

Abstract
Obeticholic acid (OCA), a potent farnesoid X receptor agonist, was studied as monotherapy in an international, randomized, double‐blind, placebo‐controlled phase 2 study in patients with primary biliary cholangitis who were then followed for up to 6 years. The goals of the study were to assess the benefit of OCA in the absence of ursodeoxycholic acid, which is relevant for patients who are intolerant of ursodeoxycholic acid and at higher risk of disease progression. Patients were randomized and dosed with placebo (n = 23), OCA 10 mg (n = 20), or OCA 50 mg (n = 16) given as monotherapy once daily for 3 months (1 randomized patient withdrew prior to dosing). The primary endpoint was the percent change in alkaline phosphatase from baseline to the end of the double‐blind phase of the study. Secondary and exploratory endpoints included change from baseline to month 3/early termination in markers of cholestasis, hepatocellular injury, and farnesoid X receptor activation. Efficacy and safety continue to be monitored through an ongoing 6‐year open‐label extension (N = 28). Alkaline phosphatase was reduced in both OCA groups (median% [Q1, Q3], OCA 10 mg −53.9% [−62.5, −29.3], OCA 50 mg −37.2% [−54.8, −24.6]) compared to placebo (−0.8% [−6.4, 8.7]; P < 0.0001) at the end of the study, with similar reductions observed through 6 years of open‐label extension treatment. OCA improved many secondary and exploratory endpoints (including γ‐glutamyl transpeptidase, alanine aminotransferase, conjugated bilirubin, and immunoglobulin M). Pruritus was the most common adverse event; 15% (OCA 10 mg) and 38% (OCA 50 mg) discontinued due to pruritus. Conclusion: OCA monotherapy significantly improved alkaline phosphatase and other biochemical markers predictive of improved long‐term clinical outcomes. Pruritus increased dose‐dependently with OCA treatment. Biochemical improvements were observed through 6 years of open‐label extension treatment. (Hepatology 2018;67:1890‐1902).
Abbreviations
- AE
adverse event
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- C4
7α‐hydroxycholest‐4‐en‐3‐one
- EOS
end of study
- ET
early termination
- FGF‐19
fibrosis growth factor‐19
- FXR
farnesoid X receptor
- GGT
γ‐glutamyl transpeptidase
- HDL‐C
high‐density lipoprotein cholesterol
- hsCRP
high sensitivity C‐reactive protein
- IgM
immunoglobulin M
- OCA
obeticholic acid
- OLE
open‐label extension
- PBC
primary biliary cholangitis
- TNF‐α
tumor necrosis factor‐α
- UDCA
ursodeoxycholic acid
- ULN
upper limit of normal
Primary biliary cholangitis (PBC), formerly known as “primary biliary cirrhosis,” is a chronic liver disease predominantly affecting women (∼10:1) and commonly diagnosed between 40 and 60 years of age.1, 2 While the pathogenesis of PBC is multifactorial and incompletely understood, the pathology, symptoms, and course of the disease are typically associated with cholestasis and increased liver concentrations of bile acids.3, 4 Untreated, PBC may progress to cirrhosis, hepatic decompensation, and liver transplantation and/or death.5, 6 Elevated alkaline phosphatase (ALP) levels and other biochemical parameters associated with liver function and cholestasis have been correlated with clinical outcomes in PBC.7 Until recently, ursodeoxycholic acid (UDCA) was the only approved therapy for PBC and has been recommended as the standard of care.3, 4 However, elevated ALP levels persist in up to 40% of UDCA‐treated patients, and their mortality risk remains higher than that for the general population, demonstrating the need for additional therapies.6, 8 While the majority of patients take UDCA, a minority are unable to tolerate UDCA, primarily due to adverse events (AEs) such as gastrointestinal symptoms.9 Patients who do not receive UDCA treatment have diminished liver transplant‐free survival.7 In addition to the clear unmet need in patients who are intolerant to standard of care, when evaluating new therapies it is critical to understand each new treatment's unique clinical profile to fully gauge its clinical utility and assess whether a new therapy may be adequate alone or serve as a second‐line therapy. As such, obeticholic acid (OCA) was evaluated as a monotherapy in the absence of UDCA in patients with PBC.
