

Abstract
Motivation
Controlling for tumor purity in molecular analyses is essential to allow for reliable genomic aberration calls, for inter-sample comparison and to monitor heterogeneity of cancer cell populations. In genome wide screening studies, the assessment of tumor purity is typically performed by means of computational methods that exploit somatic copy number aberrations.
Results
We present a strategy, called Purity Assessment from clonal MEthylation Sites (PAMES), which uses the methylation level of a few dozen, highly clonal, tumor type specific CpG sites to estimate the purity of tumor samples, without the need of a matched benign control. We trained and validated our method in more than 6000 samples from different datasets. Purity estimates by PAMES were highly concordant with other state-of-the-art tools and its evaluation in a cancer cell line dataset highlights its reliability to accurately estimate tumor admixtures. We extended the capability of PAMES to the analysis of CpG islands instead of the more platform-specific CpG sites and demonstrated its accuracy in a set of advanced tumors profiled by high throughput DNA methylation sequencing. These analyses show that PAMES is a valuable tool to assess the purity of tumor samples in the settings of clinical research and diagnostics.
Availability and implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
DNA methylation is an essential player of gene regulation and one of the most studied epigenetics mechanism. Its role in cancer initiation and progression has been extensively investigated during the last years (Esteller, 2008; Hansen et al., 2011; Lister et al., 2009). Hyper-methylation may occur in the regulatory regions of tumor suppressor genes, leading to inactivation of genes suppressing the cancer initiation or progression; alternatively demethylation may cause the formation of euchromatin states making oncogenes available to transcription factors (Esteller, 2007; Kundaje et al., 2015). Through a pan-cancer analysis of DNA methylation profiles of thousands of tumor samples available through The Cancer Genome Atlas Consortium (TCGA), we observed high-degree of methylation concordance within each tumor type. In prostate cancer patients, GSTP1 is hyper-methylated in nearly all tumors and the methylation signal suggests that such event is clonal (Lee et al., 1994); similarly, RUNX3 and RASSF1A are consistently methylated in bladder cancer (Kim et al., 2005) and in head and neck squamous cell carcinoma (Fan, 2004), respectively. While the role of these highly recurrent events in tumor initiation and progression is not completely clear, we reasoned that differential methylation, if clonal, can be considered as excellent proxy to estimate the cellularity (i.e. tumor purity) of each tumor sample.
Computational tumor purity estimation has been widely performed in most recent genomic studies including those from TCGA genomic landscape analyses. Tumor purity adjustment allows for uniform inter-sample and inter-patients comparisons of genomic profiles and helps unmask subclonal events (Yadav and De, 2015). Most commonly, purity assessment approaches exploit somatic copy number aberration (SCNA) profiles where the core of the computations is based on deviations of observed from expected values of tumor to normal ratios in aberrant genomic segments. SCNA-based methods (Carter et al., 2012; Prandi et al., 2014) have been demonstrated to be accurate; however, they rely on the availability of matched normal samples data and are limited in use in the presence of tumor genomes that do not demonstrate marked genomic changes (i.e. flat genomes, dominant somatic aberration mechanism differs from structural variants). DNA methylation-based strategies for purity assessment have also been used (Wang et al., 2016), including MethylPurify (Zheng et al., 2014), LUMP (Aran et al., 2015) and InfiniumPurify (Zhang et al., 2015), which showed purity estimates concordant with SCNAs based methods values (Carter et al., 2012; Prandi et al., 2014). MethylPurify uses differentially methylated regions to infer the purity of tumor samples in bisulfite sequencing data. Leukocytes Unmethylation for Purity (LUMP) predicts the purity by averaging the methylation level (β values) of 44 non-methylated immune-specific CpG sites. InfiniumPurify, the only DNA methylation-based method implemented as a tool and specifically developed for the most used DNA methylation platform (Infinium HumanMethylation450 (HM450) BeadChip, Illumina inc, http://www.illumina.com), calculates the purity by exploiting the statistical features of the distribution of the β values of differential DNA methylation sites requiring hundreds to thousands of cancer specific sites to obtain robust measures.
Here, we present a new tool for purity estimation of cancer samples from their DNA methylation profile, named Purity Assessment from clonal MEthylation Sites (PAMES). The method relies on the selection of up to 20 highly clonal cancer specific sites. We first tested PAMES on more than 6000 cancer and normal TCGA samples including 14 tumor types and next applied it to additional cancer datasets. The comparison with state-of-the-art methods showed high concordance. We then extended PAMES to the analysis of CpG islands (as opposed to platform specific methylation sites) to favor a platform independent approach and successfully tested it on enhanced reduced representation bisulfite sequencing (eRRBS) profiles (Garrett-Bakelman et al., 2015). PAMES is available as R package under GPLv3 license at https://github.com/cgplab/PAMES.
