

Abstract
Objective
Paraneoplastic neurological syndromes (PNS) are rare neurological disorders in which ectopic expression of neural antigens by a tumor results in an autoimmune attack against the nervous system. Onconeural antibodies not only guide PNS diagnosis but may also help detecting underlying malignancies. Our project aims to uncover new potential antibodies in paraneoplastic neuropathies (PN).
Methods
Thirty‐four patients fulfilling diagnostic criteria of possible (n = 9; 26.5%) and definite (n = 25; 73.5%) PN without onconeural antibodies and 28 healthy controls were included in our study. Sera were tested for known antibodies against neural cell adhesion molecules and screened for novel IgG and IgM reactivities against nerve components: dorsal root ganglia (DRG) neurons, motor neurons, and Schwann cells. Patients showing autoantibodies against any of these cell types were used for immunoprecipitation (IP) studies.
Results
Overall, 9 (26.5%) patients showed significant reactivity against DRG neurons, motor neurons, or Schwann cells, whereas 5 (17.9%) healthy controls only showed moderate reactivity. Compared with control sera, serum samples from patients with paraneoplastic sensory‐motor neuropathies had a higher frequency of IgM antibodies against Schwann cells (0% vs. 40%; P = 0.0028). No novel antigens were identified from our IP experiments. Antibodies against the neural adhesion molecules CNTN1, NF155, NF140, NF186, NCAM1, L1CAM, and the CNTN1/CASPR1 complex were not detected in patients with PN. One (2.9%) patient with CIDP and thymoma had CASPR2 antibodies.
Interpretation
Almost 30% of patients with PN harbor antibodies targeting neural structures, suggesting that novel neoplasm‐associated antigens remain to be discovered.
Introduction
Paraneoplastic neurological syndromes (PNS) are heterogeneous and rare neurological disorders in which the ectopic expression of a neural antigen in a neoplasm drives an autoimmune response resulting in neurological impairment.1 PNS comprise multiple clinical entities that may affect all parts of the nervous system.1 According to the PNS Euronetwork database, one third of patients with PNS have the peripheral nervous system as the primary site of involvement. Among peripheral PNS, sensory neuronopathy is the most frequent syndrome.2
In PNS, breakage of tolerance against self follows ectopic expression of tumor‐expressed antigens, which leads to the production of paraneoplastic or onconeural antibodies.1 To date, several well‐characterized onconeural antibodies have been described,3 being anti‐Hu and anti‐Yo antibodies the most frequent.2 Except for anti‐Hu antibodies, which strongly associate with pure sensory neuronopathy, most of these onconeural antibodies associate with central nervous system disorders. Since neurological symptoms generally precede tumor detection,4 the finding of circulating onconeural antibodies may be key in guiding what is often a challenging diagnosis.3 Furthermore, their detection in patients is in some cases invaluable in the discovery of an underlying malignancy, allowing the initiation of an early oncological treatment which might, in turn, contribute to the management of the PNS.5, 6, 7, 8
Contactin 1 (CNTN1) is an axonal cell adhesion molecule implicated in myelin loops fixation at the paranode of Ranvier.9 Our group described the presence of anti‐CNTN1 antibodies in small subsets of chronic inflammatory demyelinating polyneuropathy (CIDP) patients with characteristic clinical features.10 Both in vitro and passive‐transfer experiments in animal models have demonstrated anti‐CNTN1 antibodies’ pathogenicity in CIDP.11 CNTN1 has also been postulated to play a pivotal role in the development, progression, and pathogenesis of multiple tumors, being its expression considered as a marker of poor prognosis.12, 13, 14, 15, 16 Two of our CIDP patients harboring antibodies against CNTN1, respectively, developed a lymphoma and a colon adenocarcinoma in addition to their neuropathy, one of them concomitantly, and the other several years later. The role of other cell adhesion molecules such as neural cell adhesion molecule 1 (NCAM1)17, 18, 19, 20 and L1 (L1CAM)21, 22, 23 in cancer pathogenesis has been widely described. However, their possible involvement as antigens in paraneoplastic neuropathies has not been studied.
Here, we present a prospective study in which sera from a group of 34 patients fulfilling diagnostic criteria of possible (n = 9; 26.5%) and definite (n = 25; 73.5%) paraneoplastic neuropathies (PN), according to the PNS Euronetwork criteria,3 were analyzed to uncover new potential antibodies. Sera were tested for known antibodies against neural cell adhesion molecules and screened for novel reactivities against nerve components.
Materials and Methods
Patients, informed consent, and protocol approvals
In this prospective study, 34 patients followed at the Neuroimmunology‐Multiple Sclerosis Unit in Hospital Clínic de Barcelona (n = 32; 94.1%) or at the Neuromuscular Diseases Unit in Hospital de Sant Pau (n = 2; 5.9%) were included. Patients fulfilled diagnostic criteria of possible (n = 9; 26.5%) or definite (n = 25; 73.5%) PN.3 Peripheral nerve involvement due to neoplastic infiltration, infectious or metabolic complications, or from any sort of cancer treatment was appropriately ruled out. All patients lacked antibodies against well‐characterized antigens as determined by immunoblot at Hospital Clínic. Twenty‐eight healthy controls from the Neuromuscular Diseases Laboratory biobank were additionally included. Serum samples were obtained and frozen at −80° until needed.
This clinical research was conducted in compliance with the Declaration of Helsinki and was approved by the Human Ethics Committees Review Board of Hospital de la Santa Creu i Sant Pau and Hospital Clínic de Barcelona. Written informed consents were obtained from all participants.