OCA is a modified bile acid with a similar structure to chenodeoxycholic acid, the endogenous farnesoid X receptor (FXR) ligand. The addition of an ethyl group on the six‐carbon confers OCA with ∼100‐fold greater potency for FXR relative to chenodeoxycholic acid.10 FXR activation results in repression of cholesterol 7α‐hydroxylase, the enzyme catalyzing the rate‐limiting step of bile acid synthesis,11 and induces production of an ileum‐derived enterokine, fibroblast growth factor‐19 (FGF‐19), which functions as a negative regulator of bile acid synthesis.12 Additionally, FXR activation regulates the expression of key bile acid transporters; it up‐regulates organic solute transporters and the bile salt export pump, which increase bile acid flux from the hepatocytes into bile.13 The sum of these effects are reduced bile acid synthesis and improved choleresis.11, 12, 13 In previous studies, FXR activation through OCA treatment elicited anticholestatic, antifibrotic, and anti‐inflammatory effects.10, 14, 15 In a phase 3 study of OCA in patients with PBC with an inadequate response to UDCA or who could not tolerate UDCA, only 7% of patients received OCA as a monotherapy.15 Given the unmet need in this patient population, further evaluation of OCA as monotherapy is warranted.
The objectives of this phase 2 study were to investigate the safety, tolerability, and efficacy of daily OCA 10 mg and OCA 50 mg administered as a monotherapy compared to placebo. Furthermore, this study evaluated the efficacy, safety, and durability of treatment with OCA in an ongoing open‐label extension (OLE) with up to 6 years of treatment.
Participants and Methods
The study protocol was approved by regulatory authorities and institutional review boards or institutional ethics committees at each site. The study was conducted according to good clinical practice guidelines and the Declaration of Helsinki and is reported in accordance with Consolidated Standards of Reporting Trials guidelines. The double‐blind phase was conducted between November 2007 and September 2010 at 18 centers in six countries. Enrollment was stopped once results were available for the parallel phase 2 study of OCA in combination with UDCA.8 The OLE was not part of the original study design but was initiated in November 2008; as a result, some patients had varying durations between the end of the double‐blind phase and the start of the OLE (mean [SD], 58 [101] days). The OLE remains ongoing to further monitor safety and efficacy. Additional study information is available in the http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo.
STUDY DESIGN
This was an international, double‐blind, placebo‐controlled study of OCA monotherapy in patients with PBC. Doses investigated in this study were dose‐finding and different from those later investigated in phase 3 studies of OCA. At the time of this study, only phase 1 data of OCA were available, testing doses of 25‐500 mg in healthy volunteers. No substantial AEs were observed at 50 mg, which was therefore considered an appropriate potential dose. Patients meeting enrollment criteria were randomized 1:1:1 to OCA 10 mg, OCA 50 mg, or placebo, administered once daily for 3 months. The computerized randomization schedule used a block size of 3 at each center. Patients were assessed at week 2, month 1, month 2, and month 3 (end of the double‐blind phase). All patients had a follow‐up visit 2 weeks after study medication discontinuation. Patients who discontinued the study early (and thereby stopped usage of OCA) had an early termination (ET) visit. The primary endpoint of the study was the median percent change in ALP from baseline to the end of study (EOS). After completion of the double‐blind phase, patients could enroll in an OLE.
ELIGIBILITY CRITERIA
Patients were 18‐75 years old and either UDCA‐naive or not taking UDCA for ≥3 months prior to screening (http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo). Key inclusion criteria included PBC diagnosis by the American Association for the Study of Liver Diseases/European Association for the Study of the Liver guidelines3, 4 and serum ALP 1.5‐10 times the upper limit of normal range (ULN; normal ALP, ∼120 U/L) at screening. Key exclusion criteria were elevated plasma aspartate aminotransferase (AST) or alanine aminotransferase (ALT) levels >5 times the ULN; conjugated bilirubin >2 times the ULN; serum creatinine >1.5 mg/dL (133 μmol/L); the use of colchicine, methotrexate, azathioprine, or systemic corticosteroids during the 3 months prior to screening; history or presence of hepatic decompensation; and concomitant liver diseases.