2 Materials and methods
2.1 DNA methylation data
We downloaded DNA methylation data of 14 tumor types of The Cancer Genome Atlas from the GDC Legacy Archive (https://portal.gdc.cancer.gov/legacy-archive/search/f); specifically, bladder urothelial carcinoma (BLCA), breast invasive carcinoma (BRCA), colon adenocarcinoma (COAD), esophageal carcinoma (ESCA), head and neck squamous cell carcinoma (HNSC), kidney renal clear cell carcinoma (KIRC), kidney renal papillary cell carcinoma (KIRP), liver hepatocellular carcinoma (LIHC), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), pancreatic adenocarcinoma (PAAD), prostate adenocarcinoma (PRAD), thyroid carcinoma (THCA) and uterine corpus endometrial carcinoma (UCEC). DNA methylation values were originally generated using Illumina HumanMethylation450 BeadChip (http://www.illumina.com). Upon exclusion of metastatic tumors and of duplicated experimental data, our study dataset comprises a total of 5623 tumor samples and 712 normal samples (see Supplementary Table S1). The cancer cell line dataset (Iorio et al., 2016) was downloaded from the Gene Expression Omnibus (GEO) portal (GSE68379); it includes 374 samples corresponding to cell lines from 13 tumor types profiled with Illumina HumanMethylation450 BeadChip. DNA methylation data of 100 normal, cancer-free breast tissues profiled by Illumina HumanMethylation450 BeadChip (Johnson et al., 2017) were downloaded from GEO portal (GSE88883). eRBBS data of a set of 28 advanced metastatic prostate cancer samples from our previous study (Beltran et al., 2016) was also analyzed to test the CpG islands implementation.
2.2 Selection of informative CpG sites and purity prediction
DNA methylation status is usually reported as the fraction of alleles that are methylated (β value, ranging from 0 (unmethylated) to 1 (fully methylated)). At a CpG site that is completely methylated in tumor cells and completely unmethylated in non-tumor cells, tumor sample β values below 1 reflect admixture of normal cells. The same applies for unmethylated sites in tumor cells and methylated sites in normal cells. Therefore, β values of differentially methylated sites can be used as a direct estimation of the purity of a tumor sample (or 1-β of unmethylated loci). A schematic of the method is represented in Figure 1. To pinpoint tumor-specific CpG loci that could better discriminate between tumor samples and normal samples we exploited TCGA data and computed the area under curve (AUC) of a receiver operating characteristic (ROC) curve for each site in each cancer type. ROC curves therefore display the accuracy of a binary classification, which assumes hyper-methylation in tumor samples. Thus, AUC scores close to 1 identify optimal segregations between tumor and normal samples with tumor samples on average showing β values greater than normal samples (hyper-methylation). On the contrary, AUC scores close to 0 correspond to sites in which tumor samples demonstrate on average lower β values than normal samples (hypo-methylation). For each tumor type, we considered significant those probes that demonstrate an AUC score either lower than or equal to 0.2 or higher than or equal to 0.8. To enrich for clonal events, we imposed the following criteria on β range: either min(β) < 0.1 AND max(β) > 0.6 (hypo-methylated), or min(β) < 0.4 AND max(β) > 0.9 (hyper-methylated) (see Supplementary Material for total number of informative CpGs retrieved). The established range of β values reflects the expected diversity of TGCA tumor samples in terms of purity (range > 0.5). We opted for pre-defined over data-driven threshold values to avoid technology biased selection of sites. Next, we ranked them according to the AUC scores and selected the top and bottom ranking N sites, thus retaining a total of 2 N loci. To avoid redundancy in the selection of top ranking CpG sites, we retained the top-ranking one in case CpG sites mapping within 1 Mb from each other. The tumor purity of each sample is then estimated by averaging (median) β (for hyper-methylated sites) and 1-β (for hypo-methylated sites) of the selected informative sites, as summarized in Figure 1.
Fig. 1.
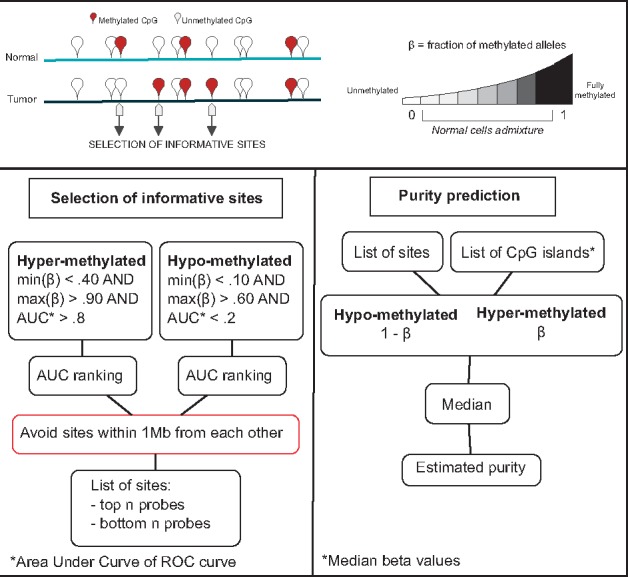
Schematic of PAMES workflow. PAMES identifies a set of tumor specific, highly clonal, CpG sites or islands (informative sites) through the differential analysis of DNA methylation levels in tumor versus normal samples (using β difference and the AUC). The β values of the selected sites are considered as optimal estimators of the admixture of tumor cells in each sample (tumor purity)
2.3 Estimation of clonal level of informative sites
To investigate the clonality of the sites selected through our strategy, we forced PAMES to consider random selections of differentially methylated sites (n = 10 for hyper-methylation and n = 10 for hypo-methylation) and then compared the original predictions and the averaged predictions of 10 PAMES-random models in the cancer cell line dataset (Iorio et al., 2016).