With regard to experiments involving rats, experimental procedures were approved by our institution's Service of Animal Experimentation at CSIC‐ICCC (Institut Català de Ciències Cardiovasculars).
Protocol overview
Sera were tested for antibodies against previously described antigens at the node of Ranvier; CNTN1,10 neurofascin 140 (NF140),24 neurofascin 155 (NF155),25 neurofascin 186 (NF186),24 contactin‐associated protein 2 (CASPR‐2)26, and the CNTN1/contactin‐associated protein 1 (CASPR1) complex10; and against other neural cell adhesion molecules (L1CAM and NCAM1). Sera were additionally screened for novel IgM and IgG reactivities against primary cultures of rat peripheral nerve components: dorsal root ganglia (DRG) neurons, motor neurons, and Schwann cells.
Patients with autoantibodies against any of these nerve structures were used for antigen discovery with immunoprecipitation (IP) studies. If any candidate antigen was detected, confirmatory experiments with transfected human embryonic kidney (HEK) 293 cells were conducted.
Autoantibody screening in peripheral nerve components: cell cultures
Rat DRG neurons,27 motor neurons,28 and Schwann cells29 were isolated and cultured as previously described with modifications. Briefly, DRG neurons were dissociated from E16 embryos and cells were cultured in neurobasal medium (Gibco BRL, NY) supplemented with B27 (Gibco BRL), Glutamax (Gibco BRL), and nerve growth factor (NGF) (Invitrogen, CA). After 24 h, cytosine arabinoside (ARA‐C) (Sigma, MO) and fluorouracil (5‐FU) (both by Sigma) were added to the medium to remove fibroblasts. Two‐thirds of the ARA‐C/5‐FU‐supplemented medium was replaced with fresh medium every other day until reaching DRG neuron full growth.
Motor neurons were obtained from spinal cords from E16 rat embryos. Cells were cultured in neurobasal medium (Gibco BRL) supplemented with B27 (Gibco BRL), Glutamax (Gibco BRL), and NGF (Invitrogen). Medium was replaced every other day until full cell growth was achieved.
Schwann cells were harvested from sciatic nerves from 4‐ to 6‐day‐old rat pups and cultured in CB‐medium overnight (O/N). After 24 h, medium was changed and fresh CB‐medium supplemented with ARA‐C (Sigma) was added. After 3 days, medium was changed to Schwann cell proliferation medium, which was replaced every other day until full proliferation was achieved. Cells were then plated in Poly‐D‐Lysine (Sigma) coated cover glasses, fixed with 4% paraformaldehyde (PFA) (Affymetrix Inc, CA), blocked with 5% goat serum (Gibco BRL) and frozen at ‐80°C until immunocytochemistry (ICC) experiments were performed.
DRG neuron, motor neuron, and Schwann cell immunocytochemistry
Live DRG and motor neurons were incubated with patients’ sera diluted 1/150 (for IgG experiments) or 1/40 (for IgM experiments) in culture medium. Cells were then fixed with 4% PFA (Affymetrix Inc) and incubated with appropriate secondary antibodies (both from Invitrogen) 1/1500. Due to the limiting scarcity of rat motor neurons extraction, only patients’ sera were tested in these experiments.
ICC experiments on Schwann cells were performed on previously fixed and blocked frozen cells, using patients’ sera diluted 1/100 (for IgG) or 1/40 (for IgM) and 1/1000 secondary antibodies (both from Invitrogen).
Coverslips were mounted with Vectashield with DAPI (Vector Laboratories, CA, USA) and fluorescence signal intensity was scored in a 0–3 scale by two independent researchers. Participants were codified as patients or controls and their ICC results in each condition were grouped in two separate categories: moderate to strong staining, including scores two and three (relevant staining), and other staining, featuring scores zero (negatives) and mild (score one, unspecific background). The latter category included sera with no immunological relevance. Images were obtained with an Olympus BX51 Fluorescence Microscope (Olympus Corporation, Tokyo, Japan) and processed with GraphPad layout editor (GraphPad Software, CA).
Immunoprecipitation
Sera showing moderate or strong (scores 2 or 3) reactivity against peripheral nerve cells were used for IP experiments using the same cell as IP substrate. Briefly, cells were cultured as described above and incubated 1 h at 37°C with sera diluted 1/100 (for IgG) or 1/40 (for IgM) in growth medium. Cells were then lysed and protein A and G agarose beads (both by Invitrogen) or anti‐human IgM agarose beads (Sigma) were added to the lysate and incubated O/N. Agarose beads were then spun down and the supernatants removed. Precipitated proteins were detached from the agarose beads resuspending the sample in Laemli buffer (Bio‐Rad, CA, USA) with 5% b‐mercaptoethanol (Merck, Germany), heated up to 100°C for 5 min. Supernatants were then collected, loaded in a polyacrylamide gel for electrophoresis and stained with Coomassie blue (Invitrogen). Bands appearing in the IP material were analyzed by mass spectrometry (MS). Protein relevance as an autoantigen was ranked by a custom software (Anaxomics Biotech, Spain) according to an algorithm taking into account MS protein features (score, number of identified peptides, sequence coverage) and protein's cellular and tissular location and function. Proteins in the IP material were selected as candidate antigens when they fulfilled any of these criteria: protein score > 100, peptide sequence coverage >5%, or two or more peptides identified with the absence of the same criteria in the control sample.