EFFICACY ANALYSES
The primary endpoint was the percent change in ALP from baseline to EOS (month 3 or ET in the case of premature discontinuations or the last observed ALP level for patients who did not complete the study). The last observation carried forward method was used for missing data for the primary endpoint only. For secondary and exploratory endpoints, month 3 values included ET data in the cases of discontinuations when ET data were available. Secondary endpoints included change in ALP, γ‐glutamyl transpeptidase (GGT), ALT, AST, conjugated bilirubin, and achieving previously developed response criteria in PBC.6 Exploratory endpoints included change from baseline in high sensitivity C‐reactive protein (hsCRP), tumor necrosis factor‐α (TNF‐α), transforming growth factor‐β, immunoglobulin M (IgM), glutathione, osteopontin, albumin, nonesterified fatty acids, FGF‐19, enhanced liver fibrosis score, PBC‐40 questionnaire, Short Form 36 survey, and achieving different thresholds of ALP decrease. Additional post hoc analyses included change in 7α‐hydroxycholest‐4‐en‐3‐one (C4), a bile acid precursor used to assess bile acid synthesis, and estimated future survival based on the UK‐PBC and Global PBC prognostic models.16, 17 Safety assessments included AEs as defined by the Medical Dictionary for Regulatory Activities, physical examinations, vital signs, pruritus visual analogue scale score, Pruritus 5D questionnaire, and electrocardiograms. Additional methods can be found in the http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo.
OPEN‐LABEL EXTENSION
Patients completing the double‐blind phase were eligible for the OLE. Placebo‐treated patients or patients who had been off therapy for ≥1 month after the double‐blind phase were started on OCA 10 mg. OCA‐treated patients completing the double‐blind phase resumed their double‐blind dose during the OLE phase. During the OLE, investigators could titrate doses up to 75 mg based on therapeutic response. The OCA dose could be reduced during the OLE based on the physician's discretion. UDCA could be initiated during the OLE phase at the investigator's discretion. The primary objectives of the OLE were to assess the long‐term safety and efficacy of OCA.
SAMPLE SIZE
The planned sample size was ∼40 patients/treatment group. Absent an a priori estimate of treatment effect and assuming a type I error of 5%, a cohort size of 40 patients/treatment arm was calculated to provide 80% power to detect an effect size of 0.6466 or 70% power to detect an effect size of 0.5770. The rarity of PBC, coupled with the inclusion criterion that patients not be taking UDCA, resulted in a final sample size of ∼20 patients/group, resulting in 49% power to detect an effect size of 0.6466 for the difference in the primary efficacy endpoint between treatment arms and placebo using a Wilcoxon‐Mann‐Whitney rank‐sum test with a 0.05 two‐sided significance level.
STATISTICAL ANALYSES
The primary efficacy endpoint was analyzed using a two‐sided Wilcoxon‐Mann‐Whitney test to perform pairwise comparisons of changes in each OCA group with the placebo group. A hierarchical testing strategy18 accounted for multiple comparisons: OCA 10 mg versus placebo was tested first at α = 0.05 and once significance was shown; then OCA 50 mg versus placebo was tested at α = 0.05.
Secondary, exploratory, and safety endpoints were summarized by treatment group using descriptive statistics. All data refer to median (Q1, Q3) values unless otherwise specified. For continuous endpoints, exploratory endpoints, safety endpoints (pruritus visual analogue scale score/5D questionnaire), and post hoc analysis of C4, pairwise comparisons of placebo versus each OCA group used the two‐sided Wilcoxon‐Mann‐Whitney test. For categorical and exploratory variables, each OCA group was compared to placebo using a Fisher's exact test. All tests were evaluated at α = 0.05 with no adjustments for multiplicity.
Post hoc comparisons between each OCA group and placebo for the UK‐PBC and Global PBC prognostic models were obtained using a rank analysis of covariance model with baseline value as a covariate.
Analyses were performed using SAS version 9.1.3 or newer. All graphs and figures were produced in GraphPad Prism version 6.0.