2.4 Selection of informative CpG islands
To allow for platform independent purity estimate approach, we aggregated the β values of CpG sites to corresponding CpG islands. The β value of each CpG island is estimated by the median of the β values of CpG sites mapping to it. Only CpG islands with at least three sites were considered for downstream analysis. We then applied our selection criteria (see Fig. 1) to select top-ranking CpG islands. The list of annotated CpG islands was downloaded from the UCSC genome browser (hg19, March 2016).
2.5 Estimation of genomic and DNA methylation alteration recurrence
Common cancer specific genomic events (mutations, SCNAs and gene translocations) were retrieved from cBio Portal (http://www.cbioportal.org) (Cerami et al., 2012; Gao et al., 2013). To estimate the recurrence of DNA methylation events, we used the following strategy. For each tumor type, the AUC of each methylation site i was computed. We classified as hyper- or hypo-methylated events with AUC < 0.2 or AUC > 0.8, respectively, indicating as nhyper and nhypo the total number of differential methylation sites identified. The mean and standard deviation of β values were calculated for each site in normal samples. We then compared the β value of each differentially methylated site in tumor samples j to the corresponding mean and standard deviation calculated in the set of normal samples (see Supplementary Table S3). Per sample fraction of supporting events (PFSE) were calculated as follows:
| (1) |
where Θ is the Heaviside function.
2.6 Comparison of purity estimates
For each sample, we computed the purity estimate using InfiniumPurify version 3.0 (Zhang et al., 2015). In addition, we considered purity estimates from the study of Aran et al., 2015, that includes purity predictions of TCGA samples from ESTIMATE (Yoshihara et al., 2013) (n = 4952), ABSOLUTE (Carter et al., 2012) (n = 2215), LUMP (Aran et al., 2015) (n = 4930), Immunohistochemistry (IHC (Aran et al., 2015)) (n = 5257) and Consensus Purity Estimate (CPE (Aran et al., 2015)) (n = 5239). In addition, purity predictions of 333 PRAD samples by ABSOLUTE were also considered. Purity levels of eRBBS data samples were computed with CLONET (Prandi et al., 2014).
2.7 Suitability of the selection of informative sites
To verify the power of our strategy to select informative sites, we both verified the effect of increasing the 2 N number of sites used to estimate purity and the outcome of a random selection of sites on PAMES predictions. Random sites were selected as follows: first, we split the set of sites into hypo-methylated and hyper-methylated depending on their AUC score ([0, 0.5) for hypo-methylation; (0.5, 1] for hyper-methylation), then, we selected an increasing number of sites from each set and we reported the Pearson’s correlation coefficient R and the Root Mean Square Deviation (RMSD) of our predictions compared with those estimated by InfiniumPurify.
2.8 Functional analysis of informative sites
Genomic regions of the 15-state model defined by ENCODE Consortium (Kundaje et al., 2015) through ChromHMM segmentation (Ernst and Kellis, 2012) were downloaded from http://egg2.wustl.edu/roadmap/data/ byFileType/chromhmmSegmentations/ChmmModels/coreMarks/ jointModel/final/STATEBYLINE/. For each 200 bp bin, we calculated the frequency of each of the 15 states across the epigenomes. State-specific genomic regions were identified as 200 bp genomic bins showing frequency greater than or equal to 0.5. Enrichment of informative sites were calculated by Fisher Exact Test considering the overlap between informative sites and each of the 15 states genomic regions by the command “fisher” of bedtools (Quinlan and Hall, 2010). Functional annotation of informative sites was performed by GREAT version 3.0.0 (McLean et al., 2010).
2.9 Accuracy evaluation of PAMES and InfiniumPurify
To compare the accuracy of PAMES and InfiniumPurify, we exploited the normal samples from TCGA and cell lines data to compute the AUC of a ROC curve for each cancer type. Here, ROC curves display the accuracy of a binary classification which assumes tumor samples purity always higher than normal samples purity, thus AUC scores close to 1 identify optimal segregations between tumor samples and normal samples.
2.10 Selection of flat genome cancer samples
We evaluated PAMES purity estimates of the TCGA PRAD samples for which neither CLONET nor ABSOLUTE could compute a score, due either to a lack of sufficient number of SCNAs or to noisy genomic profiles and selected 11 samples. We then further selected the six samples with aberrant genomic segments of at least 1 Mb in size and with an absolute Log2 Ratio greater than 0.1.
