

Abstract
In many types of CNS neurons, repetitive spiking produces a slow afterhyperpolarization (sAHP), providing sustained, intrinsically generated negative feedback to neuronal excitation. Changes in the sAHP have been implicated in learning behaviors, in cognitive decline in aging, and in epileptogenesis. Despite its importance in brain function, the mechanisms generating the sAHP are still controversial. Here we have addressed the roles of M‐type K+ current (I M), Ca2+‐gated K+ currents (I Ca(K)'s) and Na+/K+‐ATPases (NKAs) current to sAHP generation in adult rat CA1 pyramidal cells maintained at near‐physiological temperature (35 °C). No evidence for I M contribution to the sAHP was found in these neurons. Both I Ca(K)'s and NKA current contributed to sAHP generation, the latter being the predominant generator of the sAHP, particularly when evoked with short trains of spikes. Of the different NKA isoenzymes, α1‐NKA played the key role, endowing the sAHP a steep voltage‐dependence. Thus normal and pathological changes in α1‐NKA expression or function may affect cognitive processes by modulating the inhibitory efficacy of the sAHP.
Keywords: α1 Na+/K+‐ATPase, CA1, KCa3.1, KV7, pyramidal cell, slow afterhyperpolarization, sodium pump
1. INTRODUCTION
In CA1 pyramidal cells, as in many other types of CNS neurons, bouts of action potentials are followed by a slow afterhyperpolarization (sAHP) lasting tens of seconds. It is generally agreed that the sAHP is generated by a Ca2+‐gated K+ current (I K(Ca); hence, I K(Ca)‐sAHP; Alger & Nicoll, 1980; Hotson & Prince, 1980; Gustafsson & Wigström, 1981; Brown & Griffith, 1983; Madison & Nicoll, 1984; Lancaster & Adams, 1986), whose identity remains controversial to date (King et al., 2015; Turner et al., 2016; Wang et al., 2016). This mechanism may be secondarily reinforced by Ca2+‐induced Ca2+ release (Borde, Bonansco, Fernández de Sevilla, Le Ray, & Buño, 2000). Recently it has been suggested that the M‐type K+ current (I M), known to generate the medium afterhyperpolarization (mAHP) in CA1 pyramidal cells (Gu, Vervaeke, Hu, & Storm, 2005), may also contribute to sAHP generation (Tzingounis & Nicoll, 2008, Tzingounis et al., 2010, Kim, Kobayashi, Takamatsu, & Tzingounis, 2012, 2016). Furthermore, long spike trains can induce an additional sAHP component lasting tens of seconds and attributable to activation of the electrogenic Na+ “pump”, that is the Na+/K+ ATPase (NKA; hence NKA‐sAHP; Gustafsson & Wigström,1983; Gulledge et al., 2013).
In a recent study, Gulledge et al. (2013) have investigated the relative contribution of the I K(Ca)‐ and NKA‐sAHP components to the sAHP in adult mouse CA1 pyramidal cells maintained at near‐physiological temperature (35 °C). The sAHPs were evoked by 3 s‐long trains of 150 spikes (50 Hz) spike trains and lasted ∼20 s. Intriguingly, their cumulative results suggested that the sAHP is generated predominantly by NKA current; the I K(Ca)‐sAHP component was largely absent. Similar results were obtained also with sAHPs evoked by 15 spikes. Only when the experiments were repeated at room temperature (23 °C) did a I K(Ca)‐sAHP component became evident. Consequently Gulledge et al. (2013) concluded that at physiological conditions the sAHP is predominantly a NKA‐sAHP. However, in a more recent paper King et al. (2015) argue contradictorily that even at 35 °C only a small component of the sAHP in CA1 pyramidal cells is mediated by NKA.
The NKA is a ubiquitous membrane bound enzyme responsible for maintaining Na+ and K+ gradients across the plasmalemma of living cells (Matchkov & Krivoi, 2016). For each ATP hydrolyzed it exports three Na+ ions in exchange of two K+ ions, thus generating an outward pump current that increases membrane potential (Vm). The rate of cation transport by NKAs is enhanced by increases in intracellular Na+ concentration (Therien and Blostein, 2000). Consequently, intense spike activity is expected to augment NKA current and hyperpolarize the neurons until excess intracellular Na+ is pumped out of the cell. Engagement of NKA in this type of feedback inhibition was previously described in several types of neurons in addition to CA1 pyramidal cells (Jansen & Nicholls, 1973; Koike, Mano, Okada, & Oshima, 1972; Gordon, Kocsis, & Waxman, 1990; Morita, David, Barrett, & Barrett, 1993; Parker, Hill, & Grillner, 1996; Pulver & Griffith, 2010; Kim & von Gersdorff, 2012; Zhang, Picton, Li, & Sillar, 2015; Kueh, Barnett, Cymbalyuk, & Calabrese, 2016).
Here we have investigated the contribution of NKA to sAHPs evoked by spike trains of various sizes in rat CA1 pyramidal cells maintained at 35 °C. Our findings indicate that the NKA‐sAHP is the major component of the sAHP in these neurons also when the sAHP is evoked by short spike trains. We also report that, as expected for an NKA‐dependent mechanism (Rakowski, Gadsby, & De Weer, 1997), the NKA‐sAHP is steeply voltage‐dependent, strongly decreasing with hyperpolarization. Of the two main NKA isoenzymes expressed by CA1 pyramidal cells, namely, the α1‐ and α3‐NKA (Hieber, Siegel, Fink, Beaty, & Mata, 1991; McGrail, Phillips, & Sweadner, 1991; Juhaszova & Blaustein; 1997; Bøttger et al., 2011), the relatively ouabain‐insensitive α1‐NKA appears to be the major isoenzyme generating the NKA‐sAHP, and hence, the sAHP. Finally, our results indicate that the I K(Ca)‐sAHP component is largely, but not exclusively, generated by intermediate conductance Ca2+‐gated K+ channels (KCa3.1).
2. MATERIALS AND METHODS
2.1. Ethical approval
All animal experiments were conducted in accordance with the guidelines of the Animal Care Committee of the Hebrew University.
2.2. Hippocampal slice preparation
Male Wistar rats (4–5 weeks old) were decapitated under isoflurane anesthesia and transverse dorsal hippocampal slices (400 µm) were prepared with a vibratome and transferred to a storage chamber perfused with oxygenated (95% O2 and 5% CO2) artificial cerebro‐spinal fluid (aCSF) at room temperature. For recording, slices were placed one at a time in an interface chamber and superfused (flow rate 1 ml/min) with warmed (35 °C) oxygenated aCSF containing blockers of synaptic transmission. The temperature was measured with a thermal probe juxtaposed to the slice and maintained at 35 °C with a feedback controller (NPI, Tamm, Germany).
2.3. Electrophysiology
Intracellular current‐clamp recordings were performed with sharp glass microelectrodes rather than with patch‐pipettes in order to avoid inadvertent rundown of NKA activity due to washout of soluble ingredients critical for NKA function (Gadsby & Nakao, 1989; Dobretsov, Hastings, & Stimers, 1999; Wang & Huang, 2006). This also allowed us to monitor NKA activity for long periods and to assess the effects of drugs at known concentrations in single neurons. Microelectrodes were filled with 4 M K+‐acetate (90–110 MΩ). A bridge amplifier (Axoclamp 2A, Molecular Devices, Foster City, CA) was used, allowing simultaneous injection of current and measurement of membrane potential. The bridge balance was carefully monitored and adjusted before each measurement. The pyramidal cells included in this study had stable resting potentials of −60 mV or more, and overshooting action potentials. The intracellular signals were digitized at a sampling rate of 10 kHz or more, and stored by a personal computer using a data acquisition system (Digidata 1322A) and pCLAMP9 software (Molecular Devices, CA).
Apparent input resistance (RN) was measured using a series of 500 ms hyperpolarizing square current pulses of 50 pA increments. R in was provided by the slope of the relationship of voltage deviation versus current intensity in the linear part of the hyperpolarizing range. Spikes were evoked by injecting brief (2 ms), suprathreshold depolarizing current pulses via the intracellular microelectrode. Spike trains of various durations were elicited by repetitive stimulation at 50 Hz. The size of a sAHP was assessed by measuring its amplitudes at distinct time points after cessation of stimulation and its integral (“area under the curve”). Steady positive or negative currents were injected to depolarize or hyperpolarize membrane potential (Vm), as required by the experimental protocol. Intracellular application of BAPTA (200 mM in the recording microelectrode) was performed by delivering 100 ms long negative current pulses (−500 pA) at 3 Hz for 10–15 min (Chen & Yaari, 2008).
2.4. Solutions and chemicals
The normal aCSF comprised (in mM) 124 NaCl, 3.5 KCl, 1 MgCl2, 1.6 CaCl2, 26 NaHCO3, 1.25 NaH2PO4 and 10 glucose (pH 7.35; osmolarity 305 mOsm) to which 6‐cyano‐7‐nitro‐quinoxaline‐2,3‐dione (CNQX; 15 µM), 2‐amino‐5‐phosphono‐valeric acid (D‐APV; 50 µM), picrotoxin (100 µM) and 3‐Aminopropyl)(diethoxymethyl) phosphinic acid hydrate (CGP‐55845; 1 μM) were added to block synaptic transmission. We used 1.6 mM CaCl2 in the aCSF because in bicarbonate buffer system this concentration yields 1.2 mM free Ca2+, corresponding to the baseline interstitial brain Ca2+ concentration measured in vivo (e.g., Jones & Keep, 1988). In most experiments, as indicated, the aCSFs contained also the HCN channel blocker 4‐4thylphenylamino‐1,2‐dimethyl‐6‐methylamino‐pyrimidinium chloride (ZD7288; 50 μM). The ACSFs designed to block voltage‐gated Ca2+ channels contained also NiCl2 (200 µM) and CdCl2 (200 µM). In K+‐free aCSF KCl was omitted. Picrotoxin, CGP‐55845, NiCl2, CdCl2 and ouabain were obtained from Sigma‐Aldrich Ltd. (Rehovot, Israel), CNQX and APV from Alomone Labs (Jerusalem, Israel), ZD7288 and XE991 from Tocris Bioscience (Bristol, UK), and BAPTA tetra‐potassium salt from Thermo Fisher Scientific (Waltham, MA).
2.5. Data analysis
Results are presented as the mean ± SEM. Assessment of statistical significance of differences between means was performed with paired or unpaired two‐tailed Student's t test. In all tests the significance level was set to p < .05. In the figures, significance level was presented as *(p < .05), **(p < .01), or ***(p < .001).
3. RESULTS
3.1. The sAHP in rat CA1 pyramidal cells
We first examined the properties of sAHPs evoked by trains of 150 spikes (50 Hz), as used in the mouse experiments of Gulledge et al. (2013). A representative sAHP is shown in Figure 1A. Because the very early part of the sAHP coincides with an afterhyperpolarization of medium duration (mAHP; lasting 50–100 ms; Storm, 1989), we routinely assessed the size of the “pure” sAHP by measuring its amplitudes at 1 s after stimulus termination (Figure 1A). At this time point the sAHPs attained a peak amplitude of −8.4 ± 0.3 mV, and lasted up to 32.4 ± 0.6 s (n = 64 cells, 50 slices, 42 rats). Another measurement of sAHP amplitude was made at 7 s after stimulus termination (Figure 1A), a time point at which the I K(Ca) implicated in sAHP generation (commonly referred to as I sAHP) supposedly has declined to null (e.g., Lancaster & Adams, 1986; Wang et al., 2016) We also monitored the sAHP integral (“area under the curve”) to which the mAHP makes only a small contribution.
Figure 1.