HEK cell transfection and ICC
HEK 293 cells were cultured O/N at 37°C in serum‐supplemented Dulbecco's modified Eagle medium (Lonza, Switzerland) in culture dishes with poly‐D‐coated (Sigma) coverslips. Mammalian expression vectors encoding human CNTN1, CASPR1, CASPR2, NF140, NF155, NF186, NCAM1, and L1CAM (additional information in Table S1) in Opti‐MEM (Gibco BRL) were transfected with Lipofectamine 2000 (Invitrogen). Cells were then fixed with 4% PFA (Affymetrix Inc) and blocked for 1 h with either 5% goat serum (Gibco BRL) or 1/40 normal rabbit serum (Jackson Immunoresearch, PA, USA) in PBS. ICC experiments were performed as described in Table S1. Coverslips were mounted as described above.
Clinical–immunological correlations
Medical records of patients were retrospectively reviewed in order to detect any clinical association with the presence of antibody reactivity. Detailed clinical features (including phenotype at onset, time to nadir, electrophysiological features, and response to therapy) were collected in a coded database to detect clinical–immunological associations.
Statistical analysis
Results were analyzed by GraphPad Prism v5.0 (GraphPad Software). Statistical comparison was performed using contingency analysis with the application of a two‐tailed Fisher's exact test, accepting an alpha‐level <0.05 to determine significance. Whenever this analysis could not be performed due to high number of zeros in the equation, a nonapplicable (NA) status was given in place of a P value.
Results
Thirty‐four patients with a neuropathy fulfilling diagnostic criteria of possible (n = 9; 26.5%) or definite (n = 25; 73.5%) PN were included in the study, 8 (23.5%) of them were women, while 26 (76.5%) were males. Mean age was 62.9 ± 10.6 years; 57.4 ± 12.1 years for women and 64.7 ± 9.6 years for men. According to the recommended diagnostic criteria for PNS,3 9 (26.5%) patients with a possible diagnosis featured a nonclassical neurological syndrome with presence of a tumor and absence of onconeural antibodies. Patients with a definite diagnosis either had a classical syndrome with a tumor and a lack of antibodies (n = 24; 70.6%) or suffered from a tumor and nonclassical syndrome that improved after oncologic therapy (n = 1; 2.9%). Overall, 10 (29.4%) patients presented with a tumor and a neuropathy involving both sensory and motor symptoms while lacking any known antigen, while the remaining 24 (70.6%) patients presented with a tumor and a classical sensory neuronopathy. Among the latter, 7 (20.6%) patients were diagnosed with small cell lung cancer (SCLC). The rest of the 27 (79.4%) patients, regardless of their type of symptom, sensory alone or sensory‐motor, suffered from a myriad of other malignancies (Fig. S1). Clinical and demographic data are gathered in Table S2.
Screening experiments
ICC experiments with primary cultures of rat DRG neurons, motor neurons, and Schwann cells were used to identify novel IgG and IgM reactivities against neural components. Overall, 9 (26.5%) patients reacted moderately or strongly against DRG, motor neurons, or Schwann cells, whereas 5 (17.9%) healthy controls reacted only moderately. Since motor neuron staining in all patients was exclusively moderate and poorly significant (only three patients reacted, two of them moderately), and due to the limiting scarcity of rat motor neurons, no controls were tested for reactivity against motor neurons. Patients with moderate and strong staining and their clinical features are summarized in Table 1. Further details regarding the remaining patients can be found in Table S3. When comparing moderate to strong staining intensities between all patients and controls, no significant statistical differences were appreciated in any of our screening experiments (Fig. 2A; Table S4). Differences were nonetheless observed when stratifying by disease phenotype.
Table 1.
Patients with moderate or strong staining ICCs
| Patient number | NEUROPATHY | TUMOR | DRG neurons IgG | DRG neurons IgM | Schwann cells IgG | Schwann cells IgM | Motor neurons IgG | Motor neurons IgM | Clinical features |
|---|---|---|---|---|---|---|---|---|---|
| 3 | Sensory‐motor | OVARIAN | 2 | 1 | 0 | 1 | 0 | 0 | Rapidly progressive (over 1 month), predominantly distal, severe limb weakness plus ataxia. Very severe impairment of arthrokynetic and vibration sensation. Areflexia.EMG: Demyelinating features with severe axonal impairment and acute denervation |
| 4 | Sensory‐motor | LYMPHOMA | 1 | 2 | 0 | 3a | 0 | 0 | Itch and paresthesia in 3 months. Gait unsteadiness and frequent falls. Mild distal weakness, severe vibration sensation impairment. Sudden progression to severe quadriparesis. Areflexia.EMG: severe sensory‐motor axonal polyneuropathy |
| 7 | Sensory‐motor | BREAST | 0 | 0 | 0 | 3a | 0 | 0 | One week of weakness and distal paresthesia. Proximal (4/5) and distal (3/5) weakness. Severe vibration and arthrokynetic sensation impairment in all four limbs. Areflexia.EMG: axonal polyneuropathy. Seventeen cells and high protein content in CSF |
| 33 | Sensory‐motor | COLON | 0 | 0 | 0 | 2 | 0 | 0 | Mild gait impairment 1 year before. Progression in last weeks, with pan‐sensory impairment in four limbs. EMG: sensory‐motor axonal polyneuropathy. Dramatic improvement after tumor removal |
| 34 | Sensory‐motor | THYMOMA | 3 | 1 | 0 | 2 | 2 | 0 | Gait impairment, distal sensory disturbances, and distal weakness. EMG: demyelinating features, classified as CIDP. Improvement with steroids |
| 19 | Sensory | NSCLC | 2a | 0 | 1 | 0 | 0 | 0 | Left hand paresthesia. Progression to all limbs, left side of face and trunk. Gait ataxia. Abolition of vibratory sensation in all four limbs. Arthrokinetic sensation impaired in left hand and foot. No weakness. Areflexia. First EMG, normal. Second EMG severe sensory neuronopathy |
| 21 | Sensory | TONSIL | 0 | 0 | 1 | 2 | 0 | 0 | Ataxia, paresthesia, vibration sensation impairment, and global areflexia. |
| 30 | Sensory | NSCLC | 2a | 0 | 0 | 0 | 0 | 0 | Distal painful paresthesia in hands and feet. EMG: sensory axonal polyneuropathy |
| 31 | Sensory | PAROTID GLAND | 0 | 1 | 1 | 0 | 1 | 2 | Radicular pain in legs. Paresthesia in four limbs and trunk. Progression over 3 weeks to inability to walk. Severe impairment of joint position. High protein content in CSF |
NSCLC, non‐small‐cell lung cancer; EMG, electromyography; CSF, cerebrospinal fluid.