3 Results
3.1 DNA methylation across cancer types
We studied the differential DNA methylation profiles of a total of 5623 cancer samples across 14 tumor types by means of AUC, a proxy of average β differences of tumor versus normal samples (see Fig. 2A for BLCA and Supplementary Fig. S1 for the other tumor types). The box plots of Figure 2B report the distributions of the fraction of tumor-type specific differentially methylated events (by considering AUC < 0.2 or AUC > 0.8) that are supported by each tumor sample (PFSE, see Methods). We observed that of differential methylation signal is shared among all samples of a specific tumor type with values ranging from 0.10 (THCA) to 0.97 (LUSC) for hyper-methylation events and from 0.006 (THCA, n = 889 sites) to 0.91 (COAD) for hypo-methylation events (Supplementary Table S2 lists relevant cancer-specific differential methylation events per tumor type). As an example, in the PRAD dataset, we observed that more than 50% of data points are above PFSE = 0.5 both for hyper- and for hypo-methylation events. In other words, more than the half of PRAD samples (data in the box plots) supports the majority of differentially methylated events. Figure 2C reports the frequencies of the two most common somatic point mutations (SNVs) and SCNAs per tumor type (see also Supplementary Table S3); as for SNVs, TP53 and KRAS genes are the only ones that demonstrate high level of recurrence () in LUSC and PAAD, respectively, while second ranking SNVs range from about 7% (PRAD) to 70% (PAAD) frequencies. In terms of recurrence, SCNAs show a more diverse scenario in which most recurrent events ranges from 1% (THCA) to about 50% (PRAD). These observations support DNA methylation as a potential better estimation source of tumor purity. We can now compare the information reported in Figure 2B with the frequency of alteration of tumor-type specific genomic alterations. As for PRAD, in the best possible situation, we observe that about 40% of samples support one genomic event (T2-ERG rearrangement). Even assuming that it would be possible to estimate samples purity based on only one genomic event, we would able to assess the purity of less than half of the samples. Altogether this comparison highlights that alterations in DNA methylation may represent a more powerful proxy for the estimation of the purity of cancer samples.
Fig. 2.

DNA Methylation is shared within and between cancer types. (A) Scatter plot of AUC values versus β-differences for the TCGA BLCA dataset. β-differences are computed as differences between β values in tumor samples and averaged (mean) β-values in normal samples. The local regression analysis with LOESS is reported (red line). Pearson’s correlation coefficient is significant (P < 0.05). (B) Box plots report the distributions of the per sample fraction of supporting events (PFSE) for both hyper- (red) and hypo- (green) methylation in each cancer type. (C) Bar plots show the frequencies of the most recurrent (n = 2) genomic alterations for both single nucleotide variants (SNVs, green) and somatic copy number alterations (SCNAs, purple). For each tumor type, altered genes or genomic regions are reported above the corresponding bar; top to bottom terms correspond to left to right bars. (D) heat map and Ward’s hierarchical clustering using Euclidean as distance measure of the Pearson’s correlation coefficients of AUC scores of the differential methylation sites among the 14 tumor types
We next performed a pan-cancer comparative analysis of differentially methylated sites. Figure 2D summarizes the Pearson’s correlation coefficients of the AUC values calculated for each tumor type (only sites with AUC < 0.2 or AUC > 0.8 were considered). Surprisingly, we observed that a set of nine tumor types (BLCA, LUAD, HNSC, LUSC, ESCA, COAD, LIHC, BRCA and UCEC) shows similar differential methylation patterns, suggesting that, in principle, a single pan-cancer methylation signature could be built to estimate the purity of samples from multiple tumor types.
3.2 Evaluation of informative sites
To verify the relationship between the number of sites in the signature and the purity prediction, we varied the number of sites and compared purity results against multiple sets of randomly selected sites for each cancer set. Figure 4A reports the trend of the Pearson’s correlation coefficient R and root mean square deviation (RMSD) of the predictions compared with InfiniumPurify for one dataset (BLCA; all tumor types in Supplementary Fig. S2). We observed overall stability of both measures, speaking toward a robust selection strategy. We therefore reasoned that a fair balance between number of sites and high level of information for further tests would correspond to about 20 sites, equally divided between hypo- and hyper-methylated sites. We selected a 10 hyper- and 10 hypo-methylation sites for each cancer type (N = 280 in total, see Supplementary Table S4 for the list of cancer specific sites). Interestingly, in line with the clustering analysis from Figure 2D, 13 sites were selected as informative in two tumor types (six hyper-methylated and seven hypo-methylated sites), involving LUAD, LUSC, COAD, ESCA, HNSC, BLCA, BRCA and UCEC. We then studied the functional meaning of informative sites in the context of the 15 functional genome states defined by the ENCODE consortium (Ernst and Kellis, 2012; Kundaje et al., 2015) (Fig. 3B). The most significant states (log2 of odds ratio (OR) > 6) were ‘repressed polycom’ and ‘Transcription at gene 5′ and 3′’ for hyper- and hypo-methylation sites, respectively, while both set of differential methylation sites were found significantly enriched in “Enhancers” regions (). In addition, we found that informative hypo-methylation events strongly enrich regions annotated as ‘weak repressed PolyComb’ (; P-value < 10−4). Of note, all these terms remain significant when a larger selection of informative sites (N = 200, 100 for hyper and 100 for hypo, corresponding to a total of 2800 sites) is considered (Supplementary Table S5). Functional annotation of informative hyper-methylated sites reveals robust enrichment for transcription regulation related terms (i.e. for GO:0003700 and GO:0006355), while no significant enrichment was obtained from the set of hypo-methylated sites. These data suggest common functional mechanisms for informative hyper-methylated sites, while informative hypo-methylated sites might be preferentially related to cancer-specific events.
Fig. 4.