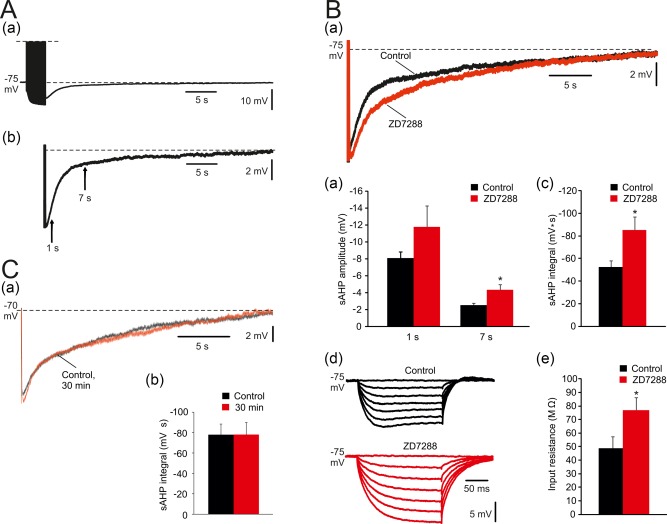
Prolonged sAHPs evoked by spike trains in CA1 pyramidal cells. (A. a) In a representative neuron, sAHPs were evoked by a 50 Hz train of 150 spikes lasting about 35 s. Each spike was evoked by a brief suprathreshold depolarizing current pulse. Here and below, each sAHP trace is the average of three consecutive recordings obtained at 3‐min intervals. (b) Enlarged trace of a sAHP depicting the time points for measuring early (1 s) and late (7 s) sAHP amplitudes (indicated by arrows). Dashed lines represent baseline Vm, used to delineate “area under the curve” representing sAHP integral. (B) Effects of blocking HCN channels on sAHPs evoked by 150 stimuli. (a) Representative traces; application of 50 μM ZD7288 caused an increase in sAHP amplitude and integral. The duration of the sAHP was not affected. (b and c) Bar diagrams depicting the pooled results (mean ± SEM; n = 8) for sAHP amplitudes and integrals, respectively. Note that only sAHP amplitude at 7 s post‐stimulus and sAHP integral were significantly increased by ZD7288. (d) Representative traces; in the same 8 neurons, application of ZD7288 caused an increase in RN and abolished the “sag” of Vm during the hyperpolarizing responses to the negative current pulses. (e) Bar diagram showing the significant RN increase in ZD7288 (mean ± SEM). (C. a) representative traces; the sAHPs evoked in CA1 pyramidal cells by trains of 150 spikes were stable for 30 min. Here and below, unless otherwise stated, all normal and modified aCSFs contained 50 μM ZD7288. (b) Bar diagram depicting the pooled (mean ± SEM; n = 10) and showing the stability of sAHPs over periods of 30 min [Color figure can be viewed at http://wileyonlinelibrary.com]
Hyperpolarizing CA1 pyramidal cells activates HCN channels (underlying the h‐current; Maccaferri, Mangoni, Lazzari, & DiFrancesco, 1993), shunting the sAHP (Kaczorowski, 2011). Indeed, adding the HCN channel blocker ZD7288 (50 μM) to the aCSF caused a marked increase in sAHP size (Figure 1Ba). The sAHP amplitude at 7 s increased significantly by 70% (from −2.5 ± 0.2 to −4.3 ± 0.6 mV; p = .02; n = 8 cells, 8 slices, 8 rats), though not at 1 s (from −8.1 ± 0.8 to −11.8 ± 2.5 mV; p = .098; Figure 1Bb). The sAHP integral also was markedly and significantly augmented (by 61%; from 52.6 ± 5.3 to 85.7 ± 11.9 mV s; p = .022; Figure 1Bc). Expectedly, these effects of ZD7288 were associated with a large and significant increase in RN (by 57%; from 48.9 ± 8.5 to 77.1 ± 9.2 MΩ; p = .029; n = 8; Figure 1Bd,e) and with a block of Vm “sagging” during prolonged hyperpolarizations (Figure 1Bd; Gasparini & DiFrancesco, 1997; Kaczorowski, 2011). To maximize sAHP size, and to enable exploration of its voltage dependence at hyperpolarized potentials, all further experiments were performed in slices bathed in ZD7288‐containing aCSFs. In this condition, hyperpolarizing the neurons down to −115 mV did not affect RN (data not shown).
No significant changes in amplitudes or integrals of the sAHPs were detected during prolonged (30–60 min) intracellular recordings (−77.5 ± 10.9 mV s at the onset of recording and −78.2 ± 11.3 mV s 30 min thereafter; n = 10 cells, 7 slices, 6 rats; Figure 1Ca,b), indicating absence of run‐down of membrane currents generating the sAHP.
3.2. Effects of blocking KV7 channels on the sAHP
Neuronal I M is generated by low voltage‐activated, noninactivating KV7 (KCNQ) channels (Shah, Mistry, Marsh, Brown, & Delmas, 2002; Brown & Passmore, 2009). These channels, having activation and deactivation kinetics of tens of milliseconds, limit the spike afterdepolarization (Yue & Yaari, 2004) and generate the mAHP in CA1 pyramidal cells (Gu et al., 2005). They have also been implicated in the generation of the sAHP in dentate granule cells and CA3 pyramidal cells (Tzingounisa & Nicoll, 2008; Tzingounisa et al., 2010; Kim et al., 2016, 2012) even though their activation range is normally more positive to resting Vm (>−60 mV; Greene & Hoshi, 2017). We used the selective I M blocker XE991 to test whether this current contributes to sAHP generation in CA1 pyramidal cells (Figure 2). The Vm of all tested cells (n = 5 cells, 5 slices, 4 rats) was maintained at −70 mV throughout these experiments using steady current injections, applied if required. sAHPs were evoked by trains of 150 suprathreshold stimuli. Adding 10 μM XE991 to the aCSF for 30 min had no significant effect on sAHP amplitudes (1 s post‐stimulus: from −8.4 ± 1.5 to −7.9 ± 1.5 mV; p = .82; 7 s post‐stimulus: from −4.7 ± 0.7 to −4.0 ± 0.6 mV; p = .55; Figure 2Aa,b) or integrals (from −93.7 ± 13.8 to −85.9 ± 14.1 mV s; p = .71; Figure 2Aa,c). Likewise, XE991 had no significant effect on RN (Figure 2Ba,b). As previously shown (Yue & Yaari, 2004), XE991 induced burst‐firing in these regular firing neurons (Figure 2C). These data indicate that KV7 channels do not contribute to sAHP generation in rat CA1 pyramidal cells.
Figure 2.
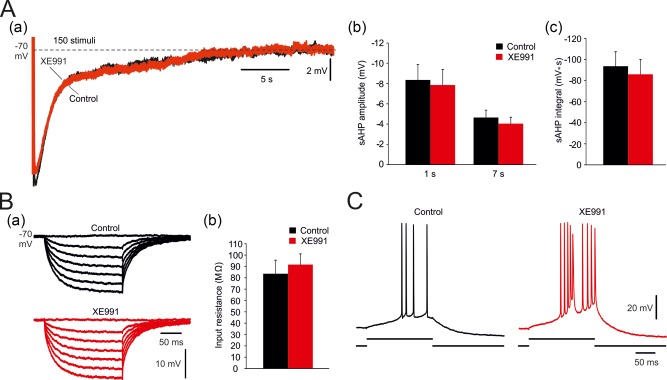
Effects of blocking I M on the sAHPs. (A. a) In a representative neuron, the traces show sAHPs evoked by trains of 150 stimuli before and 30 min after application of 10 μM XE991. (b and c) Bar diagram summarizing, respectively, the effects of XE991 on amplitudes and integrals of sAHPs evoked by 150 stimuli (mean ± SEM; n = 5). No significant changes were observed. In control condition, the number of evoked spikes equaled the number of stimuli in the train. Under XE991, the first stimulus in the train sometimes evoked a burst of 2–3 spikes. (B. a) In the same neuron, hyperpolarizing responses to negative current pulses also were not affected by XE991. (b) Bar diagram comparing RN before and after XE991 application in the same group of neurons (mean ± SEM). No significant change was observed. (C) In the same neuron, exposure to XE991 converted its regular firing in response to an excitatory current pulse (150 pA, 180 ms) to burst‐firing [Color figure can be viewed at http://wileyonlinelibrary.com]
3.3. Effects of blocking I K(ca)s on the sAHP
To resolve the contribution of a I K(Ca) to the sAHP in rat CA1 pyramidal cells, we first blocked voltage‐gated Ca2+ channels by adding Cd2+ (200 μM) and Ni2+ (200 μM) to the aCSF (Cd + Ni‐aCSF). This treatment, which prevents activation of I K(Ca) by blocking spike Ca2+ influx (Lancaster & Adams, 1986), caused a significant decrease in sAHP amplitude (Figure 3Aa), particularly in that of the early phase (1 s post‐stimulus; by 59%: from −8.7 ± 0.76 to −3.6 ± 0.7 mV; p = .0001; n = 18 cells, 18 slices, 14 rats), but also in that of the late phase (7 s post‐stimulus: by 45%; from −3.8 ± 0.3 to −2.1 ± 0.3 mV; p = .0032; Figure 3Ab). Likewise, sAHP integral significantly decreased (by 49%; from −83.2 ± 8.7 to −42.5 ± 6.2 mV s; p = .0005; Figure 3Ac). It is noteworthy, however, that roughly 50% of the sAHP persevered in Cd + Ni‐aCSF.
Figure 3.
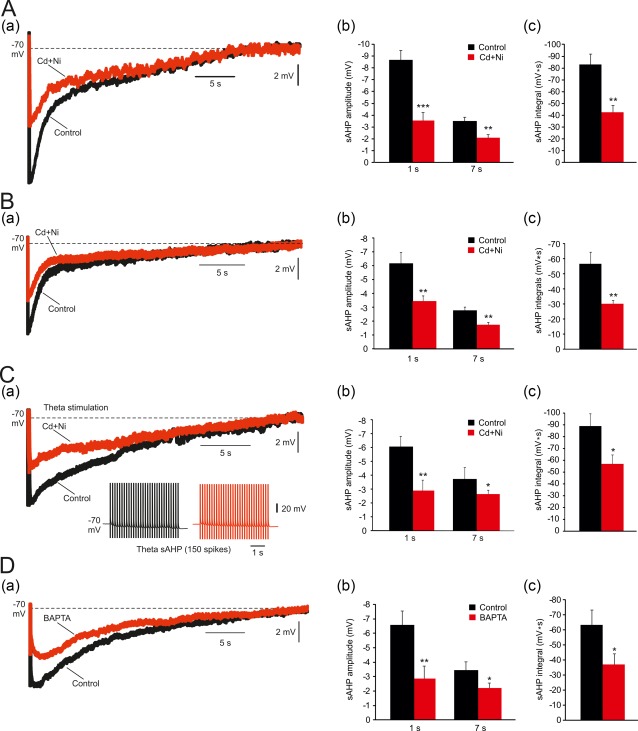
Effects of blocking I K(Ca)'s on the sAHPs. (A. a) In a representative neuron, sAHPs were evoked by 50 Hz spike trains of 150 spikes. Exchanging normal aCSF with Cd+Ni‐aCSF caused a marked reduction of the sAHP, particularly of the early phase of the sAHP. (b and c) Bar diagrams depicting the pooled results (mean ± SEM; n = 18) for sAHP amplitudes (measured at 1 and at 7 s post‐stimulus) and integrals, respectively. (B. a) Same as in Aa, but aCSFs did not contain ZD7288. Also in this condition exchanging aCSF with Cd+Ni‐aCSF caused a marked reduction of the sAHP, particularly of the early phase of the sAHP. (b and c) Bar diagrams depicting the pooled results (mean ± SEM; n = 6) for sAHP amplitudes (measured at 1 and at 7 s post‐stimulus) and integrals, respectively. (C. a) In a representative neuron, sAHP were evoked by 150 spikes delivered using a theta‐burst stimulation protocol, consisting of 30 high‐frequency (100 Hz) bursts of 5 spikes delivered at 5 Hz. Exchanging normal aCSF with Cd+Ni‐aCSF caused a marked reduction of the sAHP, particularly of the early phase of the sAHP. (b and c) Bar diagrams depicting the pooled results (mean ± SEM; n = 6) for sAHP amplitudes (measured at 1 and at 7 s post‐stimulus) and integrals, respectively. (D. a) In a representative neuron, intracellular injection of the fast Ca2+ chelator BAPTA (see Section 2) was used to block of I K(Ca) activation. BAPTA suppressed the sAHP (the traces depict the—sAHPs before and 15 min after onset of BAPTA injection, respectively). Note again the marked reduction in amplitude of the early phase of the sAHP, compared to the lack of effect on the late phase. (b and c) Bar diagrams depicting the pooled results (mean ± SEM; n = 11) for sAHP amplitudes (measured at 1 and at 7 s post‐stimulus) and integrals, respectively [Color figure can be viewed at http://wileyonlinelibrary.com]
We also examined the effects of Cd + Ni‐aCSF on the sAHP evoked in the absence of ZD7288 in the bathing solutions. This is because ZD7288, used here to block HCN channels, may affect other types of channels involved in sAHP generation, for example, voltage‐gated Na+ and Ca2+ channels (Sanchez‐Alonso et al., 2008; Wu et al., 2012). We found that also in this condition, exchanging to Cd + Ni‐aCSF significantly, but only partially, reduced the sAHP. Thus the amplitude of the early phase of the sAHP decreased by 45% (from −6.2 ± 0.8 to −3.4 ± 0.4 mV; p = .0097; n = 6 cells, 6 slices, 4 rats; Figure 3Ba), and that of the late phase by 37% (from −2.7 ± 0.2 to −1.7 ± 0.2 mV; p = .004; Figure 3Bb). Likewise, sAHP integral significantly decreased (by 47%; from −56.6 ± 7.6 to −30.1 ± 2.2 mV s; p = .007; Figure 3Bc). These results indicate that the use of ZD7288 does not modify the relative contribution of I K(Ca) to the sAHP.
We also tested the effects of Cd + Ni‐aCSF on the sAHP evoked by 150 spikes delivered using a theta‐burst stimulation protocol, thus mimicking a more physiological firing pattern of hippocampal pyramidal cells (Bland, Andersen, Ganes, & Sveen, 1980). The protocol consisted of 30 high‐frequency (100 Hz) bursts of 5 spikes delivered at 5 Hz. The resultant sAHPs were similar to those evoked by the continuous 50 Hz protocol and similarly affected by Cd + Ni‐aCSF (Figure 3C). The amplitude of the early phase of the sAHP decreased by 52% (from −6.4 ± 0.7 to −3.1 ± 0.7 mV; p = .005; n = 6 cells, 6 slices, 3 rats; Figure 3Ca), and that of the late phase by 31% (from −3.9 ± 0.5 to −2.7 ± 0.2 mV; p = .04; Figure 3Cb). Likewise, sAHP integral significantly decreased (by 36%; from −89.3 ± 10.07 to −57.0 ± 7.6 mV s; p = .02; Figure 3Cc).