Results
STUDY PATIENTS
Sixty patients were randomized into the study; 1 withdrew prior to dosing, yielding a study population of 59 patients. Patient disposition, demographics, and baseline characteristics are summarized in Table 1 and in http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo and http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo. Demographics and baseline clinical characteristics were generally similar between treatment groups. The patients were 85% female, 95% white, and 54 (48, 61) years of age. ALP and GGT were elevated at baseline in all groups (Table 1). While the reason for not receiving UDCA was not captured in this study, the median PBC disease duration ranged 1.0‐2.1 years (Table 1), suggesting that many patients may have been UDCA‐naive. While baseline liver enzymes were elevated in most patients, the short disease duration and the moderate estimated 10‐year survival suggest that most patients were early in the PBC stage. Forty‐eight patients (81%) completed the 3‐month double‐blind phase of the study (placebo, 23 patients [100%]; OCA 10 mg, 16 patients [80%]; OCA 50 mg, 9 patients [56%]). Reasons for study discontinuation were pruritus (OCA 10 mg, 3 patients [15%]; OCA 50 mg, 6 patients [38%]), consent withdrawal (OCA 10 mg, 1 patient [5%]), and major protocol violation (OCA 50 mg, 1 patient [6%]). One patient (6%) in the OCA 50 mg group had been receiving 200 mg fenofibrate orally for fatigue while in the study but discontinued due to pruritus.
Table 1.
Demographic and Baseline Characteristics of PBC Patients
|
Placebo (n = 23) |
OCA 10 mg (n = 20) |
OCA 50 mg (n = 16) |
|
|---|---|---|---|
| Female, n (%) | 20 (87) | 14 (70) | 16 (100) |
| White, n (%) | 21 (91) | 19 (95) | 16 (100) |
| Age (years) | 54 (48, 63) | 54 (47, 62) | 54 (49, 61) |
| Body weight (kg) | 78.1 (63.0, 99.9) | 77.7 (67.1, 82.5) | 59.5 (54.0, 73.5) |
| Body mass index (kg/m2) | 28.9 (23.3, 33.3) | 25.3 (24.7, 29.0) | 22.7 (20.8, 28.0) |
| Estimated 10‐year survival (Global‐PBC)a | 90.5 (85.2, 93.7) | 82.6 (74.5, 92.9) | 91.0 (85.7, 94.6) |
| Estimated 10‐year survival (UK‐PBC)b | 98.6 (97.4, 99.0) | 97.6 (92.6, 98.3) | 98.4 (96.5, 98.8) |
| PBC duration (years)c | 1.0 (0.2, 6.3) | 1.1 (0.2, 7.4) | 2.1 (0.2, 9.3) |
| Albumin (g/L) | 40.0 (37.0, 42.0) | 38.9 (34.5, 43.2) | 40.5 (38.1, 43.5) |
| Platelets (109/L) | 314 (213, 386) | 260 (226, 313) | 269 (217, 329) |
| Total cholesterol (mg/dL) | 226.5 (178.6, 259.1) | 229.3 (201.1, 282.8) | 222.0 (210.4, 242.5) |
| IgM (g/L) | 3.0 (2.7, 4.3) | 2.7 (1.9, 3.6) | 3.8 (2.7, 6.3) |
| ALP (U/L) | 321 (234, 586) | 366 (236, 623) | 379 (306, 590) |
| GGT (U/L) | 359 (242, 625) | 550 (412, 962) | 344 (258, 449) |
| AST (U/L) | 64 (42, 99) | 54 (41, 91) | 58 (52,64) |
| ALT (U/L) | 63 (50, 97) | 71 (57, 124) | 64 (45, 79) |
| Conjugated bilirubin (mg/dL) | 0.18 (0.11, 0.30) | 0.22 (0.17, 0.40) | 0.20 (0.18, 0.26) |
| Total bilirubin (mg/dL) | 0.55 (0.38, 0.80) | 0.65 (0.49, 0.95) | 0.51 (0.39, 0.82) |
Data are median (Q1, Q3) unless otherwise specified. Normal ranges: albumin, 31‐52 g/L; platelets, 121‐450 109/L; total cholesterol, 69.5‐231.7 mg/dL; IgM, 0.4‐2.3 g/L; ALP, ≤117 U/L (female), ≤129 U/L (male); GGT, ≤50 U/L (female), ≤73 U/L (male); AST, ≤50 U/L; ALT, ≤67 U/L; conjugated bilirubin: ≤0.41 mg/dL; total bilirubin: ≤1.40 mg/dL.