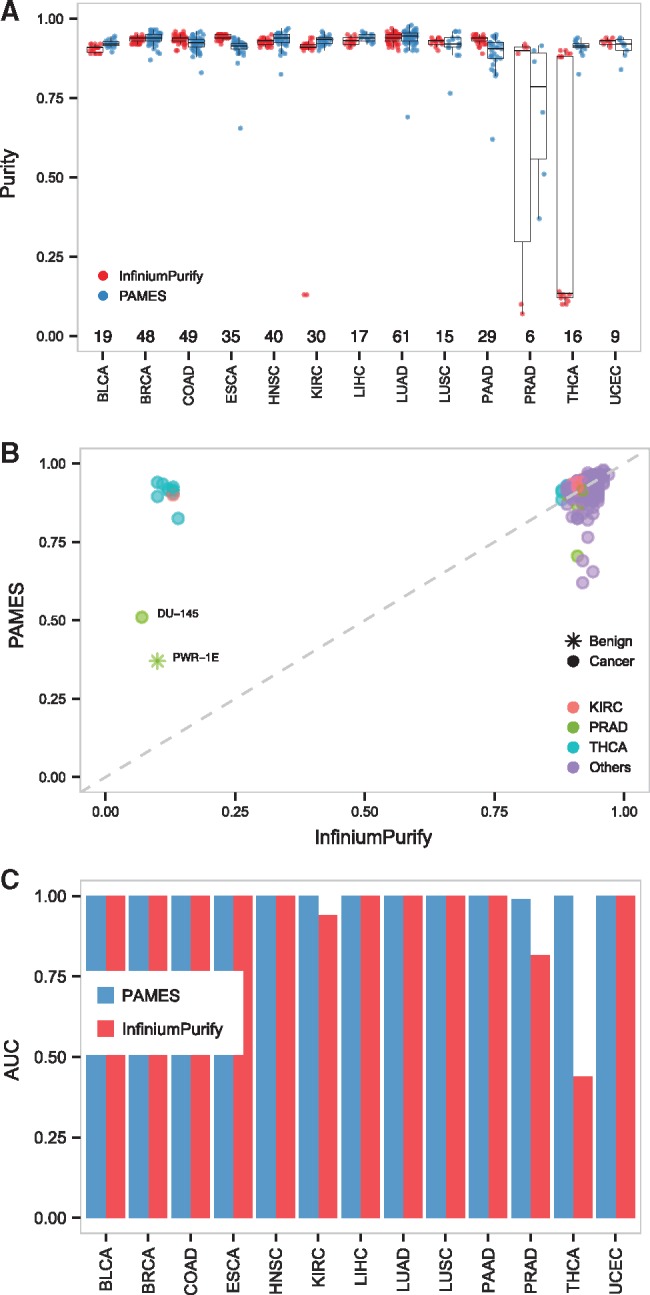
Accuracy evaluation of the purity estimates. (A) Box plots of PAMES and InfiniumPurify purity estimates on the cancer cell line dataset from Iorio et al., 2016. (B) Scatter plot of PAMES and InfiniumPurify purity estimates on cancer cell line dataset from Iorio et al., 2016. (C) AUC values of PAMES (blue) and InfiniumPurify (red) across the 14 tumor types, using cancer cell lines as positive events and normal samples from TCGA and benign cell lines as negative events
Fig. 3.

Purity estimates in TCGA datasets. (A) Correlation coefficient R and RMSD of purity estimates computed with different number of informative and random sites. Lines represent local regression (LOESS). (B) Functional enrichment analysis of the top informative sites (N = 140) from all tumor types, using the 15-state ENCODE model of the functional genome. (C) Heat map and Ward’s hierarchical clustering using Euclidean as distance measure of the Pearson’s correlation coefficients of purity estimates of the seven state-of-the-art methods in BLCA. The row annotation refers to the different strategy to estimate purity. (D) Correlation of PAMES and InfiniumPurify purity estimates on the TCGA tumor samples. R and RMSD refer to the Pearson’ s correlation coefficient and root mean square deviation averaged (mean) across the 14 tumor types
3.3 Purity prediction of TCGA datasets samples
We applied PAMES to 6335 TCGA samples and computed the Pearson’s correlation coefficient R between its predictions and other seven states-of-the-art methods (Supplementary Table S6, Supplementary Fig. S3). We observed overall concordant prediction between our method and the methylation-based InfiniumPurify method (Rmean = 0.83; Rmin = 0.46; Rmax = 0.98; see Fig. 4C for BLCA dataset and Supplementary Fig. S3 for the other datasets). In addition, PAMES gives concordant prediction with SCNA based methods. Similarly, to the other tested methods, PAMES showed the greatest deviation from IHC (Rmean = 0.70; Rmin = 0.40; Rmax = 0.95). We then focused the comparative analysis on InfiniumPurify, as it is the only DNA methylation based tool published so far and predictions were easily computable for all data included in our work. As reported in Figure 4D, the comparison between the purity estimates from our method and those from InfiniumPurify highlights a general concordance (R = 0.83, RMSD = 0.11) in the full TCGA dataset (see Supplementary Fig. S4, for single tumor type plots). We then studied the effect of threshold selection approach on informative CpG sites identification and PAMES estimations, testing two data-driven approaches (a) selection based on pan-cancer analysis and (b) selection based on tumor-type specific analysis. In both cases, thresholds were selected as the β values representing the 90% (related to high purity samples) and 10% (related to low purity samples) of the distribution of hyper and hypo methylated sites. No marked differences emerge from the comparative analyses between standard PAMES, PAMES with ’pan-cancer thresholds’ and PAMES with ’tumor-type specific thresholds’ (see Supplementary Table S7 for the resulting thresholds) as demonstrated by correlation coefficients and root mean square deviations of inferred purities (see Supplementary Fig. S6 and Supplementary Table S6), although data-driven approaches tend to mitigate the discrepancies with respect to InfiniumPurify (Supplementary Fig. S7) Relevant to computations on large datasets using precomputed sites, analysis on 6335 samples by PAMES takes about 5 min on a machine with 32 Gb RAM. A new selection of informative sites takes at most 2 h, with the most computational intensive step represented by AUC calculation.