Finally, we tested the effects of intracellularly applied BAPTA (see Section 2), which prevents activation of I K(Ca) by chelating cytosolic Ca2+ (Lancaster & Nicoll, 1987). This treatment also caused a significant decrease in sAHP amplitude (Figure 3Da), particularly in that of the early phase (1 s post‐stimulus: by 56%; from −6.6 ± 0.9 to −2.9 ± 1.0 mV; p < .0052; n = 11 cells, 7 slices, 7 rats), but also in that of the late phase (7 s post‐stimulus; by 37%; from −3.5 ± 0.6 to −2.2 ± 0.4 mV; p = .055; Figure 3Db). Similarly, sAHP integral significantly decreased (by 42%; from −63.5 ± 9.8 to −37.0 ± 7.3 mV s; p = .019; Figure 3Dc). These data indicate that sAHPs evoked by 150 spikes are only partially generated by a I K(Ca). However, roughly 50% of these sAHPs (as deduced from sAHP integrals) are generated by other currents.
3.4. Conductance changes during the sAHP
The inclusion of ZD7288 in the aCSFs allowed us to assess changes in membrane conductance generating the sAHP without the inadvertent effects of h‐current. To that end, negative current pulses (50 ms, −50 pA) were applied at 2 Hz before and during sAHPs. In normal aCSF containing ZD7288 (50 μM), the amplitudes of the hyperpolarizing voltage responses generated by these current pulses were reduced during the early phase of the sAHP (by 30% at 1 s post‐stimulus; from 57.7 ± 4.0 to 40.9 ± 3.8 mV; p = .012; n = 6 cells, 6 slices, 5 rats). A smaller (15%) decrease was noted also at 7 s post‐stimulus, but was not statistically significant (Figure 4a,b). These results point to a conductance increase limited to the early phase of the sAHP. In contrast, no change in conductance was detected in sAHPs similarly evoked in slices bathed in Cd + Ni‐aCSF (Figure 4c,d; n = 6 cells, 6 slices, 5 rats). These results further support the notion that a I K(Ca) contributes to the generation of the early phase of the sAHP. They also indicate that the large Ca2+‐independent sAHP component is not generated by a change in membrane conductance, thus pointing to a pump mechanism.
Figure 4.
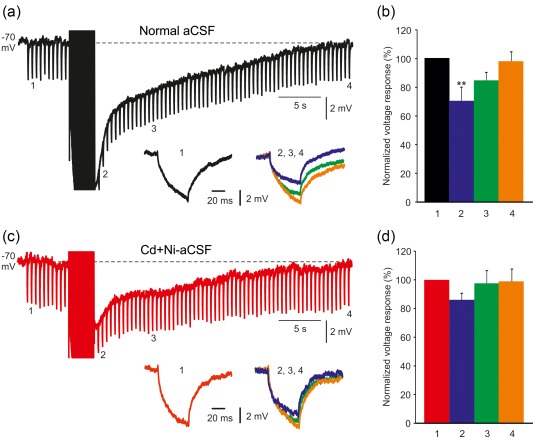
Changes in membrane conductance contributing to sAHP generation. (a) In a representative neuron, sAHPs were evoked by 50 Hz spike trains of 150 spikes in normal aCSF. To monitor changes in membrane conductance, negative current pulses (50 ms, −50 pA) were applied at 2 Hz before and during the sAHPs. Hyperpolarizing voltage responses elicited 5 s before the sAHP (1) and 1, 7, and 30 s (2, 3, 4, respectively) are shown in the inset (2, 3, and 4 are overlaid). Note the large decrease in amplitude of hyperpolarizing voltage response 2 (compared to 1) and lack of any change in that of 4, indicative of an increase in membrane conductance early after the sAHP. (b) Bar diagram showing the amplitudes of the hyperpolarizing voltage responses at the four chosen time pointes (normalized to that obtained at time point 1; mean ± SEM; n = 6). (c) Same as in a, but sAHPs were evoked in Cd+Ni‐aCSF to isolate the NKA‐sAHP component. No changes in conductance were detected during the sAHPs in this condition, pointing to an exclusive pump mechanism. (d) Same as in b, for the experiments illustrated in C (n = 6) [Color figure can be viewed at http://wileyonlinelibrary.com]
3.5. Effects of blocking NKAs on the Ca2+‐independent sAHP component
To examine whether the large Ca2+‐independent sAHP component is generated by NKA, we first tested the effects of NKA inhibition on this component isolated by bathing slices in Cd + Ni‐aCSF. Adding 10 μM ouabain depolarized Vm within 10–20 min by 11.7 ± 2.5 mV (from −70.7 ± 1.3 to −59.0 ± 3.8 mV; p = .02; n = 5 cells, 5 slices, 3 rats) and markedly reduced the sAHPs (Figure 5A). For measuring sAHP parameters, the neurons were maintained at −70 mV by injecting steady negative current via the recording microelectrode. Ouabain markedly reduced both the amplitude (1 s post‐stimulus: by 75%; from −5.9 ± 0.8 to −1.5 ± 0.3 mV; p = .002; 7 s post‐stimulus: by 68%; from −2.5 ± 0.4 to −0.8 ± 0.1 mV; p = .0015; Figure 5Ab) and the integral of the Ca2+‐independent sAHP component (by 77%; from −55.0 ± 9.8 to −12.6 ± 5.6 mV s; p = .0055; Figure 5Ac). Exposing slices to ouabain beyond 20 min caused further neuronal depolarization, spontaneous firing and decrease in spike amplitude, which precluded further monitoring of the sAHP. We also tested the effects of nominally K+‐free aCSF (0‐K‐aCSF) on the Ca2+‐independent sAHP component, as NKA activity is inhibited upon removal of extracellular K+ (Glynn & Karlish, 1975; Wang & Huang, 2006). This treatment also depolarized the neurons (within 10–20 min by 7.6 ± 1.2 mV; from −74.3 ± 1.5 to −66.7 ± 1.76 mV; p = .01; n = 5 cells, 5 slices, 4 rats) and markedly reduced the sAHPs (Figure 5Ba) measured either as amplitude (1 s post‐stimulus: by 90%; from −5.2 ± 0.6 to −0.5 ± 0.2 mV; p = .001; 7 s post‐stimulus: by 94%; from −3.6 ± 0.5 to −0.2 ± 0.1 mV; p = .0001; Figure 5Bb) or integral (by 91%; from −53.7 ± 10.7 to −5.1 ± 2.5 mV s; p = .0021; Figure 5Bc).
Figure 5.
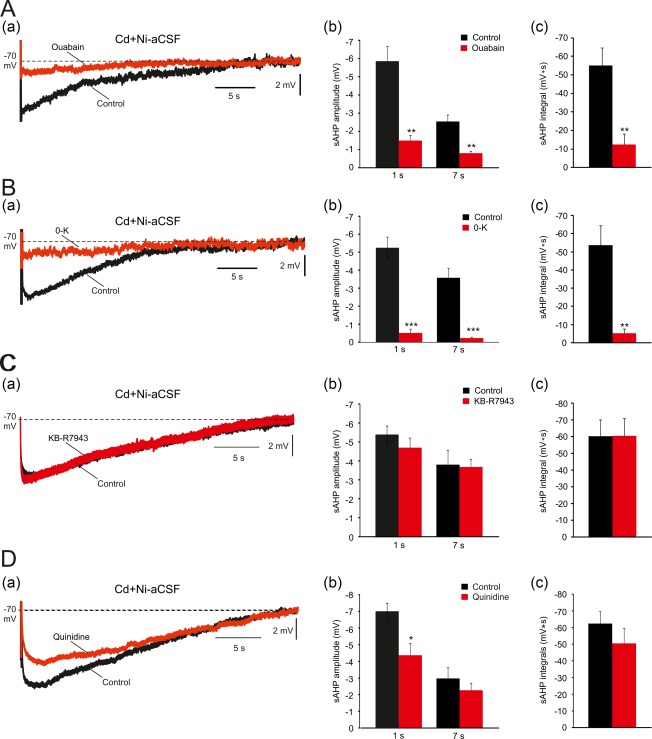
Effects of NKA and NCX inhibition on the Ca2+‐independent sAHP component. (A. a) In a representative neuron, sAHPs were evoked by 50 Hz spike trains of 150 spikes in Cd+Ni‐aCSF. Application of 10 μM ouabain markedly reduced the sAHPs (the traces depict the sAHPs before and 20 min after ouabain application, respectively). (b and c) Bar diagrams depicting the pooled results (mean ± SEM; n = 5) for sAHP amplitudes (measured at 1 and 7 s post‐stimulus) and integrals, respectively. (B. a) Same as in Aa, but NKA inhibition was attained by exchanging the standard Cd+Ni‐aCSF with K+‐free Cd+Ni‐aCSF (the traces depict the sAHPs before and 20 min after exchanging to K+‐free aCSF, respectively). (b and c) Bar diagrams depicting the pooled results (mean ± SEM; n = 5) for sAHP amplitudes (measured at 1 and 7 s post‐stimulus) and integrals, respectively. (C. a) Same as in Aa, but 10 μM KB‐R7943, a blocker of reversed Na+–Ca2+ transport, were added to the Cd+Ni‐aCSF (the traces depict the sAHPs before and 30 min after adding KB‐R7943, respectively). (b and c) Bar diagrams depicting the pooled results of 5 experiments (mean ± SEM; n = 5) for sAHP amplitudes (measured at 1 and 7 s post‐stimulus) and integrals, respectively. No significant change in the sAHP was imposed by this drug. (D. a) Same as in Aa, but 1 mM quinidine, a KNa channel blocker, was added to the Cd+Ni‐aCSF. Quinidine mildly reduced the sAHPs (the traces depict the sAHPs before and 30 min after adding quinidine, respectively). (b and c) Bar diagrams depicting the pooled results (mean ± SEM; n = 5) for sAHP amplitudes (measured at 1 and 7 s post‐stimulus) and integrals, respectively. The drug effect was significant only when measured at 1 s post‐stimulus amplitudes [Color figure can be viewed at http://wileyonlinelibrary.com]
Another source of neuronal hyperpolarization consequent to repetitive firing and massive Na+ entry may be the electrogenic Na+/Ca2+ exchanger (NCX) operating in reverse mode (Dietz et al., 2007). Indeed, this mechanism has been suggested to contribute to the sAHP increase in aging snail neurons (Scutt, Allen, Kemenes, & Yeoman, 2015). However, adding KB‐R7943 (10 μM), a NCX inhibitor (Iwamoto & Kita, 2004), to the Ni‐Cd‐aCSF had no effect on the Ca2+‐independent sAHP component (Figure 5Ca), measured either as amplitude (1 s post‐stimulus: from −5.4 ± 0.5 to −4.7 ± 0.5 mV; p = .33; 7 s post‐stimulus: from −3.8 ± 0.8 to −3.7 ± 0.4 mV; p = .89; Figure 5Cb) or integral (from −60.2 ± 9.7 to −60.4 ± 10.3 mV s; p = .99; n = 5 cells, 5 slices, 5 rats; Figure 5Cc).
Finally, we assessed the possibility that the Ca2+‐independent sAHP component is driven by Na+‐gated K+ channels, that are highly expressed in CA1 pyramidal cells (Bhattacharjee, von Hehn, Mei, & Kaczmarek, 2005; Rizzi, Knaus, & Schwarzer, 2016), and were shown to contribute to sAHPs in other neuron types (Schwindt, Spain, & Crill, 1989; Wallen et al., 2007). Adding quinidine (1 mM), a KNa inhibitor (Bhattacharjee et al., 2003), to the Ni‐Cd‐aCSF mildly but significantly reduced the amplitude of the sAHP at 1 s post‐stimulus (by 37% reduction; from −7.0 ± 0.5 to −4.4 ± 0.7 mV; p = .03), but not at 7 s post‐stimulus (from −3.7 ± 0.8 to −2.8 ± 0.5 mV; p = .18; Figure 5Da,b). Quinidine also had no significant effect on its integral (from −62.4 ± 7.2 to −50.4 ± 9.1 mV s; p = .33; n = 5 cells, 5 slices, 4 rats; Figure 5Dc). Thus, despite the small observed effect that may be due to quinidine itself reducing NKA activity (Samaha, 1967), these data do not support a role for Na+‐gated K+ channels in generating the Ca2+‐independent sAHP.
Together, these results strongly indicate that the Ca2+‐independent sAHP component in rat CA1 pyramidal cells is generated by NKAs.
