and molecular data. Mol Phylogenet Evol 78:386–398. doi: 10.1016/j.ympev.2014.04.024. [DOI] [PubMed] [Google Scholar]
- 82.Di Fiore A, Chaves PB, Cornejo FM, Schmitt CA, Shanee S, Cortés-Ortiz L, Fagundes V, Roos C, Pacheco V. 2015. The rise and fall of a genus: complete mtDNA genomes shed light on the phylogenetic position of yellow-tailed woolly monkeys, Lagothrix flavicauda, and on the evolutionary history of the family Atelidae (Primates: Platyrrhini). Mol Phylogenet Evol 82:495–510. doi: 10.1016/j.ympev.2014.03.028. [DOI] [PubMed] [Google Scholar]
- 83.Kimura Y, Hawkins MT, McDonough MM, Jacobs LL, Flynn LJ. 2015. Corrected placement of Mus-Rattus fossil calibration forces precision in the molecular tree of rodents. Sci Rep 5:14444. doi: 10.1038/srep14444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cushion MT, Linke MJ, Ashbaugh A, Sesterhenn T, Collins MS, Lynch K, Brubaker R, Walzer PD. 2010. Echinocandin treatment of pneumocystis pneumonia in rodent models depletes cysts leaving trophic burdens that cannot transmit the infection. PLoS One 5:e8524. doi: 10.1371/journal.pone.0008524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Clinical information and demographic data of the groups of individuals involved in the study (as determined using WGS). (B) Published data sets used in this study (WGS). (C) Clinical information and demographic data of the groups of individuals involved in the study (next-generation sequencing [NGS]-based amplicon multilocus sequence typing [MLST]). (D) Clinical information and demographic data of the groups of individuals involved in the acquisition of Pneumocystis jirovecii mitochondrion full-genome study data. Download TABLE S1, DOCX file, 0.03 MB (34.3KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Initial analysis of Pneumocystis murina samples. The dendrogram was constructed from results of a hierarchical cluster analysis of 29,692 biallelic SNPs using z scores and an outlier threshold value of 5 to split the nodes. The population level of inbreeding was measured using coancestry coefficients, which are measures of relatedness between individuals in a population. Coancestry is expressed as the probability of identity by descent (IBD). Individual dissimilarities are calculated for each pair of individuals. The graph shows that most of the isolates were from clonal populations as defined by dissimilarity and coancestry coefficient values above 1 and below 0.5, respectively. The calculated kinship coefficient values, i.e., those corresponding to twins, a first sibling (FS), a half-sibling (HS), and a first cousin (FC), which are probabilistic measures of relatedness, are presented on the right axis. Download FIG S1, TIF file, 0.3 MB (278.1KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Description of the methodology used for the analysis of genetic subdivisions, phylodynamics, within-individual populations, multilocus sequencing and analysis, evolutionary rates, and Pneumocystis macacae transcriptome sequencing. Download TEXT S1, DOCX file, 0.1 MB (103.1KB, docx) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Random distribution of SNPs at linkage equilibrium. Polymorphic site (SNP) positions were analyzed using a Monte Carlo test implemented in the R package adegenet to determine if they are randomly distributed across chromosomes. The significance data are based on 500 replicates. Density plots of representative chromosomes, which clearly indicated a random distribution of SNPs (P value = 0.0019), are shown. The first chromosome (“chr1”) in each species corresponds to the following NCBI accession numbers: LFWA01000001 (P. jirovecii), LFVZ01000001 (P. carinii), and NW_006920852 (P. murina). Download FIG S2, TIF file, 1.1 MB (1.1MB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Principal-component analysis. Results of analysis of SNP sites at linkage equilibrium indicate a lack of population structure in three Pneumocystis species. (A and B) PCA of P. jirovecii isolates with (A) and without (B) outliers. The following codes are used: DEN (Denmark), CIN (Cincinnati, OH, USA), IND (Indianapolis, IN, USA), Bethesda (MD, USA), and SWZ (Switzerland). The P. murina samples for which whole-genome sequences could be obtained were all from Maryland (USA), and the color codes correspond to collection dates. There was no geographic clustering for P. jirovecii and P. carinii species and no apparent temporal clustering for P. murina populations. Download FIG S3, TIF file, 0.4 MB (452.2KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