Baseline 10‐year survival estimated by the Global‐PBC score.
Baseline 10‐year survival estimated by the UK‐PBC score.
PBC duration is based on the day of diagnosis from individual medical histories.
PRIMARY ENDPOINT
Both doses of OCA were superior to placebo in achieving the primary endpoint at EOS (Fig. 1A). ALP was significantly reduced in the OCA 10 mg and OCA 50 mg groups by 53.9% (−62.5, −29.3) and 37.2% (−54.8, −24.6), respectively, compared to 0.8% (−6.4, 8.7) in the placebo group (P < 0.0001).
Figure 1.
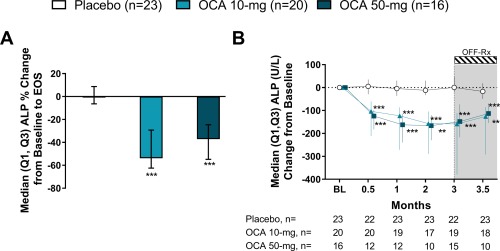
OCA reduces ALP. (A) Percent change in ALP at EOS. (B) ALP change from baseline. Data are median (Q1, Q3). ** P < 0.01, *** P < 0.0001; P values determined by pairwise comparisons using a two‐sided Wilcoxon‐Mann‐Whitney test comparing changes in each OCA group with the placebo group. Note: the primary endpoint includes ET values or last observation carried forward values. ALP change from baseline values at month 3 include the ET visit data for patients who discontinued where available. Abbreviation: BL, baseline.
SECONDARY ENDPOINTS
ALP (units per liter) was significantly reduced from baseline in both OCA groups compared to placebo (P ≤ 0.0001) throughout double‐blind treatment (Fig. 1B; http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo). At month 3/ET, the change from baseline in ALP in the OCA 10 mg and OCA 50 mg groups was −159 (−379, −112) and −148 (−217, −74), respectively, compared with 1 (−18, 49) in the placebo group (P < 0.0001). At the follow‐up visit (2 weeks after terminating treatment), ALP levels trended toward baseline values; however, they remained reduced in the OCA 10 mg (P < 0.0001) and OCA 50 mg (P = 0.0092) groups compared to placebo.
With OCA treatment, GGT (units per liter) decreased significantly from baseline (P < 0.0001) throughout the double‐blind phase (Fig. 2A; http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo). AST (units per liter) levels in the OCA 10 mg group and the OCA 50 mg group also decreased from baseline to month 3/ET (Fig. 2B; http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo). Changes in AST at month 3/ET were not different between treatment groups; however, baseline values were only modestly elevated from normal (Table 1), and levels in the OCA 10 mg group approached normal range (AST ≤50 U/L) by week 2 and fell within normal range for the remainder of the study. Significant reductions in ALT (units per liter) from baseline to month 3/ET were observed in both OCA groups compared to placebo (OCA 10 mg, P = 0.0150; OCA 50 mg, P = 0.0476) (Fig. 2C; http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo). Conjugated bilirubin (milligrams per deciliter) was decreased from baseline in both OCA 10 mg (P = 0.0177) and OCA 50 mg (P = 0.0266) groups compared to an increase at month 3/ET in the placebo group (Fig. 2D; http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo). At the follow‐up visit, GGT, AST, ALT, and conjugated bilirubin levels trended toward baseline values following withdrawal of OCA.
Figure 2.