3.6. Effects of blocking NKAs on the sAHP in normal aCSF
We also examined how NKAs inhibition affects the sAHP recorded in normal aCSF. Adding 10 μM ouabain to the normal aCSF depolarized Vm by 9.5 ± 0.4 mV (from 70.3 ± 1.2 to 60.8 ± 1.3 mV; p = .0003; n = 6 cells, 6 slices, 4 rats) within 10–20 min. It also markedly depressed the sAHP (Figure 6Aa), particularly the amplitude of the late phase (7 s post‐stimulus: by 92%; from −4.8 ± 0.7 to −0.4 ± 0.1 mV; p = .0001), but also that of the early phase (1 s post‐stimulus: by 48%; from −9.2 ± 0.4 to −4.8 ± 0.5 mV; p = .0001; Figure 6Ab). Likewise, sAHP integral markedly decreased (by 85%; from −100.1 ± 14.3 to −14.8 ± 5.0 mV s; p = .0001; Figure 6Ac). Washing the slices with 0‐K‐aCSF depolarized the neurons by 5.3 ± 0.7 mV (from −71.3 ± 1.3 to −66.0 ± 1.2 mV; p = .016; n = 8 cells, 8 slices, 5 rats). The effects of this treatment on the sAHP were more complex (Figure 6Ba). While it markedly depressed the amplitude of its late phase (7 s post‐stimulus: by 71%; from −4.8 ± 0.6 to −1.4 ± 0.4 mV; p = .0002), it actually increased the amplitude of its early phase (1 s post‐stimulus: by 43%; from −7.5 ± 1.1 to −10.7 ± 0.9 mV; p = .05; Figure 6Bb). Despite the latter effect, the sAHP integral was markedly reduced in 0‐K‐aCSF (by 50%; from −99.7 ± 12.0 to −49.7 ± 9.6 mV s; p = .0057; Figure 6Bc). The differential increase in the amplitude of the early sAHP phase in 0‐K‐aCSF (also reported Gulledge et al., 2013), is consistent with the notion that this phase is generated in part by a I K(Ca), which expectedly increases as K+ driving force increases due to the decrease in extracellular K+ concentration.
Figure 6.
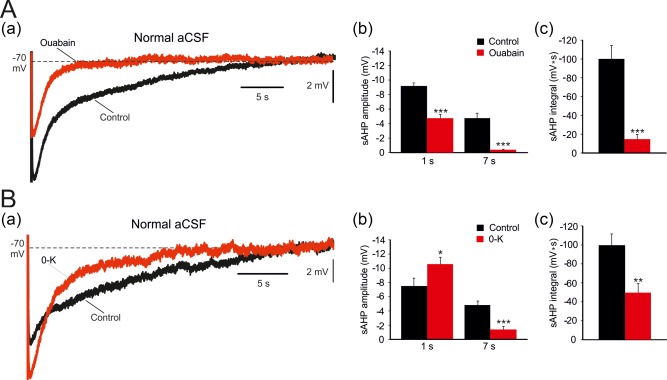
Effects of NKA inhibition on the entire sAHP. (A. a) In a representative neuron, sAHPs were evoked by 50 Hz spike trains of 150 spikes in a normal aCSF. Application of 10 μM ouabain reduced the sAHPs, particularly the late phase (the traces depict the sAHPs before and 20 min after ouabain application, respectively). (b and c) Bar diagrams depicting the pooled results (mean ± SEM; n = 6) for sAHP amplitudes (measured at 1 and at 7 s post‐stimulus) and integrals, respectively. (B. a) Same as in Aa, but NKA inhibition was attained by exchanging the normal aCSF with 0‐K‐aCSF. Note that the early phase of the sAHP increases in 0‐K‐aCSF whereas the late phase is reduced. (b and c), Bar diagrams depicting the pooled results (mean ± SEM; n = 8) for sAHP amplitudes (measured at 1 and at 7 s post‐stimulus) and integrals, respectively [Color figure can be viewed at http://wileyonlinelibrary.com]
3.7. Voltage dependence of the sAHP
We next examined the voltage dependence of the sAHP by hyperpolarizing the neuron from −65 to −115 mV in steps of 10 mV. The sAHP decreased with hyperpolarization but not in a uniform manner (Figure 7a). The early phase displayed clear reversal at Vm equal to or more negative than −105 mV. The late phase of the sAHP also decreased with hyperpolarization but did not reverse even at −115 mV (Figure 7a,b; n = 10 cells, 8 slices, 7 rats). These findings are congruent with the notion that the early sAHP phase is generated in part by a I K(Ca), whereas the late sAHP phase is due predominantly to NKA. Furthermore, they also indicate that operation of the NKA is strongly voltage‐dependent, decreasing with hyperpolarization. To test the latter notion, we repeated these experiments in slices bathed in Cd + Ni‐aCSF to isolate the NKA‐sAHP component. Consistent with the above findings, NKA‐sAHP amplitudes decreased with hyperpolarization, but did not display any reversal even at −115 mV (Figure 7c; n = 9 cells, 8 slices, 6 rats). At 1 s post‐stimulus the NKA‐sAHP decreased from −8.0 ± 1.2 mV at −65 mV to −0.8 ± 0.2 at −115 mV (Figure 7c,d), an average decrease of −1.45 mV (18%) for a 10 mV hyperpolarization.
Figure 7.
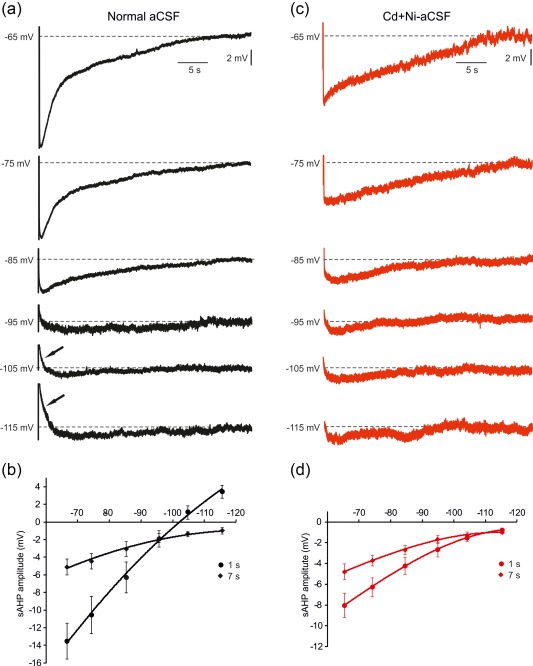
Voltage dependence of the sAHP. (A) Modulation of the sAHP by Vm in slices bathed in normal aCSF. (a) In a representative neuron, sAHPs were evoked by 50 Hz trains of 150 spikes at different Vm values, altered by current injection from −65 to −115 mV in steps of 10 mV. The size of the sAHP decreased with hyperpolarization. The amplitude of the early phase (measured at 1 s post‐stimulus) reversed at Vm values more negative than −100 mV (indicated by arrows), whereas the later phase did not reverse even at −115 mV. (b) Plots of the sAHP amplitudes versus Vm are depicted for the two measured time points (1 and 7 s). Each point represents data from 10 experiments (mean ± SEM). (c) Same as in a, but for slices bathed in Cd+Ni‐aCSF to isolate the NKA‐sAHP component. Note in the representative experiment the marked decrease in NKA‐sAHP size with hyperpolarization, but without any reversal even at the most negative Vm values. (d) Plots of the sAHP amplitudes versus Vm are depicted for the two measured time points (1 and 7 s). Each point represents data from 9 experiments (mean ± SEM) [Color figure can be viewed at http://wileyonlinelibrary.com]
3.8. Effects of varying the number of spikes on sAHP components
The results described above clearly show that sAHPs evoked by trains of 150 spikes (at 50 Hz) can be separated into two major components, namely, I K(Ca)‐sAHP and NKA‐sAHP. The two components contribute differentially to the waveform of the sAHP. Whereas the I K(Ca)‐sAHP contributes mostly, albeit partially, to the first few seconds of the sAHP, the NKA‐sAHP is the predominant sAHP component, contributing both to its early and later phases. We questioned whether this NKA‐sAHP predominance is a function of the number of spikes evoking the sAHP. To explore this issue, we employed stimulation protocols of different durations triggering 5 up to 150 spikes at 50 Hz. Expectedly, sAHPs amplitudes and integrals increased with the length of the spike train (Figure 8a). We then compared the sAHPs evoked by these stimulation protocols before and after exchanging normal aCSF with Cd + Ni‐aCSF. Intriguingly, the small sAHPs evoked by short spike trains (up to 20–40 spikes) were not significantly reduced by Cd + Ni‐aCSF (Figure 8a–d; n = 12 cells, 12 slices, 10 rats), indicating the absence of an I K(Ca)‐sAHP component. With longer spike trains (>40 spikes) a significant reduction in the measured parameters of the sAHPs upon exposure to Cd + Ni‐aCSF became evident (Figure 8a–d). These data indicate that generating a significant I K(Ca)‐sAHP component requires more than 20–40 spikes, whereas only 5 spikes are sufficient to evoke a noticeable, albeit small, NKA‐sAHP. Furthermore, the NKA‐sAHP is the dominant sAHP component irrespective of the number of spikes triggering this potential.
Figure 8.
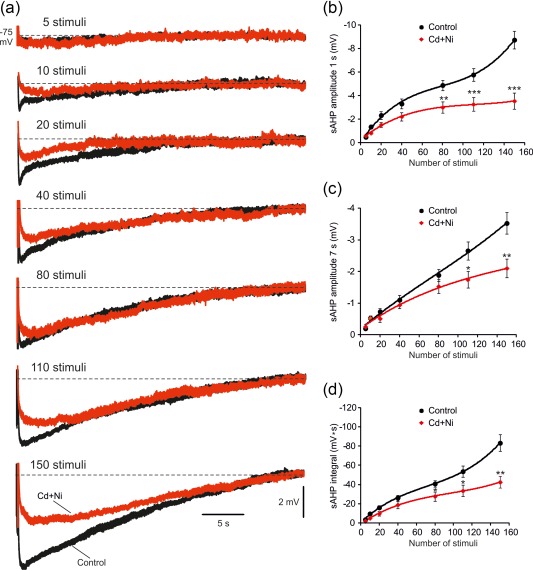
Contribution of I K(Ca)s to sAHPs evoked by spike trains of different lengths. (a) In a representative neuron, recordings of sAHPs evoked by 50 Hz spike trains of different lengths (from 5 to 150 spikes) were made in a slice bathed in normal aCSF, which was then replaced with Cd+Ni‐aCSF to block I K(Ca)s (the traces depict the sAHPs in normal aCSF and 30 min after adding Cd+Ni, respectively). (b) Plots of sAHP amplitudes measured at the 1 s time point versus number of spikes in the train in the two experimental conditions. Each point represents mean ± SEM (n = 12). (c) Same as in b, but for the 7 s time point. (d) Same as in b, but for the sAHP integral. Note that on average, only sAHPs evoked by spike trains longer than 40 spikes were significantly reduced by Cd+Ni, indicating a negligible contribution of I K(Ca)s to the generation of small sAHPs [Color figure can be viewed at http://wileyonlinelibrary.com]
3.9. Effects of low ouabain concentration
Rat CA1 pyramidal cells express predominantly α1‐ and α3‐NKAs (Hieber et al., 1991; Bøttger et al., 2011; McGrail et al., 1991; Juhaszova & Blaustein; 1997). Τhe two isoenzymes differ in their sensitivity to inhibition by ouabain, the α1‐NKA being over two orders of magnitude more resistant (IC50 ≥ 30 μM) than α3‐NKA (IC50 ≤ 100 nM; Sweadner, 1985; Urayama et al., 1988; Munzer, Daly, Jewell‐Motz, Lingrel, and Blostein, 1994; Chakraborty, 2017). To assess the roles of α1‐ and α3‐NKAs in NKA‐sAHP generation, we exploited the fact that 1 μM ouabain completely inhibits α3‐NKA while sparing α1‐NKA activity (Monteith & Blaustein, 1998; Richards, Bommert, Szabo, & Miles, 2007; Azarias et al., 2013). Slices were bathed in Cd + Ni‐aCSF and NKA‐sAHPs were evoked by 50 Hz trains of 10, 40, and 150 stimuli. Adding 1 μM ouabain for 20 min depolarized the neurons by −4.9 ± 0.9 mV (from −69.5 ± 0.7 to −64.6 ± 1.3 mV; p = .006; n = 7 cells, 7 slices, 3 rats), suggesting that α3‐NKA is operative at rest. However, the peak amplitudes and integrals of NKA‐sAHPs evoked by trains of 10 spikes (amplitude: from −0.8 ± 0.1 to −0.7 ± 0.2 mV, p = .4; integral: from −11.5 ± 1.2 to −9.1 ± 0.4 mV ms, p = .081; Figure 9Aa–c) or 40 spikes (amplitude: from −2.1 ± 0.3 to −1.7 ± 0.2 mV; p = .264; integral: from −35.7 ± 6.2 to −32.7 ± 5.4 mV s; p = .715; Figure 9Ba–c), were not significantly altered by 1 μM ouabain. Only NKA‐sAHPs evoked by 150 spikes were significantly reduced in amplitude (by 27%; from −4.3 ± 0.3 to −3.1 ± 0.3 mV; p = .021) and in integral (by 28%; from −58.8 ± 2.7 to −42.8 ± 5.0 mV s; p = .048; Figure 9Ca–c) by 1 μM ouabain. Increasing ouabain concentration to ≥10 μM completely suppressed all NKA‐sAHPs (data not shown). These results suggest that the NKA‐sAHP is generated mostly by α1‐NKA, and that α3‐NKA may be additionally recruited in conditions of sustained spike discharge.
Figure 9.