Indicate sera used for antigen discovery with immunoprecipitation (IP) studies.
Figure 2.
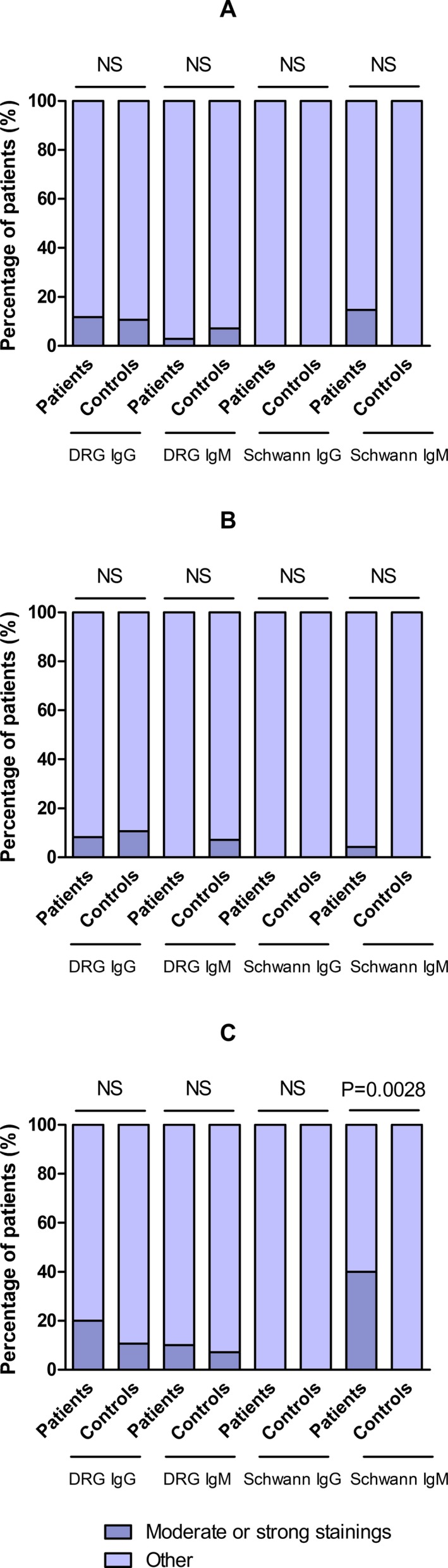
Statistical comparison between patients and controls. Statistical analysis of DRG and Schwann cells ICC considering moderate or strong staining in all patients (A); patients with sensory neuronopathy (B) and patients with sensory‐motor (S‐M) neuropathy (C).
Patients with sensory‐motor neuropathy
In IgM experiments, 12 (35.3%) out of the total of patients reacted against rat Schwann cells; 5 (14.7%) of them featured strong or moderate staining (Fig. 1A). Four (11.8%) out of these five patients, belonged in the cohort of patients with sensory‐motor neuropathy (n = 10), representing 40.0% of these patients. A higher proportion of patients with sensory‐motor neuropathy reacted against Schwann cells in IgM experiments compared to healthy controls (P = 0.0028) (Fig. 2; Table S4) and to patients with pure sensory neuronopathy (P = 0.0190) (Fig. 3; Table S5). Three (8.8%) of these patients presented with a sensory‐motor axonal polyneuropathy with prominent vibratory and joint position sensation involvement. However, shared specific clinical features among them could not be identified. The fourth patient presented with a sensory‐motor neuropathy with demyelinating features in the EMG. Clinical features are detailed in Table 1. Sera from two of these patients (with strong reactivity against Schwann cells) and a normal control were used to identify target antigens using Schwann cells as the IP substrate. Unfortunately, no relevant antigens were identified in comparison to the control serum.
Figure 1.
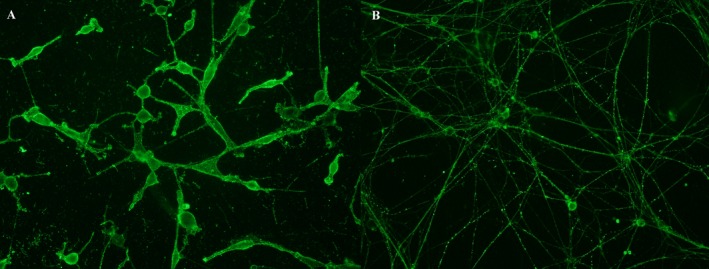
Positive reactivities in screening ICC experiments. Strong IgM staining in Schwann cells (A) and moderate IgG staining in DRG neurons (B).
Figure 3.