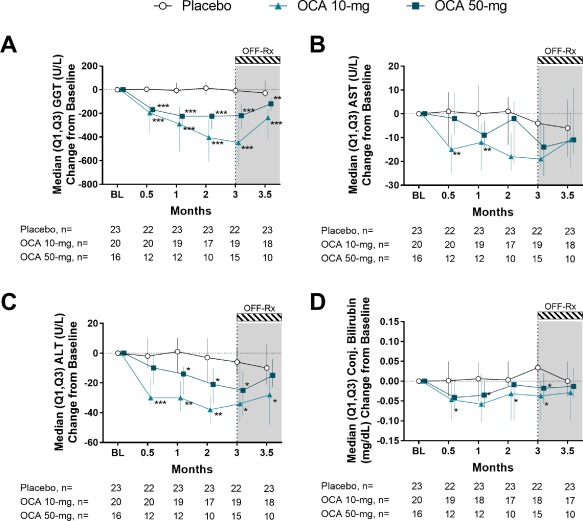
Effect of OCA on liver biochemistries. (A) GGT change from baseline. (B) AST change from baseline. (C) ALT change from baseline. (D) Conjugated bilirubin change from baseline. Data are median (Q1, Q3). Change from baseline values at month 3 include the ET visit data for patients who discontinued. * P < 0.05, ** P < 0.01, *** P < 0.0001; P values determined by pairwise comparisons using a two‐sided Wilcoxon‐Mann‐Whitney test comparing changes in each OCA group with the placebo group. Abbreviation: BL, baseline.
Various biochemical disease severity criteria have been derived, and achieving these criteria was prognostic of improved outcomes in PBC.5 A significantly greater percentage of OCA‐treated patients met these biochemical disease severity criteria compared to placebo at month 3/ET with the exception of 1 (ALP ≤3 times ULN, AST ≤2 times ULN, and total bilirubin ≤1 mg/dL, Paris I criterion) (http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo). The Barcelona criterion (ALP >40% decrease or normal ALP) was the most discriminating; no patients in the placebo group met this criterion at month 3/ET compared with 70% in OCA 10 mg (P < 0.0001) and 44% in OCA 50 mg (P = 0.0006) groups.
EXPLORATORY ENDPOINTS
The ALP levels in both OCA groups decreased substantially by month 3/ET, with 50% of patients in each of the OCA groups (P < 0.01) achieving ALP <1.67 times ULN (http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo). In the OCA 10 mg group 25% of patients (P = 0.0155) achieved normal ALP, although no patients in the OCA 50 mg or placebo group achieved normal ALP.
Dose‐dependent increases in FGF‐19 (nanograms per liter) were observed, confirming FXR activation. The change from baseline to month 3/ET was 176.5 (39.0, 8898.7) in the OCA 50 mg group (P = 0.0154) but not significant in the OCA 10 mg group (106.4 [−0.8, 132.2], P = 0.2857) compared to placebo (10.7 [−10.3, 81.7]) (Fig. 3A). Baseline IgM levels (grams per liter) were above ULN in all treatment groups and significantly reduced at month 3/ET in both OCA treatment groups (OCA 10 mg, −0.6 [−0.9, −0.1], P = 0.0205; OCA 50 mg, −1.0 [−1.9, −0.6], P = 0.0033) versus placebo (0.0 [−0.3, 0.2]) (Table 2). Changes from baseline to month 3/ET in TNF‐α and osteopontin were significant only in the OCA 50 mg group, with a decrease in TNF‐α (P = 0.0061) and an increase in osteopontin (P = 0.0348). Glutathione increased from baseline to month 3/ET in the OCA 10 mg group (P = 0.0151) compared to placebo, with no significant change observed in the OCA 50 mg group. There were no differences between treatment groups from baseline to month 3/ET in change in hsCRP, transforming growth factor‐β, or enhanced liver fibrosis score (and all enhanced liver fibrosis components) (Table 2). There were no changes from baseline to month 3/ET in albumin, platelets, international normalized ratio, or nonesterified fatty acids with OCA treatment; all remained in the normal range. Baseline serum bile acid levels (micromoles per liter) were in the normal range; however, OCA treatment resulted in reductions from baseline (OCA 10 mg, −1.5 [−2.6, 3.6], P = 0.5203; OCA 50 mg, −0.5 [−8.6, 0.4], P = 0.5940) but not significantly compared to placebo (0.9 [−1.1, 7.5]).
Figure 3.