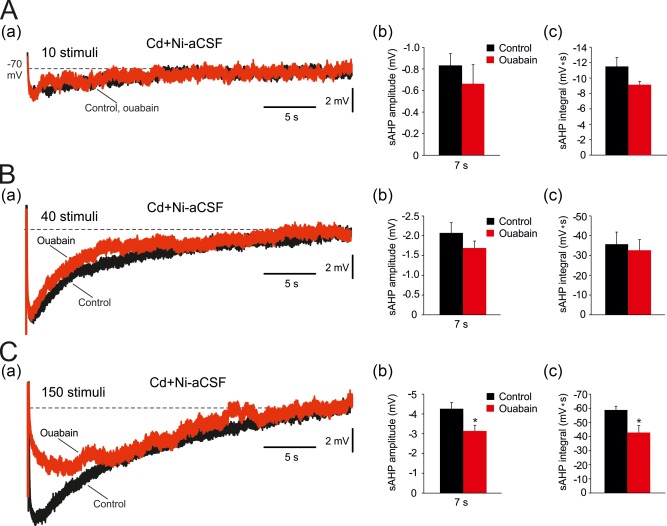
Effects of 1 μM ouabain on NKA‐sAHPs evoked by spike trains of different lengths. (A. a) In a representative neuron, sAHPs were evoked by 50 Hz spike trains of 10 spikes in Cd+Ni‐aCSF to isolate the NKA‐sAHP component. Bath application of 1 μM ouabain, a concentration that selectively blocks α3‐NKAs, only marginally reduced the NKA‐sAHPs (the traces depict the sAHPs before and 30 min after ouabain application, respectively). (b and c) Bar diagrams depicting the pooled results (mean ± SEM; n = 7) for sAHP amplitudes (measured at 1 and at 7 s post‐stimulus) and integrals, respectively. (B. a) Same as in Aa, but for spike trains of 40 spikes. (b and c) Bar diagrams depicting the pooled results (mean ± SEM; n = 7) for sAHP amplitudes (measured at 1 and at 7 s post‐stimulus) and integrals, respectively. (C. a) Same as in Aa, but for spike trains of 150 spikes. (b and c) Bar diagrams depicting the pooled results (mean ± SEM; n = 7) for sAHP amplitudes (measured at 1 and at 7 s post‐stimulus) and integrals, respectively. Only the NKA‐sAHPs evoked by 150 spikes were significantly reduced by 1 μM ouabain [Color figure can be viewed at http://wileyonlinelibrary.com]
3.10. Effects of apamin, charybdotoxin and TRAM‐34 on the sAHP
The identity of the channels contributing to the I K(Ca)‐sAHP component in CA1 pyramidal cells is currently under debate. The insensitivity of the sAHP to apamin, a selective blocker of small conductance (SK) Ca2+‐activated K+ channels (Blatz & Magleby, 1986), has ruled out a role for these channels in sAHP generation (Lancaster & Nicoll, 1987). We have also found that exposing slices to 100 nM apamin for 30 min had no effects on sAHPs evoked by trains of 150 spikes (1 s post‐stimulus: from −9.3 ± 0.5 to −7.9 ± 0.6 mV; p = .09; 7 s post‐stimulus: from −5.2 ± 0.5 to −5.0 ± 0.4 mV; p = .78; integral: from −97.4 ± 9.7 to −84.0 ± 8.1 mV s; p = .31; n = 7 cells, 7 slices, 6 rats).
Recently, it has been argued by one group KCa3.1 channels generate the sAHP in mouse CA1 pyramidal cells (King et al., 2015; Turner et al., 2016), but another group displayed evidence to the contrary (Wang et al., 2016). We tested this notion using two structurally unrelated KCa3.1 channel blockers charybdotoxin (ChTx) and TRAM‐34 (Wulff et al., 2001). Because ChTx also blocks big conductance (BK) Ca2+‐gated K+ channels (Miller, Moczydlowski, Latorre, & Phillips, 1985), we conducted the ChTx experiments in aCSFs containing 10 μM paxilline, a selective BK channel blocker (Sanchez & McManus, 1996). When tested on sAHPs evoked by trains of 150 spikes, ChTx (500 nM) reduced the early phase of the sAHP by 35% (1 s post‐stimulus: from −8.5 ± 0.9 to −5.5 ± 0.5 mV; p = .02), and the late phase by 38% (7 s post‐stimulus: from −4.8 ± 0.6 to −3.0 ± 0.3 mV; p = .01; Figure 10Aa,b). The sAHP integral also significantly decreased by 39% (from −96.1 ± 9.7 to −59.0 ± 5.8 mV s; p = .01; Figure 10Aa,c).
Figure 10.
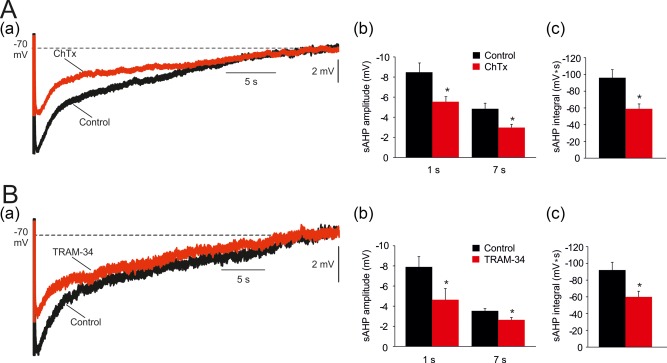
Effects of blocking KCa3.1 on the sAHPs. (A. a) A representative sAHP evoked by 50 Hz spike trains of 150 spikes in a normal aCSF. Application of 500 nM charybdotoxin (ChTx; 500 nM) reduced the sAHPs, particularly early phase (the traces depict the sAHPs before and 30 min after ChTx application, respectively). (b and c) Bar diagrams depicting the pooled results of 5 experiments (mean ± SEM; n = 5) for sAHP amplitudes (measured at 1 and at 7 s post‐stimulus) and integrals, respectively. (B. a) Same as in Aa, but KCa3.1 inhibition was attained by another blocker TRAM‐34 (5 µM) which is structurally unrelated to ChTx. Likewise ChTx, application of TRAM‐34 reduced the early phase of the sAHP. (b and c) Bar diagrams depicting the pooled results of 10 experiments (mean ± SEM; n = 10) for sAHP amplitudes (measured at 1 and at 7 s post‐stimulus) and integrals, respectively [Color figure can be viewed at http://wileyonlinelibrary.com]
Adding 5 μM TRAM‐34, a more widely used KCa3.1 channel blocker, reduced the early phase of the sAHP by 35% (1 s post‐stimulus: from −7.9 ± 1.0 to −5.1 ± 1.1 mV; p = .043), and the late phase by 34% (7 s post‐stimulus: from −3.5 ± 0.2 to −2.3 ± 0.2 mV; p = .014; Figure 10Ba,b). The sAHP integral also significantly decreased by 35% (from −92.0 ± 9.3 to −60.1 ± 6.6 mV s; p = .012; Figure 10Ba,c). As was the case with Ni‐Cd aCSF (Figure 8), TRAM‐34 caused a significant decrease in the size of sAHPs evoked by long spike trains (>40 spikes), while sAHPs evoked by short spike trains were not significantly affected (n = 10 cells, 10 slices, 6 rats; Figure 11a–d).
Figure 11.
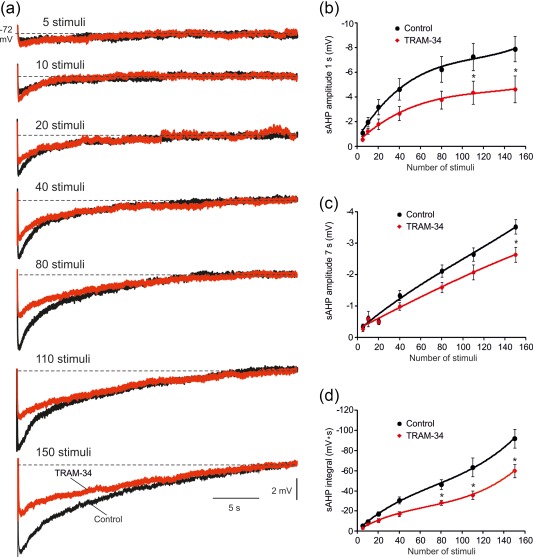
Contribution of KCa3.1 to sAHPs evoked by spike trains of different lengths. (a) In a representative neuron, sAHPs were evoked by 50 Hz spike trains of different lengths (from 5 to 150 spikes), after which 5 μM TRAM‐34 was added to the slice to block KCa3.1 (the traces depict the sAHPs in normal aCSF and 30 min after adding TRAM‐34, respectively). (b) Plots of sAHP amplitudes measured at the 1 s time point versus number of spikes in the train in the two experimental conditions. Each point represents mean ± SEM (n = 10). (c) Same as in b, but for the 7 s time point. (d) Same as in b, but for the sAHP integral. Note that on average, only sAHPs evoked by spike trains longer than 60 spikes were significantly reduced by TRAM‐34, indicating a negligible contribution of I K(Ca)s to the generation of small sAHPs [Color figure can be viewed at http://wileyonlinelibrary.com]
Adding TRAM‐34 (5 μM) to slices bathed in Ni + Cd‐aCSF for 30 min did not modify the isolated NKA‐sAHP component (7 s post‐stimulus: from −1.9 ± 0.3 to −1.8 ± 0.2, p = .76; integral: from −33.3 ± 7.2 to 26.9 ± 5.4 mV s, p = .5; n = 5 cells, 4 slices, 4 rats), indicating that the TRAM‐34‐induced depression of the sAHP is due exclusively to its effect on the I K(Ca)‐sAHP component.
Noteworthy, the sAHP reduction by ChTx and TRAM‐34 were significantly lesser than that induced by Ni + Cd (1 s post‐stimulus amplitude: 33.5 ± 4.0% and 35.0 ± 5.7%, respectively, versus 59.1 ± 7.1%, p = .03 and p = .01; 7 s post‐stimulus amplitude: 37.7 ± 3.9% and 34.7 ± 4.0%, respectively, versus 46.9 ± 4.0%, p = .04 and p = .13; integral: 36.9 ± 2.6% and 34.9 ± 2.7%, respectively, versus 52.2 ± 6.0%, p = .02 and p = .04). These data suggest that KCa3.1 channels are the major, but likely not the sole, generators of the I K(Ca)‐sAHP component in rat CA1 pyramidal cells.
3.11. Effects of TRAM‐34 on the isolated I K(ca)‐sAHP
To further examine the role of KCa3.1 in generating the I K(Ca)‐sAHP, we isolated this component by perfusing slices in aCSF containing 0.5 μM TTX, 3 mM 4‐aminopyridine, 50 μM ZD7288, 10 μM XE991 (blocker of KV7 channels generating M‐current) and 100 nM apamin (blocker of small conductance Ca2+‐gated K+ channels). In this condition, applying 90‐ms long depolarizing pulses of 400–500 pA evoked Ca2+ spikes, which were followed by sAHPs lasting up to ∼6 s, generated solely by I K(Ca). Application of TRAM‐34 affected neither Ca2+ spike threshold current (control: 475.0 ± 41.2 pA; TRAM‐34: 512.5 ± 58.1 pA; p = .28; n = 8; Figure 12Aa,b) nor its amplitude (control: 65.2 ± 1.7 mV; TRAM‐34: 67.3 ± 1.9 mV; p = .42; Figure Aa,c), assuring that Ca2+ influx is not affected by this drug. Yet TRAM‐34 markedly reduced sAHP amplitude by 56% (from −9.49 ± 0.69 mV to −4.16 ± 0.69 mV; p = .0003; Figure 12Ba,b) and integral by 64% (from −7.6 ± 1.0 mV to −2.7 ± 0.7 pA; p = .003; Figure 12Ba,c). These results attribute KCa3.1 channels a major, but not exclusive, role in generating the I K(Ca)‐sAHP in CA1 pyramidal cells.
Figure 12.
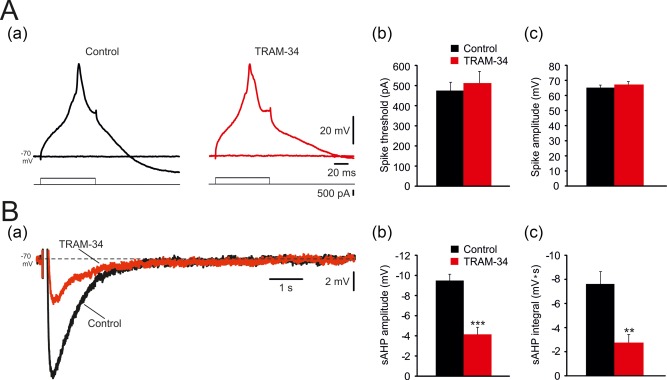
Effects of TRAM‐34 on the isolated I K(Ca)‐sAHP. (A. a) In a representative neuron bathed in aCSF containing 0.5 μM TTX, 50 μM ZD7288, 10 μM XE991 and 100 nM apamin, 90 ms‐long, 400–500 pA depolarizing pulses evoked Ca2+ spikes (left panel; top traces are voltage recordings, bottom traces are current protocols), which were not affected by adding 5 µM TRAM‐34 to the aCSF (right panel). (b and c) Bar diagrams depicting the pooled results (mean ± SEM; n = 8) for Ca2+ spikes threshold (rheobase current) and peak amplitudes, respectively. (B. a) Evoked Ca2+ spikes were followed a sAHP, generated purely by I K(Ca), lasting up to ∼6 s, which was markedly reduced by TRAM‐34 (overlaid traces—sAHPs before and 30 min after adding 5 µM TRAM‐34, respectively). (b and c) Bar diagrams depicting the pooled results (mean ± SEM; n = 8) for sAHPs measured as peak amplitudes and integrals, respectively [Color figure can be viewed at http://wileyonlinelibrary.com]
4. DISCUSSION
Despite the indisputable importance of the sAHP in controlling intrinsic neuronal excitability, the identity of its underlying mechanisms is still controversial even with respect to one of the most thoroughly investigated neurons, the CA1 pyramidal cell. Here we have addressed the relative contributions of I M, I K(Ca)'s and NKA current to sAHP generation at near‐physiological temperature (35 °C). No contribution of I M to the sAHP was noted. Our findings show that the NKA‐sAHP is the major sAHP component in this condition, requiring a smaller number of spikes for elicitation and lasting much longer than the I K(Ca)‐sAHP component. We further show that the NKA‐sAHP is steeply voltage‐dependent, decreasing with hyperpolarization. Finally, we suggest that α1‐NKA is the major isoenzyme generating the NKA‐sAHP, and hence, the sAHP in CA1 pyramidal cells.