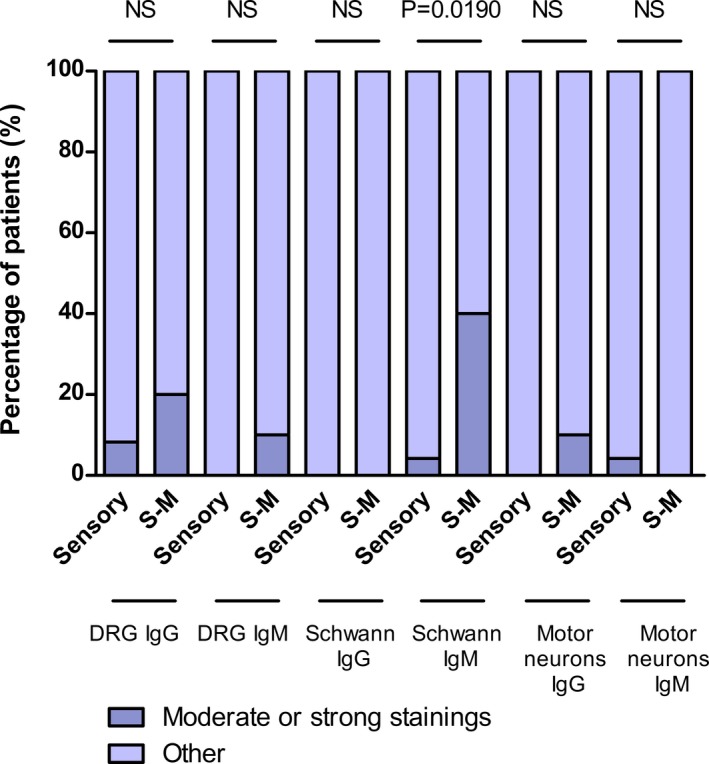
Statistical comparison between PN patients with sensory neuronopathy and patients with sensory‐motor neuropathy. Statistical analysis of DRG neurons, motor neurons, and Schwann cells ICC considering moderate or strong staining in patients with sensory neuronopathy and patients with sensory‐motor (S‐M) neuropathy.
Four patients (11.8%) with sensory‐motor neuropathy featured IgG reactivity against DRG neurons, 2 (5.9%) of them moderate, while 3 (8.8%) featured IgM reactivity, only 1 (2.9%) of them with moderate staining. Just 1 (2.9%) patient with sensory‐motor neuropathy featured moderate IgG motor neurons’ staining. These results were not significant when compared to healthy controls (Fig. 2; Table S4) or patients with sensory neuronopathy (Fig. 3; Table S5).
Patients with pure sensory neuronopathy
Eight patients (23.5%) with sensory neuronopathy, presented IgG reactivity against DRG neurons, 2 (5.9%) of them of a moderate kind (Fig. 1B), while only 2 (5.9%) showed mild IgM reactivity. Sera from these two patients showing moderate IgG reactivity against DRG neurons and a normal control were used to identify target antigens using DRG neurons as the IP substrate. These experiments did not provide any candidate antigens in comparison to the control serum.
Only 1 (2.9%) patient reacting moderately by IgM against Schwann cells and another patient equally reacting against motor neurons were identified in this cohort. No cross‐reactivity was found between motor neurons and DRG neurons or Schwann cells. Significant differences were not observed in these patients when compared to healthy controls (Fig. 2; Table S4) or patients with sensory‐motor neuropathy (Fig. 3; Table S5).
Reactivity against cell adhesion molecules
No sera reacting against CNTN1, NF155, NF140, NF186, NCAM1, L1CAM, or the CNTN1/CASPR1 complex were identified in our study. Only 1 (2.9%) patient reacted against CASPR2 (Fig. S2). This patient had a thymoma and presented with a sensory‐motor rapidly progressing chronic polyradiculoneuropathy with demyelinating features in the EMG, fulfilling diagnostic criteria of CIDP. This patient did not show any evidence of neuromyotonia (frequently associated with anti‐CASPR2 antibodies and thymoma26) and also reacted against DRG neurons and Schwann cells.
Discussion
Although our study describes a comprehensive autoantibody screening approach aimed at identifying clinically relevant antigens in PN and provides evidence of the immunopathological diversity in these patients, our experiments failed to identify novel antigens in PN and strong clinical‐immunological associations could neither be drawn from our study. Disease heterogeneity and reduced number of patients might be accountable for these results.
Patients with sensory‐motor PN featured IgM antibodies targeting Schwann cells more frequently than healthy controls (40% vs. 0%, P = 0.0028). Two of these patients with a lymphoma and a breast adenocarcinoma were used for IP experiments. IgM autoantibodies are not systematically screened in most autoimmune diseases. However, several relevant autoantibodies in nonparaneoplastic inflammatory neuropathies are of the IgM isotype. This includes anti‐MAG antibodies associated to monoclonal gammopathy of unknown significance and plasma cell dyscrasias,30 anti‐GM1 antibodies in multifocal motor neuropathy31, and anti‐disialosyl antibodies in CANOMAD syndrome.32 Four out of 10 patients with sensory‐motor neuropathy stained Schwann cells. All of them had an aggressive onset, three of them with predominantly axonal features in the EMG. However, clear clinical–immunological correlations could not be inferred from our findings. Technical caveats limit the efficiency of IP experiments with IgM antibodies and patients with sensory‐motor PN are extremely rare. However, this specific association will deserve attention and dedicated antigen search using different approaches in future studies.
Almost 30% of patients reacted moderately or strongly against neural structures; DRG neurons, motor neurons, or Schwann cells. This may imply that the immune response in PN is heterogeneous and directed toward diverse antigens. However these results might reflect the heterogeneity of cancer types and neuropathies present in our cohort. A high proportion of healthy controls (17.9%) reacted moderately against the same structures, precluding any further interpretation of these findings.