(A) Haplotype network of Pneumocystis jirovecii in 13 patients based on the internal transcribed spacer 1 (ITS1) locus. Major haplotypes are m4_seq4, m3_seq2, and m19_seq1. (B) Geographic population structure of Pneumocystis jirovecii isolates based on internal transcribed spacer (ITS1) locus. An integer neighbor-joining network of 189 ITS1 sequences, including 48 sequences from this study and 141 previously reported in GenBank, is presented. For both panel A and panel B, each circle corresponds to a haplotype and its size is proportional to the total frequency. Circles are colored according to the isolate (A) or to geographic origin (B). Small circles represent single sequences, and line lengths represent pairwise nucleotide differences. Download FIG S4, PDF file, 0.4 MB (380.8KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Population structure of Pneumocystis determined by analysis using whole-genome sequence data. Results of analysis of genetic clustering on segregating sites identified in surveyed strains are presented. The estimated individual admixture was inferred using genotype likelihoods. Each isolate is represented by a single vertical bar, which is partitioned into K colored segments that represent the isolate’s estimated ancestry proportion in each of the K clusters. A single color represents an absence of admixture, whereas stacked colors show proportions of admixtures in each isolate. The color codes differ between the plots because the samples from each species were analyzed separately. Genetic clusters are ordered according to the geographic locations using the following codes: DEN (Denmark), CIN (Cincinnati, OH, USA), IND (Indianapolis, IN, USA), MD (Maryland, USA), and SWZ (Switzerland). Rat samples from CIN samples correspond to two distinct whole-genome sequence data sets corresponding to sequences previously published by Slaven and colleagues (31) and to the latest release of the genome in 2010 (http://pgp.cchmc.org). We assume that these data sets were obtained from at least two infected animals, while the possibility cannot be ruled out that they might represent multiple aliquots from the same animal. The IND sample was a generous gift from Chao-Hung Lee at Indiana University (USA). The P. murina samples for which whole-genome sequences could be obtained were all from Maryland (USA), so the genetic clusters are organized by collection dates instead of geographic locations. The population structure analysis does not show a clear separation of lineages in all three Pneumocystis species. Download FIG S5, TIF file, 0.5 MB (474.7KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Scatterplot of genome-wide multiplicity of infection (MOI) estimates from whole-genome sequencing data excluding multicopy gene families across Pneumocystis isolates. The MOI value refers to the number of different genotypes coinfecting a single host, and each dot represents an individual isolate. P. jirovecii isolates (n = 7) are shown in green, P. carinii isolates (n = 9) in orange, and P. murina isolates (n = 23) in blue. Multiplicity of infection (MOI) values are plotted against the fraction of the genome covered per sample (x axis). Download FIG S6, TIF file, 0.3 MB (294.9KB, tif) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
(A) Time-calibrated phylogeny of Pneumocystis species and their mammalian hosts. A maximum clade credibility tree of Pneumocystis species was obtained from 10 orthologous nuclear genes using BEAST2. The 95% highest-posterior-density (HPD) intervals (i.e., confidence intervals) are presented at the nodes, and the blue bands are proportional to confidence intervals. The scale bar at the bottom depicts time in millions of years back in time toward the left. The following codes are used: MACA (Macaque), PCAR (P. carinii), PMUR (P. murina), PJIR (P. jirovecii), and PMK (P. macacae). (B) Time-calibrated phylogeny of Pneumocystis species. A maximum clade credibility tree of Pneumocystis species was obtained from 43 orthologous nuclear genes using BEAST2. The 95% highest-posterior-density (HPD) intervals (i.e., confidence intervals) are presented at the nodes, and the blue bands are proportional to confidence intervals. The scale bar at the bottom depicts time in millions of years back in time toward the left. Download FIG S7, PDF file, 0.3 MB (300.6KB, pdf) .
This is a work of the U.S. Government and is not subject to copyright protection in the United States. Foreign copyrights may apply.
Data Availability Statement
All sequence data have been linked to NCBI Umbrella project PRJNA385300. The mitochondrial genomes have been deposited into GenBank under accession codes MH010437 to MH010446. Data sets used for molecular clocks and phylodynamic analyses are available at https://doi.org/10.5281/zenodo.1215631.