4.1. KV7 channels do not contribute to sAHP generation in rat CA1 pyramidal cells
It has been suggested that KV7 channels contribute to the generation of the sAHP in dentate granule cells and CA3 pyramidal cells (Tzingounis & Nicoll, 2008, 2010; Kim et al., 2012). Given that Kv7 channels activate at membrane Vm more positive to −60 mV and deactivate upon hyperpolarization within tens of milliseconds (Wang et al., 1998; Greene & Hoshi, 2017) it is difficult to envisage how they could generate a sAHP component. One hypothetical possibility is that their voltage of activation is shifted negatively due to increases in cellular PIP2 associated with repetitive firing (Kim, Duignan, Hawryluk, Soh, & Tzingounis, 2016). Here we show that blocking KV7 channels with XE991, whose voltage‐dependent efficacy in blocking these channels dynamically matches their voltage range of activation (Greene, Kang, & Hoshi, 2017), does not affect the sAHP in CA1 pyramidal cells. Therefore we conclude that KV7 channels do not contribute to the sAHP in these neurons.
4.2. NKA strongly contributes to sAHP generation in rat CA1 pyramidal cells
Our results provide strong evidence that NKA current is a major generator of sAHP in CA1 pyramidal cells maintained at near‐physiological temperature (35 °C) and bathed in aCSF containing 1.2 mM free Ca2+. First, when long spike trains (150 spikes at 50 Hz) were used to evoke large sAHPs, blocking voltage‐gated Ca2+ channels with Cd + Ni or chelating intracellular Ca2+ with BAPTA, only partially reduced the sAHP, affecting more strongly its early phase. The remaining sAHP component, roughly 50% of the sAHP integral, was not associated with a conductance increase and could not be reversed by hyperpolarization to −115 mV, two observations that exclude the operation of I Na(K) and support a pump mechanism. This Ca2+‐insensitive sAHP component was unaffected by KB‐R7943, indicating that reversed NCX current is not involved in its generation, but was strongly reduced by two procedures that inhibit NKA, namely, adding ouabain to the aCSF or replacing normal aCSF with 0‐K‐aCSF. When tested in normal aCSF, both procedures also markedly suppressed the sAHP, except that 0‐K‐aCSF enhanced the early sAHP phase. This is readily explained by the two opposing effects of the latter treatment, that is NKA inhibition that reduces both early and late phases of the sAHP, and increase in K+ driving force that augments the early phase of the sAHP. The latter observation thus contradicts the possibility that upon NKA inhibition the reduction in the Ca2+‐insensitive sAHP is secondary to a decrease in K+ driving force (Schwindt, Spain, Foehring, Chubb, & Crill, 1988).
4.3. Differential sensitivity of the I K(ca) and NKA to neuronal spiking
Gulledge et al. (2013) analyzed the mechanisms of the sAHP in mouse CA1 pyramidal cells also using 150 spike trains (50 Hz) to evoke large sAHPs. They reported that neurons bathed in Cd2+‐containing aCSF or injected with BAPTA manifested sAHPs that were only slightly reduced in peak amplitude compared to control neurons, whereas no differences were seen in sAHP integrals. Accordingly, they concluded that I K(Ca) makes only a small and brief contribution to the sAHP. Our results demonstrate a larger contribution of a I K(Ca) to the sAHP evoked by similar long spike trains (including to its late phase; ∼50% of the sAHP integral). This difference in the results of the two studies may reflect the use of different species of rodents (rats vs. mice) or experimental procedures (e.g., sharp microelectrodes vs. patch pipettes). Notwithstanding, we found that when shorter spike trains were used to evoke sAHPs, the NKA‐sAHP component became increasingly dominant. Indeed, trains of up to 20–40 spikes (at 50 Hz) evoked sAHPs that were not significantly reduced by Cd + Ni‐aCSF, indicating that they are generated almost purely by the NKA. Gulledge et al. (2013) also found that a train of 15 spikes (mimicking physiological activity of a CA1 place cell) can evoke a NKA‐sAHP. It is worthy of note that both in this study and in that of Gulledge et al. (2013), spike trains were generated nonsynaptically (i.e., by current injections). It has yet to be determined whether synaptically evoked sAHPs, which are associated with influxes of Ca2+ and Na+ also through ligand‐gated channels, may recruit the two sAHP components in different proportions.
4.4. The NKA‐sAHP is voltage‐dependent
Here we show that the sAHP is strongly modulated by Vm. The decrease in the I K(Ca)‐sAHP component with hyperpolarization is due simply to the decrease in its driving force as Vm approaches K+ electrochemical equilibrium potential (VK), and explains the reversal of the early sAHP at about −100 mV. The VK in CA1 pyramidal cells was estimated to be −95 mV (Jensen et al., 1995). The more negative reversal of the early phase of the sAHP likely is due to co‐presence of the NKA‐sAHP component, which, although reduced at these potentials, did not reverse even at −115 mV. We found an average 18% decrease in NKA‐sAHP amplitude for every 10 mV of hyperpolarization. This decrease occurred despite the increased Na+ influx imposed by heightened spike amplitudes at hyperpolarized Vm, which would be expected to more strongly activate NKA. A decrease in NKA current with hyperpolarization (and an increase with depolarization) was previously demonstrated in different cell types (e.g., Rakowski et al., 1977; Schweigert, Lafaire, and Schwarz, 1988; Gadsby & Nakao, 1989), including neurons (Hamada et al., 2003). It is attributed mainly to the transit of Na+ ions through a channel‐like structure connecting their external binding sites with the extracellular space (Nakao & Gadsby, 1986; Gadsby, Rakowski, & De Weer, 1993; Sager & Rakowski, 1994); negative transmembrane voltages inhibit the NKA by forcing Na+ ions back into the access channel (Holmgren et al., 2000). This voltage‐dependence varies with the identity of the α subunit, α1‐NKA manifesting a steep dependence whereas α3‐NKA does not (Crambert et al., 2000). It likely plays a homeostatic role in stabilizing neuronal excitability; as neurons become increasingly depolarized and more excitable, the NKA becomes more efficient in their repolarizing and inhibition.
4.5. Predominant role of α1‐NKA in NKA‐sAHP generation
A widely held hypothesis posits that the α1‐NKA serves a “housekeeping” function in maintaining resting Na+ and K+ gradients across the neuronal plasma membrane, whereas the α3 isoenzyme is a “reserve” pump mobilized in conditions of excess Na+ influx, for example, during long spike trains (Monteith & Blaustein, 1998; Dobretsov & Stimers, 2005; Azarias et al., 2013). Our finding that applying 1 μM ouabain, a concentration that preferentially and completely inhibits α3 NKA (Monteith & Blaustein, 1998; Richards et al., 2007; Azarias et al., 2013; Chakraborty, 2017), only partially (by 28%) reduces the NKA‐sAHP evoked by intense spike discharge, indicates that α1‐NKA alone can generate most of the NKA‐sAHP. Further evidence that the NKA‐sAHP is generated predominantly by α1‐NKA is provided by its steep voltage‐dependence, as it was shown that α1‐NKA pump current is highly voltage‐dependent, whereas that of α3‐NKA is not (Crambert et al., 2000).
The dominant role of the α1‐NKA in generating the NKA‐sAHP may be due to a higher apparent affinity for intracellular Na+ compared to α3‐NKA (Jewel & Lingrel, 1991; Munzer et al., 1994; Therien, Nestor, Ball, and Blostein, 1996; Zahler, Zhang, Manor, and Boron, 1997; Segall, Daly, and Blostein, 2001; Hamada et al., 2003). Alternatively or additionally, α1‐NKA may be more abundantly expressed in pyramidal cell plasmalemma than α3‐NKA, as suggested by previous studies (Juhaszova & Blaustein, 1997; Richards et al., 2007), or differentially distributed in the proximal region of the neuron where spike‐associated Na+ influx would be greater than in distal dendrites. Despite its putative smaller abundance in CA1 pyramidal cells, α3‐NKA may serve additional unique functions due its high affinity to endogenous ouabain‐like compounds and other neuromediators (Dobretsov & Stimers, 2005) and its intimate association with the endoplasmic reticulum at discrete plasma membrane microdomains (Monteith & Blaustein, 1998; Song, Thompson, & Blaustein, 2013).
4.6. Ca2+‐activated K+ channels generating the I K(ca)‐sAHP component
Despite decades of research, the identity of the Ca2+‐activated K+ channels generating the I K(Ca)‐sAHP component remains enigmatic. Using KCa3.1–/– mice and an arsenal of pharmacological KCa3.1 channel modifiers, King et al (2015) provided strong evidence that KCa3.1 channels are expressed by CA1 pyramidal cells and generate the IsAHP. However, their key findings could not be replicated by Wang et al. (2016). This discrepancy has yet to be resolved. In our experimental conditions, which differ from those studies in multiple respects (e.g., species, recording technique, ambient temperature) we found that both ChTx and TRAM‐34 significantly reduced the sAHPs evoked by long spike trains in a manner similar to, but not amounting to, that of Ni‐Cd aCSF. Furthermore, when pure I K(Ca)‐sAHPs were evoked by Ca2+‐spikes, without the confounding presence of an NKA‐sAHP component, they were reduced by ∼60% by TRAM‐34. Our results support the notion that KCa3.1 channels considerably contribute to the generation of the I K(Ca)‐sAHP component in rat CA1 pyramidal cells, but other types of Ca2+‐activated K+ channels, yet unidentified, may cooperate in this process.
4.7. Functional implications
Altogether, our findings support the concept that the sAHP is generated by two mechanisms, namely, an I K(Ca) contributing mainly to its early phase of the sAHP, and an NKA contributing to both early and late phases of the sAHP. The latter mechanism predominates in sAHPs generated by short spike trains. Intriguingly, classical studies of the sAHP mechanism in CA1 pyramidal cells have all focused on I K(Ca) as the predominant mechanism (e.g., Alger and Nicoll, 1980; Hotson and Prince, 1980; Gustafsson and Wigström, 1981; Brown and Griffith, 1983; Madison and Nicoll, 1984; Lancaster and Adams, 1986). Methodological differences between the previous and present study, when exercised together, may account for this astounding discrepancy. Here we used a higher ambient temperature (35 °C vs. ∼30 °C or ∼22 °C), which would enhance NKA activity and may reduce I K(Ca) by facilitating intracellular Ca2+ removal. A large ouabain‐sensitive sAHP following long spike trains was previously described in CA3 pyramidal cells maintained at ∼36.5 °C (Gustafsson & Wigström, 1983). We also used a lower Ca2+ concentration in the aCSF (1.6 mM vs. 2.0–2.5 mM), which would be less effective in activating I K(Ca). We also used spike trains, triggered by brief depolarizing pulses, to evoke sAHPs, which may lead to less Ca2+ influx than stimulation paradigms used in previous studies—spike bursts overriding long depolarizing pulses, or Ca2+ spikes, or synaptically triggered spikes. Further studies are required to determine the relative contribution of the two sAHP mechanisms to sAHP generation in the intact brain.
Because CA1 pyramidal cells are the main output channel from the hippocampus to the cortex (Witter, Wouterlood, Naber, & Van Haeften, 2000), their intrinsic excitability likely determines multiple brain functions. Indeed, changes in sAHP amplitude in these neurons have been implicated in learning and memory (Disterhoft, Wu, & Ohno, 2004; Brosh, Rosenblum, & Barkai, 2006; Zelcer et al., 2006; Zhang, Ouyang, Ganellin, & Thomas, 2013; Thomas, 2015), in cognitive decline in aging (Moyer, Power, Thompson, & Disterhoft, 2000; Disterhoft et al., 2004; Matthews, Linardakis, & Disterhoft, 2009) and in epileptogenesis (Brehme, Kirschstein, Schulz, & Köhling, 2014; Tamir, Daninos, & Yaari, 2017). Assuming an important role of ΝΚΑs in sAHP generation in vivo, it is very likely that these changes involve modulation of NKA activity through multiple signaling pathways (Ewart & Klip, 1995). Indeed, partial deletion or inhibition of NKAs were shown to cause learning and memory deficits and neuronal hyperexcitability (Zhan, Tada, Nakazato, Tanaka, & Hongo, 2004; Moseley et al., 2007), and changes in NKA expression or function have been described in aging (de Lores Arnaiz & Ordieres, 2014) and in multiple brain pathologies (Benarroch, 2011; Toustrup‐Jensen et al., 2014). Such changes certainly would affect the sAHP, thereby modifying the negative feedback to neuronal excitation and contributing to brain dysfunction.