Our study did not identify patients harboring antibodies against neural cell adhesion molecules other than the previously described CASPR2. Although the role of neural cell adhesion molecules has been widely described to contribute to cancer pathogenesis17, 18, 19, 20, 21, 22, 23 and nonparaneoplastic autoimmune neuropathies,10, 24, 25 the results in our study suggest that nerve damage in our cohort cannot be attributed to a humoral autoimmune attack targeting L1CAM and NCAM1 or the nodal/paranodal CNTN1/CASPR1 complex, CNTN1, NF140, NF155, or NF186. However, this lack of antibodies should be confirmed in further studies involving other cohorts of PN. Anti‐CASPR2 antibodies associate with neuromyotonia, limbic encephalitis, and Morvan syndrome in patients with or without thymoma.33 Our CASPR2‐positive patient presented a sensory‐motor neuropathy with demyelinating features in the EMG and no evidence of central nervous system involvement or neuromyotonia. This patient also showed relevant IgG staining in motor and DRG neurons experiments and IgM staining in Schwann cells, probably reflecting the promiscuous immune response associated with the thymoma.
In PNS, 70–80% of cases of subacute sensory neuronopathy associate with SCLC, most of them boasting anti‐Hu antibodies. However, small subsets of patients may present with anti‐amphyphisin or anti‐CV2 antibodies (with or without anti‐Hu antibodies) or with no antibodies at all.34 In our study, 24 (70.6%) patients with a definite diagnosis of PNS presented with a classical sensory neuronopathy with a tumor and a lack of onconeural antibodies. As most paraneoplastic pure sensory neuropathies (neuronopathies) result from damage to the DRG,4 our hypothesis in such patients were focused on depicting impaired IgG/IgM autoimmune reactivity against DRG neurons as previously described.35, 36 However, interestingly, statistical results showed no significant differences in such cells between those patients and healthy controls or patients with sensory‐motor neuropathy in any of the tested conditions. Two of our patients with pure sensory neuronopathy and non‐small‐cell lung cancer showed moderate IgG staining against DRG neurons and were used for IP experiments. Unfortunately, no target antigens were identified from those experiments.
Since the description of anti‐Hu antibodies in 1985,35 and following the identification of other highly specific onconeural antibodies in the serum of PNS patients,37, 38, 39, 40, 41, 42, 43 current hypotheses suggest an autoimmune origin in PNS.1 According to the location of the antigen, two types of antibodies have been described in these conditions: antibodies targeting cell membrane antigens or antibodies against intracellular antigens, being the latter defined as onconeural.44 In our study, antibody screening ICC experiments and IP experiments were conducted in nonpermeabilizing conditions. Consequently, our search for novel antigens was exclusively catered toward the detection of cell surface molecules, which was further confirmed by the membrane‐staining patterns observed in our results. Although no surface antigens were identified in our IP experiments, a role for intracellular antigens in our patients cannot be ruled out and further comprehensive studies involving intracellular antigen screening techniques should be conducted to address that search. Another limitation in our study is the lack of inclusion of a control group with sera from patients with cancer but without neurologic involvement. Such approach was not taken into consideration in this study due to the exploratory nature of our work and to the general negativity of our results when compared against healthy controls.
The use of antigens of nonhuman origin might be considered as a limitation in our study. Nonetheless, the use of both mammalian expression vectors encoding nodal proteins and neuronal primary cultures of murine origin has previously provided us with relevant results.10, 25 Despite major sequence homology, interspecies variability may be relevant enough to hinder novel antigen identification. Thus, exploring novel viable sources of obtaining differentiated human neural cells would greatly benefit the search of new antigens, as has been proven in nonparaneoplastic autoimmune neuropathies.45, 46 Another limitation in our study includes the lack of identification of antigens of lipidic or glucidic nature. Last, identification of relevant protein antigens using these techniques relies on completeness and accuracy of existing databases, which may be biased in terms of describing novel candidates. We used a custom software setting a priori criteria to overcome this bias, however no novel antigens could be identified from our IP studies.
In summary, we provide experimental evidence of the heterogeneous IgG and IgM reactivity profile in PN patients. Immunopathological diversity and reduced numbers in our cohorts may have hindered identification of novel antigens. However, a significant proportion of PN patients in our study harbor antibodies targeting neural structures, which may suggest that novel neoplasm‐associated antigens remain to be discovered.
Author contributions
AMS, II, FG, and LQ designed the study; AMS, JA, and LQ conducted the experiments; EG provided technical support, experimental advice and reagents; EM, JD, RR, and FG provided characterized samples; AMS, LQ, and II wrote the manuscript and all authors revised it.
Conflict of Interest
The authors report no conflict of interest.
Supporting information
Table S1. HEK293 cell transfection and ICC conditions.
Table S2. Patients’ clinical and demographic data.
Table S3. All ICC results.
Table S4. Statistical analysis of DRG neurons and Schwann cells ICC in PN patients and healthy controls.
Table S5. Statistical analysis of DRG neurons, Schwann cells, and motor neurons ICC in PN patients with sensory neuronopathy or sensory‐motor neuropathy.
Figure S1. Study cohorts.
Figure S2. Positive CASPR2 ICC.
Acknowledgments
The authors thank Dr. Laura Casaní and Dr. Sergi Florit at the CSIC‐ICCC (Institut Català de Ciències Cardiovasculars) for their help with the handling of the animal samples. We also thank Dr Jerome Devaux, at the CNRS‐Aix‐Marseille Université for providing us with the NF140 and NF186 vectors used in our ICC experiments. The authors would like to acknowledge the Department of Medicine at the Universitat Autònoma de Barcelona. This project was supported by Fondo de Investigaciones Sanitarias (FIS), Instituto de Salud Carlos III, Spain and FEDER under grant FIS16/00627, principal investigator Luis Querol.