3.4 Effect of tumor microenvironment on purity estimates
Given that the microenvironment of cancer tissues can be markedly different than the microenvironment of normal tissues, we verified the possibility that informative sites selected by PAMES are differentially methylated in cancer cells and in the associated microenvironment (tumor adjacent normal samples). To do that, we first compared the PAMES estimates in tumor samples and normal samples across the 14 TCGA datasets. Results are reported in Supplementary Figure S8 and show that for 12 out of 14 tumor types normal samples PAMES estimates were below 0.5 (median values), and for 7 out of 14 purity estimates were below or about 0.25. Also, we applied PAMES to a set of 100, cancer-free, normal breast tissues (Johnson et al., 2017) and compared the purity estimates with those made in tumor-adjacent and tumor tissues (BRCA from TCGA). Results are reported in Supplementary Figure S9 and show that for normal and adjacent tissues, purity estimates were comparable and both were significantly lower than those in tumor tissues (Wilcoxon-Mann-Whitney P-value < 10–50 for both normal versus tumor samples and adjacent versus tumor samples). This analysis shows that PAMES estimates are not affected by microenvironment of tumor cells.
3.5 Purity prediction of cancer cell line dataset samples
We evaluated the purity predictions from methylation data of a set of independent samples, the cancer cell line set from Iorio et al., 2016. Figure 4A (top) shows the distributions of the purity predictions for each cancer type obtained by PAMES and InfiniumPurify. As expected, both methods provide high levels of purity estimates for the majority of cancer types (PAMES: purity > 0.87 in 95% of samples; InfiniumPurify: purity > 0.89 in 95% of samples; collectively: purity > 0.88 in 95% of samples), with the exception of PRAD, KIRC and THCA. Inspection of the prostate cancer cell lines revealed the presence of one benign cell line (PWR-1E) and and one adenocarcinoma prostate cancer cell line (DU-145) characterized by uncommon independence from Androgen Receptor signalling potentially confounding for purity predictions by both methods. As for KIRC and THCA, we obtained low purity estimates by InfiniumPurify for one and nine cell lines, respectively, but not by our method. Other modest purity predictions include LUSC, PRAD, LUAD, ESCA, PAAD by PAMES (purity = 0.765–0.620) (Fig. 4B bottom, Supplementary Table S8). To evaluate the clonality of informative sites, we compared the purity estimates by PAMES and averaged estimations of 10 PAMES-random models (see Methods for more details). We observed that purity estimates by standard PAMES are significantly greater (Wilcoxon-Mann-Whitney P-value < 10−90, Supplementary Fig. S10) than those using the random selection of differentially methylated sites. This analysis demonstrates that informative sites selected by PAMES are significantly more clonal than PAMES-random, supporting our definition of informative sites as “clonal” events. To statistically assess the accuracy of PAMES and InfiniumPurify, we exploited cell lines data and normal samples from TCGA (n = 712). Even though this approach can be considered too lenient to truly assess accuracy in the absence of real gold standard, this analysis attempts to estimate the performance of PAMES through a comparison with InfiniumPurify that relies on much larger informative sites. The results reported in Figure 4C demonstrated high accuracy for both methods (AUCmean > 0.99 for PAMES and AUCmean = 0.94 for InfiumiumPurify). Due to the low purity estimates made on THCA, InfiumiumPurify performs worse for this tumor type (AUC = 0.44).
3.6 Purity prediction on flat genomic samples
We tested the capability of our method to predict the purity of cancer samples with epigenetic changes and flat genomic profiles. These instances are of relevance as SCNA-based methods such as ABSOLUTE and CLONET cannot provide reliable estimates, as their copy number information is inappropriate. To this end, we run PAMES on six TCGA PRAD cases for which both ABSOLUTE and CLONET were not able to estimate the purity (see Supplementary Fig. S11) and for which we identified few putative SCNAs via ad-hoc data investigation (see Methods for details). The analysis shows that, especially for very low purity samples, PAMES estimates are compatible with the log2-ratios of the rare sample specific SCNAs that we reviewed manually. This analysis suggests that PAMES can be applied on SCNA poor tumor types.
3.7 CpG island generalization
To test the capability of PAMES to infer the purity across different platforms, we implemented a CpG island version that considers methylation regions and is therefore independent from the experimental platform (see Supplementary Table S9 for the list of informative CpG islands). First, we studied the effect of summarizing CpG islands by the median of β values. Supplementary Figure S12 shows that the distribution of the summarized β values using the first quartile, the median, and the fourth quartile are very similar. We compared purity predictions by CpG sites and CpG islands in the TCGA dataset. Results are reported in Figure 5A (see Supplementary Fig. S13, for single tumor type plot) and show that the two strategies estimate concordant predictions (R = 0.95, RMSD = 0.08). We then tested the CpG island version of PAMES on 28 advanced metastatic prostate cancer profiled with eRBBS (Garrett-Bakelman et al., 2015) generated in a previous study (Beltran et al., 2016). Figure 5B reports the comparison between the PAMES methylation-based approach results and purity inferred by a SCNA-based approach, CLONET (Prandi et al., 2014). Results highlight the suitability of the CpG island-based method in predicting the purity of samples data generated by high throughput sequencing assay.