ACKNOWLEDGMENTS
This work was supported by the Henri and Erna Leir Foundation and the Deutsch‐Israelische Projectkooperation program of the Deutsche Forschungsgemeinschaft (DIP).
Tiwari MN, Mohan S, Biala Y, Yaari Y. Differential contributions of Ca2+‐activated K+ channels and Na+/K+‐ATPases to the generation of the slow afterhyperpolarization in CA1 pyramidal cells. Hippocampus. 2018;28:338–357. https://doi.org/10.1002/hipo.22836
REFERENCES
- Alger, B. E. , & Nicoll, R. A. (1980). Epileptiform burst afterhyperpolarization: Calcium‐dependent potassium potential in hippocampal CA1 pyramidal cells. Science, 210, 1122–1124. [DOI] [PubMed] [Google Scholar]
- Azarias, G. , Kruusmägi, M. , Connor, S. , Akkuratov, E. E. , Liu, X. L. , Lyons, D. , … Aperia, A. (2013). A specific and essential role for Na,K‐ATPase α3 in neurons co‐expressing α1 and α3. Journal of Biological Chemistry, 288, 2734–2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch, E. E. (2011). Na+, K+‐ATPase: Functions in the nervous system and involvement in neurologic disease. Neurology, 76, 287–293. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee, A. , Joiner, W. J. , Wu, M. , Yang, Y. , Sigworth, F. J. , & Kaczmarek, L. K. Slick (Slo2.1), a rapidly‐gating sodium‐activated potassium channel inhibited by ATP. Journal of Neuroscience, 23, 11681–11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee, A. , von Hehn, C. A. , Mei, X. , & Kaczmarek, L. K. (2005). Localization of the Na+‐activated K+ channel Slick in the rat central nervous system. Journal of Comparative Neurology, 484, 80–92. [DOI] [PubMed] [Google Scholar]
- Bland, B. H. , Andersen, P. , Ganes, T. , & Sveen, 0. (1980). Automated analysis of rhythmicity of physiologically identified hippocampal formation neurons. Experimental Brain Research, 38, 205–219. [DOI] [PubMed] [Google Scholar]
- Blatz, A. L. , & Magleby, K. L. (1986). Single apamin‐blocked Ca‐activated K+ channels of small conductance in cultured rat skeletal muscle. Nature, 323, 718–720. [DOI] [PubMed] [Google Scholar]
- Borde, M. , Bonansco, C. , Fernández de Sevilla, D. , Le Ray, D. , & Buño, W. (2000). Voltage‐clamp analysis of the potentiation of the slow Ca2+‐activated K+ current in hippocampal pyramidal neurons. Hippocampus, 10, 198–206. [DOI] [PubMed] [Google Scholar]
- Bøttger, P. , Tracz, Z. , Heuck, A. , Nissen, P. , Romero‐Ramos, M. , & Lykke‐Hartmann, K. (2011). Distribution of Na/K‐ATPase alpha 3 isoform, a sodium‐potassium P‐type pump associated with rapid‐onset of dystonia parkinsonism (RDP) in the adult mouse brain. Journal of Comparative Neurology, 519, 376–404. [DOI] [PubMed] [Google Scholar]
- Brehme, H. , Kirschstein, T. , Schulz, R. , & Köhling, R. (2014). In vivo treatment with the casein kinase 2 inhibitor 4,5,6,7‐tetrabromotriazole augments the slow afterhyperpolarizing potential and prevents acute epileptiform activity. Epilepsia, 55, 175–183. [DOI] [PubMed] [Google Scholar]
- Brosh, I. , Rosenblum, K. , & Barkai, E. (2006). Learning‐induced reversal of the effect of noradrenalin on the postburst AHP. Journal of Neurophysiology, 96, 1728–1733. [DOI] [PubMed] [Google Scholar]
- Brown, D. A. , & Griffith, W. H. (1983). Calcium‐activated outward current in voltage‐clamped hippocampal neurones of the guinea‐pig. Journal of Physiology, 337, 287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, D. A. , & Passmore, G. M. (2009). Neural KCNQ (Kv7) channels. British Journal of Pharmacology, 156, 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty, D. , Fedorova, O. V. , Bagrov, A. Y. , & Kaphzan, H. (2017). Selective ligands for Na+/K+‐ATPase α isoforms differentially and cooperatively regulate excitability of pyramidal neurons in distinct brain regions. Neuropharmacology, 117, 338–351. [DOI] [PubMed] [Google Scholar]
- Chen, S. , & Yaari, Y. (2008). Spike Ca2+ influx upmodulates the spike afterdepolarization and bursting via intracellular inhibition of KV7/M channels. Journal of Physiology, 586, 1351–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crambert, G. , Hasler, U. , Beggah, A. T. , Yu, C. , Modyanov, N. N. , Horisberger, J. D. , … Geering, K. (2000). Transport and pharmacological properties of nine different human Na, K‐ATPase isozymes. Journal of Biological Chemistry, 275(3), 1976–1986. [DOI] [PubMed] [Google Scholar]
- de Lores Arnaiz, G. R. , & Ordieres, M. G. (2014). Brain Na+, K+‐ATPase activity in aging and disease. International Journal of Biomedical Sciences, 10, 85–102. [PMC free article] [PubMed] [Google Scholar]
- Disterhoft, J. F. , Wu, W. W. , & Ohno, M. (2004). Biophysical alterations of hippocampal pyramidal neurons in learning, ageing and Alzheimer's disease. Ageing Research Reviews, 3, 383–406. [DOI] [PubMed] [Google Scholar]
- Dobretsov, M. , Hastings, S. L. , & Stimers, J. R. (1999). Functional Na+/K+ pump in rat dorsal root ganglia neurons. Neuroscience, 93, 723–729. [DOI] [PubMed] [Google Scholar]
- Ewart, H. S. , & Klip, A. (1995). Hormonal regulation of the Na+‐K+‐ATPase: Mechanisms underlying rapid and sustained changes in pump activity. American Journal of Physiology, 269, C295–C311. [DOI] [PubMed] [Google Scholar]
- Gadsby, D. C. , & Nakao, M. (1989). Steady‐state current‐voltage relationship of the Na/K pump in guinea pig ventricular myocytes. Journal of General Physiology, 94, 511–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby, D. C. , Rakowski, R. F. , & De Weer, P. (1993). Extracellular access to the Na,K pump: Pathway similar to ion channel. Science, 260, 100–103. [DOI] [PubMed] [Google Scholar]
- Gasparini, S. , & DiFrancesco, D. (1997). Action of the hyperpolarization‐activated current (Ih) blocker ZD 7288 in hippocampal CA1 neurons. Pflugers Archives, 435, 99–106. [DOI] [PubMed] [Google Scholar]
- Glynn, I. M. , & Karlish, S. J. (1975). The sodium pump. Annual Review of Physiology, 37, 13–55. [DOI] [PubMed] [Google Scholar]
- Gordon, T. R. , Kocsis, J. D. , & Waxman, S. G. (1990). Electrogenic pump (Na+/K+‐ATPase) activity in rat optic nerve. Neuroscience, 37, 829–837. [DOI] [PubMed] [Google Scholar]
- Greene, D. L. , & Hoshi, N. (2017). Modulation of Kv7 channels and excitability in the brain. Cellular and Molecular Life Sciences, 74(3), 495–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene, D. L. , Kang, S. , & Hoshi, N. (2017). XE991 and linopirdine are state‐dependent inhibitors for Kv7/KCNQ channels that favor activated single subunits. Journal of Pharmacology and Experimental Therapeutics, 362, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, N. , Vervaeke, K. , Hu, H. , & Storm, J. F. (2005). Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after‐hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. Journal of Physiology, 566, 689–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge, A. T. , Dasari, S. , Onoue, K. , Stephens, E. K. , Hasse, J. M. , & Avesar, D. (2013). A sodium‐pump‐mediated afterhyperpolarization in pyramidal neurons. Journal of Neuroscience, 33, 13025–13041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson, B. , & Wigström, H. (1981). Evidence for two types of afterhyperpolarization in CA1 pyramidal cells in the hippocampus. Brain Research, 206, 462–468. [DOI] [PubMed] [Google Scholar]
- Gustafsson, B. , & Wigström, H. (1983). Hyperpolarization following long‐lasting tetanic activation of hippocampal pyramidal cells. Brain Research, 275, 159–163. [DOI] [PubMed] [Google Scholar]
- Hamada, K. , Matsuura, H. , Sanada, M. , Toyoda, F. , Omatsu‐Kanbe, M. , Kashiwagi, A. , & Yasuda, H. (2003). Properties of the Na+/K+ pump current in small neurons from adult rat dorsal root ganglia. British Journal of Pharmacology, 138, 1517–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieber, V. , Siegel, G. J. , Fink, D. J. , Beaty, M. W. , & Mata, M. (1991). Differential distribution of (Na, K)‐ATPase alpha isoforms in the central nervous system. Cellular and Molecular Neurobiology, 11, 253–262. [DOI] [PubMed] [Google Scholar]
- Holmgren, M. , Wagg, J. , Bezanilla, F. , Rakowski, R. F. , De Weer, P. , & Gadsby, D. C. (2000). Three distinct and sequential steps in the release of sodium ions by the Na+/K+‐ATPase. Nature, 403, 898–901. [DOI] [PubMed] [Google Scholar]
- Hotson, J. R. , & Prince, D. A. (1980). A calcium‐activated hyperpolarization follows repetitive firing in hippocampal neurons. Journal of Neurophysiology, 43, 409–419. [DOI] [PubMed] [Google Scholar]
- Iwamoto, T. , & Kita, S. (2004). Development and application of Na+/Ca2+ exchange inhibitors. Molecular and Cellular Biochemistry, 259, 157–161. [DOI] [PubMed] [Google Scholar]
- Jansen, J. K. , & Nicholls, J. G. (1973). Conductance changes, an electrogenic pump and the hyperpolarization of leech neurones following impulses. Journal of Physiology, 229, 635–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen, M. S. , Cherubini, E. , & Yaari, Y. (1993). Opponent effects of potassium on GABAA‐mediated postsynaptic inhibition in the rat hippocampus. Journal of Neurophysiology, 69, 764–771. [DOI] [PubMed] [Google Scholar]
- Jewell, E. A. , & Lingrel, J. B. (1991). Comparison of the substrate dependence properties of the rat Na,K‐ATPase alpha 1, alpha 2, and alpha 3 isoforms expressed in HeLa cells. Journal of Biological Chemistry, 266, 16925–16930. [PubMed] [Google Scholar]
- Jones, H. C. , & Keep, R. F. (1988). Brain fluid calcium concentration and response to acute hypercalcaemia during development in the rat. Journal of Physiology, 402, 579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhaszova, M. , & Blaustein, M. P. (1997). Na+ pump low and high ouabain affinity alpha subunit isoforms are differently distributed in cells. Proceedings of the National Academy of Sciences of the United States of America, 94, 1800–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowski, C. C. (2011). Bidirectional pattern‐specific plasticity of the slow afterhyperpolarization in rats: Role for high‐voltage activated Ca2+ channels and Ih. European Journal of Neuroscience, 34, 1756–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. H. , & von Gersdorff, H. (2012). Suppression of spikes during posttetanic hyperpolarization in auditory neurons: The role of temperature, Ih currents, and the Na+‐K+‐ATPase pump. Journal of Neurophysiology, 108, 1924–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K. S. , Duignan, K. M. , Hawryluk, J. M. , Soh, H. , & Tzingounis, A. V. (2016). The voltage activation of cortical KCNQ channels depends on global PIP2 levels. Biophysical Journal, 110, 1089–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, K. S. , Kobayashi, M. , Takamatsu, K. , & Tzingounis, A. V. (2012). Hippocalcin and KCNQ channels contribute to the kinetics of the slow afterhyperpolarization. Biophysical Journal, 103, 2446–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, B. , Rizwan, A. P. , Asmara, H. , Heath, N. C. , Engbers, J. D. , Dykstra, S. , … Turner, R. W. (2015). IKCa channels are a critical determinant of the slow AHP in CA1 pyramidal neurons. Cell Reports, 11, 175–182. [DOI] [PubMed] [Google Scholar]
- Koike, H. , Mano, N. , Okada, Y. , & Oshima, T. (1972). Activities of the sodium pump in cat pyramidal tract cell studied with intracellular injection of sodium ions. Experimental Brain Research, 14, 449–462. [DOI] [PubMed] [Google Scholar]
- Kueh, D. , Barnett, W. H. , Cymbalyuk, G. S. , & Calabrese, R. L. (2016). Na+/K+ pump interacts with the h‐current to control bursting activity in central pattern generator neurons of leeches. eLife, 5, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster, B. , & Adams, P. R. (1986). Calcium‐dependent current generating the afterhyperpolarization of hippocampal neurons. Journal of Neurophysiology, 55, 1268–1282. [DOI] [PubMed] [Google Scholar]
- Lancaster, B. , & Nicoll, R. A. (1987). Properties of two calcium‐activated hyperpolarizations in rat hippocampal neurones. Journal of Physiology, 389, 187–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccaferri, G. , Mangoni, M. , Lazzari, A. , & DiFrancesco, D. (1993). Properties of the hyperpolarization‐activated current in rat hippocampal CA1 pyramidal cells. Journal of Neurophysiology, 69, 2129–2136. [DOI] [PubMed] [Google Scholar]