Funding Statement
This work was funded by Fondo de Investigaciones Sanitarias (FIS) grant ; Instituto de Salud Carlos III, Spain grant ; FEDER grant FIS16/00627.
References
- 1. Darnell R, Posner J. Paraneoplastic syndromes involving the nervous system. N Engl J Med 2003;349:1543–1554. [DOI] [PubMed] [Google Scholar]
- 2. Giometto B, Grisold W, Vitaliani R, et al. Paraneoplastic neurologic syndrome in the PNS Euronetwork database: a European study from 20 centers. Arch Neurol 2010;67:330–335 [DOI] [PubMed] [Google Scholar]
- 3. Graus F, Delattre JY, Antoine JC, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry 2004;75:1135–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Darnell RB, Posner JB. Paraneoplastic syndromes affecting the nervous system. Semin Oncol 2006;33:270–298. [DOI] [PubMed] [Google Scholar]
- 5. Vedeler CA, Antoine JC, Giometto B, et al. Management of paraneoplastic neurological syndromes: report of an EFNS task force. Eur J Neurol 2006;13:682–690. [DOI] [PubMed] [Google Scholar]
- 6. Graus F, Dalmau J. Paraneoplastic neuropathies. Curr Opin Neurol 2013;26:489–495. [DOI] [PubMed] [Google Scholar]
- 7. Didelot A, Honnorat J. Paraneoplastic disorders of the central and peripheral nervous systems. Handb Clin Neurol 2014;121:1156–1179. [DOI] [PubMed] [Google Scholar]
- 8. Muppidi S, Vernino S. Paraneoplastic neuropathies. Continuum (Minneap. Minn). 2014;20(5 Peripheral Nervous System Disorders):1359–1372. [DOI] [PubMed] [Google Scholar]
- 9. Boyle MET, Berglund EO, Murai KK, et al. Contactin orchestrates assembly of the septate‐like junctions at the paranode in myelinated peripheral nerve. Neuron 2001;30:385–397. [DOI] [PubMed] [Google Scholar]
- 10. Querol L, Nogales‐Gadea G, Rojas‐Garcia R, et al. Antibodies to contactin‐1 in chronic inflammatory demyelinating polyneuropathy. Ann Neurol 2013;73:370–380. [DOI] [PubMed] [Google Scholar]
- 11. Manso C, Querol L, Mekaouche M, et al. Contactin‐1 IgG4 antibodies cause paranode dismantling and conduction defects. Brain 2016;139(Pt 6):1700–1712. [DOI] [PubMed] [Google Scholar]
- 12. Wu HM, Cao W, Ye D, et al. Contactin 1 (CNTN1) expression associates with regional lymph node metastasis and is a novel predictor of prognosis in patients with oral squamous cell carcinoma. Mol Med Rep 2012;6:265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yu JW, Wu SH, Lu RQ, et al. Expression and significances of contactin‐1 in human gastric cancer. Gastroenterol Res Pract 2013;2013:210205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang R, Yao W, Qian P, et al. Increased sensitivity of human lung adenocarcinoma cells to cisplatin associated with downregulated contactin‐1. Biomed Pharmacother 2015;71:172–184. [DOI] [PubMed] [Google Scholar]
- 15. Shi K, Xu D, Yang C, et al. Contactin 1 as a potential biomarker promotes cell proliferation and invasion in thyroid cancer. Int J Clin Exp Pathol 2015;8:12473–12481 [PMC free article] [PubMed] [Google Scholar]
- 16. Yan J, Ojo D, Kapoor A, et al. Neural cell adhesion protein CNTN1 promotes the metastatic progression of prostate cancer. Cancer Res 2016;76:1603–1614. [DOI] [PubMed] [Google Scholar]
- 17. Perl A‐K, Dahl U, Wilgenbus P, et al. Reduced expression of neural cell adhesion molecule induces metastatic dissemination of pancreatic β tumor cells. Nature 1999;5:286–291. [DOI] [PubMed] [Google Scholar]
- 18. Cavallaro U, Niedermeyer J, Fuxa M, Christofori G. N‐CAM modulates tumour‐cell adhesion to matrix by inducing FGF‐receptor signalling. Nat Cell Biol 2001;3:650–657. [DOI] [PubMed] [Google Scholar]
- 19. Bussolati B, Grange C, Bruno S, et al. Neural‐cell adhesion molecule (NCAM) expression by immature and tumor‐derived endothelial cells favors cell organization into capillary‐like structures. Exp Cell Res 2006;312:913–924 [DOI] [PubMed] [Google Scholar]
- 20. Deborde S, Omelchenko T, Lyubchik A, et al. Schwann cells induce cancer cell dispersion and invasion. J Clin Invest 2016;126:1538–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kiefel H, Bondong S, Hazin J, et al. L1CAM: A major driver for tumor cell invasion and motility. Cell Adhes Migr 2012;6:374–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Valiente M, Obenauf AC, Jin X, et al. Serpins promote cancer cell survival and vascular Co‐option in brain metastasis. Cell 2014;156:1002–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Samatov TR, Wicklein D, Tonevitsky AG. L1CAM: cell adhesion and more. Prog Histochem Cytochem 2016;51:25–32 [DOI] [PubMed] [Google Scholar]
- 24. Delmont E, Manso C, Querol L, et al. Autoantibodies to nodal isoforms of neurofascin in chronic inflammatory demyelinating polyneuropathy. Brain 2017;140:1851–1858. [DOI] [PubMed] [Google Scholar]