Fig. 5.
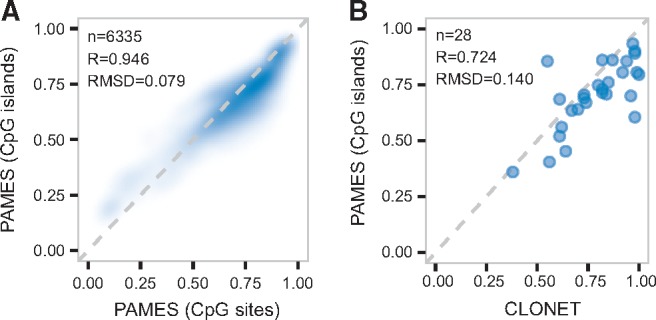
Platform independent version of PAMES. (A) Density plot of sample purities for all cancer types estimated using β values from informative sites and beta values obtained through CpG island transformation. (B) Plot of the purity estimates from PAMES (y-axis) versus CLONET (x-axis) on the eRRBS data of metastatic prostate cancer from Beltran et al., 2016
3.8 Effect of dynamic range of data on PAMES estimates
In cancer cell line data, we observed that most cell lines present 80–90% purity estimates (Fig. 4A) but systematically less than 100%. This effect can be accounted to limited dynamic range of microarrays. To verify this, we applied PAMES to a set of N = 7 normal matched benign prostate samples profiled with eRRBS (Lin et al., 2013) and compared results with N = 50 matched normal prostate samples profiles through arrays (TCGA). Results of this comparison are reported in the box plots of Supplementary Figure S14 and show that PAMES performs better when a technique with higher dynamic range is considered, such as eRRBS (purity estimates are reported in and Supplementary Table S10).
4 Discussion
A reliable estimation of the proportion of cancer cells in the admixture of cells constituting tumor microenvironment is essential to perform inter-sample analyses. Confounding effects of tumor purity on several genomic analyses have been demonstrated to be a major issue in cancer genomics studies (Aran et al., 2015). In the clinical settings, the evaluation of tumor purity allows for controlling false negative events, especially for tumor samples with low cellularity (Yoshihara et al., 2013). Through the study of thousands of tumor samples, we observed that DNA methylation alterations are markedly shared within each tumor type (Fig. 2B), suggesting that, conversely to SNVs and SCNAs, matched benign samples are not necessary to estimate the majority of DNA methylation alterations (with the exception of those sufficient to perform differential analysis). Therefore, tissue purity estimation from DNA methylation data represents a good alternative to genomics based method. The tool presented in this work, named PAMES, is able to quantify the purity of a tumor sample using few dozens of CpG informative sites. By exploiting the DNA methylation profile of thousands of tumor samples from TCGA and independent cancer sets, we demonstrated that PAMES purity estimates are largely concordant with other state-of-the-art tools. We also exploited cancer cell line dataset to investigate the clonality of selected informative sites. Even though cell lines sub-clonal events are less represented than in human tumor tissue samples, this analysis supports our definition of informative sites as ‘clonal’ events. However, for certain tumor types including KIRC, KIRP and THCA, we obtained purity estimates that significantly deviate from InfiniumPurify ones. Interestingly, these datasets show ‘anomalous’ distributions of purity estimates from both PAMES and InfiniumPurify, characterized by overall higher values and by lower dispersion when compared to the other datasets considered in this study (Supplementary Fig. S5 and Supplementary Table S6). Similarly, the clustering results of Figure 2D show dissimilar and more pronounced tumor type specific methylation profiles for four datasets, including KIRP, KIRC, and THCA. Altogether, we speculate that the selected informative sites are suboptimal for these datasets, possibly due to little availability of clonal DNA methylation alterations.
To widen its applicability, we also extended the capability of PAMES to the analysis CpG islands instead of sites; when applied to a set of advanced metastatic tumors profiled by eRRBS technique (Beltran et al., 2016), the CpG island version showed high concordance with SCNA based estimates. Also, the comparison between PAMES estimates in normal prostate sample profiled by arrays and eRRBS (Supplementary Fig. S14) suggest that PAMES performs well in the presence of higher dynamic range as provided by currently utilized DNA methylation sequencing techniques. Relevant to the clinical and the research setting, PAMES can assess tumor purity both from low throughput platforms data, such as targeted analysis (PCR, ddPCR) and from high-throughput techniques, such as microarrays and high-throughput sequencing platforms, in a platform independent manner. As a shared resource we also provide the ranked list of tumor specific CpG sites and islands, which averaged β values can be used to estimate the purity of tumor samples without the need of a matched benign control. Importantly, this could enable the evaluation of hundreds to thousands of tumor samples at low cost and the cautious selection of samples for downstream, more expensive, investigations. The informative CpG sites/islands used by PAMES to estimate samples purity were selected based on mainly untreated cancer samples from TCGA and successfully used on a limited set of advanced/treated tumor samples (see Fig. 5B). Given that in principle DNA methylation status of certain sites might change during cancer progression, the selection might result suboptimal for specific disease states in a clinical setting and should be fine-tuned by adapting the parameters (i.e., threshold values to select informative sites).