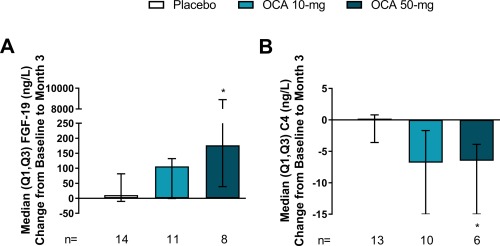
Markers of FXR activation. (A) FGF‐19 change from baseline to month 3/ET. Baseline FGF‐19 was 77.5 (27.6, 85.5), 109.7 (86.3, 228.3), and 74.7 (48.8, 144.3) for placebo, OCA 10 mg, and OCA 50 mg groups, respectively. (B) C4 change from baseline to month 3/ET. Baseline C4 was 18.2 (8.9, 22.3), 8.6 (4.3, 19.0), and 9.1 (6.5, 27.4) for placebo, OCA 10 mg and OCA 50 mg groups, respectively. Data are median (Q1, Q3). Change from baseline values at month 3 include the ET visit data for patients who discontinued. * P < 0.05; P values determined by pairwise comparisons using a two‐sided Wilcoxon‐Mann‐Whitney test comparing changes in each OCA group with the placebo group.
Table 2.
Exploratory Biomarkers
| Placebo | OCA 10 mg | OCA 50 mg | ||||||
|---|---|---|---|---|---|---|---|---|
| Baseline | Change From Baseline | Baseline | Change From Baseline | P | Baseline | Change From Baseline | P | |
| Inflammatory and immunomodulatory biomarkers | ||||||||
| hsCRP (mg/L) |
11.2 (5.6, 16.1) n = 21 |
0.0 (−5.7, 2.1) n = 19 |
5.6 (4.0, 7.8) n = 17 |
−3.0 (−5.3, −1.5) n = 15 |
0.1105 |
5.0 (3.6, 9.6) n = 13 |
−1.4 (−4.5, 0.3) n = 11 |
0.3016 |
| TNF‐α (ng/L) |
15.1 (12.6,17.4) n = 18 |
0.4 (−1.0, 3.6) n = 15 |
14.9 (12.0, 19.4) n = 14 |
−1.1 (−3.3, 0.0) n = 13 |
0.1119 |
16.1 (11.7, 21.0) n = 12 |
−2.2 (−3.7, −0.6) n = 9 |
0.0061 |
| TGF−β (μg/L) |
19.0 (16.2, 28.4) n = 10 |
1.0 (0.3, 1.4) n = 10 |
24.0 (19.2, 26.8) n = 8 |
−4.1 (−6.5, 7.0) n = 7 |
0.7327 |
21.5 (19.3, 28.1) n = 6 |
1.9 (0.1, 4.7) n = 6 |
0.7042 |
| IgM (g/L) |
3.0 (2.7, 4.3) n = 17 |
0.0 (−0.3, 0.2) n = 13 |
2.7 (1.9, 3.6) n = 14 |
−0.6 (−0.9, −0.1) n = 11 |
0.0205 |
3.8 (2.7, 6.3) n = 11 |
−1.0 (−1.9, −0.6) n = 9 |
0.0033 |
|
Osteopontin (μg/L) |
229 (169, 324) n = 8 |
60 (−19, 91) n = 7 |
320 (166, 357) n = 7 |
15 (−63, 125) n = 7 |
0.8983 |
217 (180, 264) n = 7 |
181 (134, 252) n = 5 |
0.0348 |
|
Glutathione (μmol/L) |
2.3 (2.0, 4.7) n = 9 |
0.2 (−0.5, 0.7) n = 7 |
2.3 (1.2, 3.1) n = 7 |
1.9 (0.4, 3.0) n = 7 |
0.0151 |
4.2 (1.3, 5.9) n = 7 |
0.2 (0.1, 2.1) n = 5 |
0.6261 |
| Noninvasive measures of liver fibrosis | ||||||||
| ELF score |
9.2 (8.6, 9.8) n = 22 |
−0.1 (−0.2, 0.4) n = 21 |
9.3 (8.2, 9.7) n = 20 |
0.2 (−0.1, 0.5) n = 19 |
0.2334 |
9.2 (8.9, 9.3) n = 14 |
0.0 (−0.4, 0.3) n = 14 |
0.6738 |
| HA (ng/mL) |
49.6 (24.2, 78.0) n = 22 |
0.2 (−11.5, 12.9) n = 21 |
45.8 (24.9, 71.9) n = 20 |
10.2 (−3.2, 30.4) n = 19 |
0.1436 |
40.8 (27.5, 77.2) n = 14 |
1.8 (−12.6, 10.5) n = 14 |
0.8796 |
|
P3NP (ng/mL) |
6.5 (5.8, 11.4) n = 22 |
0.3 (−0.8, 1.3) n = 21 |