- 25. Querol L, Nogales‐Gadea G, Rojas‐Garcia R, et al. Neurofascin IgG4 antibodies in CIDP associate with disabling tremor and poor response to IVIg. Neurology 2014;82:879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lancaster E, Huijbers MGM, Bar V, et al. Investigations of caspr2, an autoantigen of encephalitis and neuromyotonia. Ann Neurol 2011;69:303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malin SA, Davis BM, Molliver DC. Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nat Protoc 2007;2:152–160. [DOI] [PubMed] [Google Scholar]
- 28. Gingras M, Gagnon V, Minotti S, et al. Optimized protocols for isolation of primary motor neurons, astrocytes and microglia from embryonic mouse spinal cord. J Neurosci Methods 2007;163:111–118. [DOI] [PubMed] [Google Scholar]
- 29. Brockes JP, Fields KL, Raff MC. Studies on cultured rat Schwann cells. I. Establishment of purified populations from cultures of peripheral nerve. Brain Res 1979;165:105–118. [DOI] [PubMed] [Google Scholar]
- 30. Magy L, Kaboré R, Mathis S, et al. Heterogeneity of polyneuropathy associated with Anti‐MAG antibodies. J Immunol Res 2015;2015:450391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pestronk A, Cornblath DR, Ilyas AA, et al. A treatable multifocal motor neuropathy with antibodies to GM1 ganglioside. Ann Neurol 1988;24:73–78. [DOI] [PubMed] [Google Scholar]
- 32. Willison HJ, O'Leary CP, Veitch J, et al. The clinical and laboratory features of chronic sensory ataxic neuropathy with anti‐disialosyl IgM antibodies. Brain 2001;124(Pt 10):1968–1977. [DOI] [PubMed] [Google Scholar]
- 33. van Sonderen A, Arino H, Petit‐Pedrol M, et al. The clinical spectrum of Caspr2 antibody‐associated disease. Neurology 2016;87:521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Antoine J, Camdessanche J. Paraneoplastic neuropathies. Curr Opin Neurol 2017;30:513–520. [DOI] [PubMed] [Google Scholar]
- 35. Graus F, Cordon‐Cardo C, Posner JB. Neuronal antinuclear antibody in sensory neuronopathy from lung cancer. Neurology 1985;35:538–543. [DOI] [PubMed] [Google Scholar]
- 36. Dalmau J, Furneaux HM, Rosenblum MK, et al. Detection of the anti‐Hu antibody in specific regions of the nervous system and tumor from patients with paraneoplastic encephalomyelitis/sensory neuronopathy. Neurology 1991;41:1757–1764. [DOI] [PubMed] [Google Scholar]
- 37. Luque FA, Furneaux HM, Ferziger R, et al. Anti‐Ri: an antibody associated with paraneoplastic opsoclonus and breast cancer. Ann Neurol 1991;29:241–251. [DOI] [PubMed] [Google Scholar]
- 38. Peterson K, Rosenblum MK, Kotanides H, Posner JB. Paraneoplastic cerebellar degeneration. I. A clinical analysis of 55 anti‐Yo antibody‐positive patients. Neurology 1992;42:1931–1937. [DOI] [PubMed] [Google Scholar]
- 39. Folli F, Solimena M, Cofiell R, et al. Autoantibodies to a 128‐kd synaptic protein in three women with the stiff‐man syndrome and breast cancer. N Engl J Med 1993;328:546–551. [DOI] [PubMed] [Google Scholar]
- 40. Honnorat J, Antoine JC, Derrington E, et al. Antibodies to a subpopulation of glial cells and a 66 kDa developmental protein in patients with paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry 1996;61:270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Voltz R, Gultekin SH, Rosenfeld MR, et al. A serologic marker of paraneoplastic limbic and brain‐stem encephalitis in patients with testicular cancer. N Engl J Med 1999;340:1788–1795. [DOI] [PubMed] [Google Scholar]
- 42. Bernal F, Shams'ili S, Rojas I, et al. Anti‐Tr antibodies as markers of paraneoplastic cerebellar degeneration and Hodgkin's disease. Neurology 2003;60:230–234. [DOI] [PubMed] [Google Scholar]
- 43. Sabater L, Titulaer M, Saiz A, et al. SOX1 antibodies are markers of paraneoplastic Lambert‐Eaton myasthenic syndrome. Neurology 2008;70:924–928. [DOI] [PubMed] [Google Scholar]
- 44. Steck A, Yuki N, Graus F. Antibody testing in peripheral nerve disorders. Handb Clin Neurol 2013;115:189–212.https://doi.org/10.1016/b978-0-444-52902-2.00011-4 [DOI] [PubMed] [Google Scholar]
- 45. Harschnitz O, van den Berg LH, Johansen LE, et al. Autoantibody pathogenicity in a multifocal motor neuropathy induced pluripotent stem cell‐derived model. Ann Neurol 2016;80:71–88. [DOI] [PubMed] [Google Scholar]
- 46. Clark AJ, Kaller MS, Galino J, et al. Co‐cultures with stem cell‐derived human sensory neurons reveal regulators of peripheral myelination. Brain 2017;140:898–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. HEK293 cell transfection and ICC conditions.
Table S2. Patients’ clinical and demographic data.
Table S3. All ICC results.
Table S4. Statistical analysis of DRG neurons and Schwann cells ICC in PN patients and healthy controls.
Table S5. Statistical analysis of DRG neurons, Schwann cells, and motor neurons ICC in PN patients with sensory neuronopathy or sensory‐motor neuropathy.
Figure S1. Study cohorts.
Figure S2. Positive CASPR2 ICC.