Circulating tumor DNA methylation in serum and plasma has been shown to be effective for diagnostic or prognostic biomarkers detection in different tumor types (Board et al., 2008; Diaz and Bardelli, 2014; Kawakami, 2000; Laird, 2003; Lecomte et al., 2002; Lee et al., 2002). Exploitation of clonal, tumor-type specific methylation CpG sites/islands and the methodology from this study could represent a valuable resource to trace clone dynamics in association with the use of the new targeted therapies.
Supplementary Material
Acknowledgements
The authors would like to thank the members of the Demichelis laboratory for fruitful comments, Emma Dann for layout contribution to Figure 1 and Alessio Locallo for testing the R package and the anonymous reviewers for their valuable comments that contributed to significantly improve the quality of this manuscript.
Funding
This work was supported by the NCI R01CA125612-05A1 (F.D.) and by European Research Council ERCCoG648670 (F.D.).
Conflict of Interest: none declared.
References
- Aran D. et al. (2015) Systematic pan-cancer analysis of tumour purity. Nature Commun., 6, 8971.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H. et al. (2016) Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med., 22, 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Board R.E. et al. (2008) DNA methylation in circulating tumour DNA as a biomarker for cancer. Biomarker Insights, 2, 307–319. [PMC free article] [PubMed] [Google Scholar]
- Carter S.L. et al. (2012) Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol., 30, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E. et al. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov., 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz L.A., Bardelli A. (2014) Liquid biopsies: genotyping circulating tumor DNA. J. Clini. Oncol., 32, 579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J., Kellis M. (2012) ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods, 9, 215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M. (2007) Cancer epigenomics: DNA methylomes and histone-modification maps. Nat. Rev.. Genet., 8, 286–298. [DOI] [PubMed] [Google Scholar]
- Esteller M. (2008) Epigenetics in cancer. N. Eng. J. Med., 358, 1148–1159. [DOI] [PubMed] [Google Scholar]
- Fan C.-Y. (2004) Epigenetic alterations in head and neck cancer: prevalence, clinical significance, and implications. Curr. Oncol. Rep., 6, 152–161. [DOI] [PubMed] [Google Scholar]
- Gao J. et al. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal., 6, pl1.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett-Bakelman F.E. et al. (2015) Enhanced reduced representation bisulfite sequencing for assessment of DNA methylation at base pair resolution. J. Visual. Exp., e52246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen K.D. et al. (2011) Increased methylation variation in epigenetic domains across cancer types. Nature Genetics, 43, 768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio F. et al. (2016) A landscape of pharmacogenomic interactions in cancer. Cell, 166, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K.C. et al. (2017) Normal breast tissue DNA methylation differences at regulatory elements are associated with the cancer risk factor age. Breast Cancer Res.,19, 81.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami K. (2000) Hypermethylated APC DNA in plasma and prognosis of patients with esophageal adenocarcinoma. J. Natl. Cancer Inst., 92, 1805–1811. [DOI] [PubMed] [Google Scholar]
- Kim W.-J. et al. (2005) RUNX3 inactivation by point mutations and aberrant DNA methylation in bladder tumors. Cancer Res., 65, 9347–9354. [DOI] [PubMed] [Google Scholar]
- Kundaje A. et al. (2015) Integrative analysis of 111 reference human epigenomes. Nature, 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird P.W. (2003) The power and the promise of DNA methylation markers. Nature Rev. Cancer, 3, 253–266. [DOI] [PubMed] [Google Scholar]
- Lecomte T. et al. (2002) Detection of free-circulating tumor-associated DNA in plasma of colorectal cancer patients and its association with prognosis. Int. J. Cancer, 100, 542–548. [DOI] [PubMed] [Google Scholar]
- Lee T.-L. et al. (2002) Detection of gene promoter hypermethylation in the tumor and serum of patients with gastric carcinoma. Clin. Cancer Res., 8, 1761–1766. [PubMed] [Google Scholar]
- Lee W.H. et al. (1994) Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc. Natl. Acad. Sci. U.S.A, 91, 11733–11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P.-C. et al. (2013) Epigenomic alterations in localized and advanced prostate cancer. Neoplasia, 15, 373–IN5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R. et al. (2009) Human DNA methylomes at base resolution show widespread epigenomic differences. Nature, 462, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean C.Y. et al. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nature Biotechnol., 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prandi D. et al. (2014) Unraveling the clonal hierarchy of somatic genomic aberrations. Genome Biol., 15, 439.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F. et al. (2016) Tumor purity and differential methylation in cancer epigenomics. Brief. Funct. Genom., 15, 408–419. [DOI] [PubMed] [Google Scholar]
- Yadav V.K., De S. (2015) An assessment of computational methods for estimating purity and clonality using genomic data derived from heterogeneous tumor tissue samples. Brief. Bioinform., 16, 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara K. et al. (2013) Inferring tumour purity and stromal and immune cell admixture from expression data. Nature Commun.., 4, Article number 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N. et al. (2015) Predicting tumor purity from methylation microarray data. Bioinformatics (Oxford, England), 31, 3401–3405. [DOI] [PubMed] [Google Scholar]
- Zheng X. et al. (2014) MethylPurify: tumor purity deconvolution and differential methylation detection from single tumor DNA methylomes. Genome Biol., 15, 419.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.