- Madison, D. V. , & Nicoll, R. A. (1984). Control of the repetitive discharge of rat CA1 pyramidal neurones in vitro. Journal of Physiology, 354, 319–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matchkov, V. V. , & Krivoi, I. I. (2016). Specialized functional diversity and interactions of the Na,K‐ATPase. Frontiers in Physiology, 7, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews, E. A. , Linardakis, J. M. , & Disterhoft, J. F. (2009). The fast and slow afterhyperpolarizations are differentially modulated in hippocampal neurons by aging and learning. Journal of Neuroscience, 29, 4750–4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrail, K. M. , Phillips, J. M. , & Sweadner, K. J. (1991). Immunofluorescent localization of three Na,K‐ATPase isozymes in the rat central nervous system: Both neurons and glia can express more than one Na,K‐ATPase. Journal of Neuroscience, 11, 381–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, C. , Moczydlowski, E. , Latorre, R. , & Phillips, M. (1985). Charybdotoxin, a protein inhibitor of single Ca2+‐activated K+ channels from mammalian skeletal muscle. Nature, 13, 316–318. [DOI] [PubMed] [Google Scholar]
- Monteith, G. R. , & Blaustein, M. P. (1998). Different effects of low and high dose cardiotonic steroids on cytosolic calcium in spontaneously active hippocampal neurons and in co‐cultured glia. Brain Research, 795, 325–340. [DOI] [PubMed] [Google Scholar]
- Morita, K. , David, G. , Barrett, J. N. , & Barrett, E. F. (1993). Posttetanic hyperpolarization produced by electrogenic Na+‐K+ pump in lizard axons impaled near their motor terminals. Journal of Neurophysiology, 70, 1874–1884. [DOI] [PubMed] [Google Scholar]
- Moseley, A. E. , Williams, M. T. , Schaefer, T. L. , Bohanan, C. S. , Neumann, J. C. , Behbehani, M. M. , … Lingrel, J. B. (2007). Deficiency in Na,K‐ATPase alpha isoform genes alters spatial learning, motor activity, and anxiety in mice. Journal of Neuroscience, 27, 616–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer, J. R. , Power, J. M. , Thompson, L. T. , & Disterhoft, J. F. (2000). Increased excitability of aged rabbit CA1 neurons after trace eyeblink conditioning. Journal of Neuroscience, 20, 5476–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzer, J. S. , Daly, S. E. , Jewell‐Motz, E. A. , Lingrel, J. B. , & Blostein, R. (1994). Tissue‐ and isoform‐specific kinetic behavior of the Na,K‐ATPase. Journal of Biological Chemistry, 269, 16668–16676. [PubMed] [Google Scholar]
- Nakao, M. , & Gadsby, D. C. (1986). Voltage dependence of Na translocation by the Na/K pump. Nature, 323, 628–630. [DOI] [PubMed] [Google Scholar]
- Parker, D. , Hill, R. , & Grillner, S. (1996). Electrogenic pump and a Ca2+‐ dependent K+ conductance contribute to a posttetanic hyperpolarization in lamprey sensory neurons. Journal of Neurophysiology, 76, 540–553. [DOI] [PubMed] [Google Scholar]
- Pulver, S. R. , & Griffith, L. C. (2010). Spike integration and cellular memory in a rhythmic network from Na+/K+ pump current dynamics. Nature Neuroscience, 13, 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakowski, R. F. , Gadsby, D. C. , & De Weer, P. (1997). Voltage dependence of the Na/K pump. Journal of Membrane Biology, 155, 105–112. [DOI] [PubMed] [Google Scholar]
- Richards, K. S. , Bommert, K. , Szabo, G. , & Miles, R. (2007). Differential expression of Na+/K+‐ATPase alpha‐subunits in mouse hippocampal interneurones and pyramidal cells. Journal of Physiology, 585, 491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzi, S. , Knaus, H. G. , & Schwarzer, C. (2016). Differential distribution of the sodium‐activated potassium channels slick and slack in mouse brain. Journal of Comparative Neurology, 524, 2093–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar, A. , & Rakowski, R. F. (1994). Access channel model for the voltage dependence of the forward‐running Na+/K+ pump. Journal of General Physiology, 103, 869–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaha, F. J. (1967). Studies on Na+‐K+‐stimulated ATPase of human brain. Journal of Neurochemistry, 14, 333–341. [DOI] [PubMed] [Google Scholar]
- Sanchez, M. , & McManus, O. B. (1996). Paxilline inhibition of the alpha‐subunit of the high‐conductance calcium‐activated potassium channel. Neuropharmacology, 35, 963–968. [DOI] [PubMed] [Google Scholar]
- Sánchez‐Alonso, J. L. , Halliwell, J. V. , & Colino, A. (2008). ZD 7288 inhibits T‐type calcium current in rat hippocampal pyramidal cells. Neuroscience Letters, 439, 275–280. [DOI] [PubMed] [Google Scholar]
- Schweigert, B. , Lafaire, A. V. , & Schwarz, W. (1988). Voltage dependence of the Na‐K ATPase: Measurements of ouabain‐dependent membrane current and ouabain binding in oocytes of Xenopus laevis. Pflugers Archives, 412, 579–588. [DOI] [PubMed] [Google Scholar]
- Schwindt, P. C. , Spain, W. J. , Foehring, R. C. , Chubb, M. C. , & Crill, W. E. (1988). Slow conductances in neurons from cat sensorimotor cortex in vitro and their role in slow excitability changes. Journal of Neurophysiology, 59, 450–467. [DOI] [PubMed] [Google Scholar]
- Schwindt, P. C. , Spain, W. J. , & Crill, W. E. (1989). Long‐lasting reduction of excitability by a sodium‐dependent potassium current in cat neocortical neurons. Journal of Neurophysiology, 61, 233–244. [DOI] [PubMed] [Google Scholar]
- Scutt, G. , Allen, M. , Kemenes, G. , & Yeoman, M. (2015). A switch in the mode of the sodium/calcium exchanger underlies an age‐related increase in the slow afterhyperpolarization. Neurobiology Aging, 36, 2838–2849. [DOI] [PubMed] [Google Scholar]
- Segall, L. , Daly, S. E. , & Blostein, R. (2001). Mechanistic basis for kinetic differences between the rat alpha 1, alpha 2, and alpha 3 isoforms of the Na,K‐ATPase. Journal of Biological Chemistry, 276, 31535–31541. [DOI] [PubMed] [Google Scholar]
- Shah, M. , Mistry, M. , Marsh, S. J. , Brown, D. A. , & Delmas, P. (2002). Molecular correlates of the M‐current in cultured rat hippocampal neurons. Journal of Physiology, 544, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, H. , Thompson, S. M. , & Blaustein, M. P. (2013). Nanomolar ouabain augments Ca2+ signaling in rat hippocampal neurones and glia. The Journal of Physiology, 591, 1671–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm, J. F. (1989). An after‐hyperpolarization of medium duration in rat hippocampal pyramidal cells. The Journal of Physiology, 409(1), 171–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweadner, K. J. (1985). Enzymatic properties of separated isozymes of the Na,K‐ATPase. Substrate affinities, kinetic cooperativity, and ion transport stoichiometry. Journal of Biological Chemistry, 260, 11508–11513. [PubMed] [Google Scholar]
- Tamir, I. , Daninos, M. , & Yaari, Y. (2017). Plasticity of intrinsic firing response gain in principal hippocampal neurons following pilocarpine‐induced status epilepticus. Neuroscience, 357, 325–337. [DOI] [PubMed] [Google Scholar]
- Therien, A. G. , & Blostein, R. (2000). Mechanisms of sodium pump regulation. American Journal of Physiology Cell Physiology, 279, C541–C566. [DOI] [PubMed] [Google Scholar]
- Therien, A. G. , Nestor, N. B. , Ball, W. J. , & Blostein, R. (1996). Tissue‐specific versus isoform‐specific differences in cation activation kinetics of the Na,K‐ATPase. Journal of Biological Chemistry, 271, 7104–7112. [DOI] [PubMed] [Google Scholar]
- Thomas, S. A. (2015). Neuromodulatory signaling in hippocampus‐dependent memory retrieval. Hippocampus, 25, 415–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toustrup‐Jensen, M. S. , Einholm, A. P. , Schack, V. R. , Nielsen, H. N. , Holm, R. , Sobrido, M. J. , … Vilsen, B. (2014). Relationship between intracellular Na+ concentration and reduced Na+ affinity in Na+,K+‐ATPase mutants causing neurological disease. Journal of Biological Chemistry, 289, 3186–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, R. W. , Asmara, H. , Engbers, J. D. , Miclat, J. , Rizwan, A. P. , Sahu, G. , & Zamponi, G. W. (2016). Assessing the role of IKCa channels in generating the sAHP of CA1 hippocampal pyramidal cells. Channels (Austin), 10, 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzingounis, A. V. , Heidenreich, M. , Kharkovets, T. , Spitzmaul, G. , Jensen, H. S. , Nicoll, R. A. , & Jentsch, T. J. (2010). The KCNQ5 potassium channel mediates a component of the afterhyperpolarization current in mouse hippocampus. Proceedings of the National Academy of Sciences of the United States of America, 107, 10232–10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzingounis, A. V. , & Nicoll, R. A. (2008). Contribution of KCNQ2 and KCNQ3 to the medium and slow afterhyperpolarization currents. Proceedings of the National Academy of Sciences of the United States of America, 105, 19974–19979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urayama, O. , Shutt, H. , & Sweadner, K. J. (1989). Identification of three isozyme proteins of the catalytic subunit of the Na,K‐ATPase in rat brain. Journal of Biological Chemistry, 264, 8271–8280. [PubMed] [Google Scholar]
- Wallén, P. , Robertson, B. , Cangiano, L. , Löw, P. , Bhattacharjee, A. , Kaczmarek, L. K. , & Grillner, S. (2007). Sodium‐dependent potassium channels of a Slack‐like subtype contribute to the slow afterhyperpolarization in lamprey spinal neurons. Journal of Physiology, 585, 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H. S. , Pan, Z. , Shi, W. , Brown, B. S. , Wymore, R. S. , Cohen, I. S. , … McKinnon, D. (1998). KCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M‐channel. Science, 282, 1890–1893. [DOI] [PubMed] [Google Scholar]
- Wang, K. , Mateos‐Aparicio, P. , Hönigsperger, C. , Raghuram, V. , Wu, W. W. , Ridder, M. C. , … Adelman, J. P. (2016). IK1 channels do not contribute to the slow afterhyperpolarization in pyramidal neurons. Elife, 5, e11206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. C. , & Huang, R. C. (2006). Effects of sodium pump activity on spontaneous firing in neurons of the rat suprachiasmatic nucleus. Journal of Neurophysiology, 96, 109–118. [DOI] [PubMed] [Google Scholar]
- Witter, M. P. , Wouterlood, F. G. , Naber, P. A. , & Van Haeften, T. (2000). Anatomical organization of the parahippocampal‐hippocampal network. Annals of the New York Academy of Sciences, 911, 1–24. [DOI] [PubMed] [Google Scholar]
- Wu, X. , Liao, L. , Liu, X. , Luo, F. , Yang, T. , & Li, C. (2012). Is ZD7288 a selective blocker of hyperpolarization‐activated cyclic nucleotide‐gated channel currents? Channels (Austin), 6, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff, H. , Miller, M. J. , Hansel, W. , Grissmer, S. , Cahalan, M. D. , & Chandy, K. G. (2000). Design of a potent and selective inhibitor of the intermediate‐conductance Ca2+‐activated K+ channel, IKCa1: A potential immunosuppressant. Proceedings of the National Academy of Sciences of the United States of America, 97, 8151–8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue, C. , & Yaari, Y. (2004). KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. Journal of Neuroscience, 24, 4614–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler, R. , Zhang, Z. T. , Manor, M. , & Boron, W. F. (1997). Sodium kinetics of Na,K‐ATPase alpha isoforms in intact transfected HeLa cells. Journal of General Physiology, 110, 201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelcer, I. , Cohen, H. , Richter‐Levin, G. , Lebiosn, T. , Grossberger, T. , & Barkai, E. (2006). A cellular correlate of learning‐induced metaplasticity in the hippocampus. Cerebral Cortex, 16, 460–468. [DOI] [PubMed] [Google Scholar]
- Zhan, H. , Tada, T. , Nakazato, F. , Tanaka, Y. , & Hongo, K. (2004). Spatial learning transiently disturbed by intraventricular administration of ouabain. Neurology Research, 26, 35–40. [DOI] [PubMed] [Google Scholar]
- Zhang, H. Y. , Picton, L. , Li, W. C. , & Sillar, K. T. (2015). Mechanisms underlying the activity‐dependent regulation of locomotor network performance by the Na+ pump. Science Reports, 5, 16188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, L. , Ouyang, M. , Ganellin, C. R. , & Thomas, S. A. (2013). The slow afterhyperpolarization: A target of β1‐adrenergic signaling in hippocampus‐dependent memory retrieval. Journal of Neuroscience, 33, 5006–5016. [DOI] [PMC free article] [PubMed] [Google Scholar]