7.5 (5.0, 9.0) n = 20 |
−0.2 (−1.2, 2.76) n = 19 |
0.6648 |
8.2 (4.8, 10.7) n = 14 |
−0.4 (−1.2, 1.3) n = 14 |
0.8531 |
|
TIMP‐1 (ng/mL) |
767 (601, 906) n = 22 |
−33 (−133, 73) n = 21 |
799 (661, 897) n = 20 |
−80 (−197, −34) n = 19 |
0.2229 |
785 (689, 854) n = 14 |
−69 (−121, −21) n = 14 |
0.7491 |
| Additional measures of liver function | ||||||||
| Albumin (g/L) |
40.0 (37.0, 42.0) n = 23 |
−1.3 (3.0, 0.0) n = 22 |
38.9 (34.5, 43.2) n = 20 |
−0.6 (−2.1, 1.5) n = 19 |
0.4404 |
40.5 (38.1, 43.5) n = 16 |
−1.0 (−2.8, 1.5) n = 15 |
0.6205 |
| Platelets (109/L) |
314 (213, 386) n = 23 |
−6 (−34, 21) n = 23 |
260 (226, 313) n = 20 |
−6 (−34, 22) n = 19 |
0.9798 |
269 (217, 329) n = 16 |
2 (−10, 35) n = 14 |
0.4154 |
| Serum bile acids (μmol/L) |
8.1 (6.9, 12.9) n = 9 |
0.9 (−1.1, 7.5) n = 6 |
9.4 (7.2, 11.4) n = 7 |
−1.5 (−2.6, 3.6) n = 7 |
0.5203 |
6.0 (5.8, 23.9) n = 7 |
−0.5 (−8.6, 0.4) n = 4 |
0.5940 |
| INR |
1.00 (0.98, 1.07) n = 23 |
−0.02 (−0.05, 0.07) n = 23 |
1.01 (0.98, 1.05) n = 20 |
−0.04 (−0.07, 0.01) n = 19 |
0.1606 |
0.99 (0.96, 1.05) n = 16 |
0.00 (−0.11, 0.07) n = 15 |
0.6977 |
|
NEFA (mmol/L) |
0.5 (0.3, 0.6) n = 9 |
−0.1 (−0.3, 0.1) n = 6 |
0.5 (0.3, 0.9) n = 7 |
0.1 (−0.7, 0.3) n = 7 |
0.7206 |
0.4 (0.1, 0.6) n = 7 |
0.1 (−0.1, 0.5) n = 4 |
0.3359 |
Data are median (Q1, Q3); P values are for median change from baseline to month 3 based on a Wilcoxon‐Mann‐Whitney comparison. Change from baseline values at month 3 include the ET visit data for patients who discontinued. Normal ranges: hsCRP, ≤3.0 mg/L; TNF‐α, ≤8.1 ng/L; TGF‐β, 21.0‐82.0 μg/L; IgM, 0.4‐2.3 g/L; osteopontin, 171‐667 μg/L; glutathione, 2.4‐4.4 μmol/L; albumin, 31.0‐52.0 g/L; platelets, 121‐450 109/L; bile acids, ≤10 μmol/L; INR, 0.76‐1.27; NEFA, 0.1‐0.45 mmol/L (female), 0.1‐0.60 mmol/L (male).
Abbreviations: ELF, enhanced liver fibrosis; HA, hemagglutinin; INR, international normalized ratio; NEFA, nonesterified fatty acids; P3NP, procollagen type 3 N‐terminal peptide; TGF‐β, transforming growth factor‐β; TIMP‐1, tissue inhibitor of metalloproteinase 1.
Assessment of quality of life using the Short Form 36 survey showed no changes between treatment groups from baseline to month 3/ET in any of the Short Form 36 domains (http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo). With the exception of increased itch (OCA 50 mg, P = 0.0172) and reduced fatigue (OCA 10 mg, P = 0.0451), no significant changes were observed in any of the remaining PBC‐40 domains (http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo).
POST HOC ASSESSMENTS
FXR activation was further assessed post hoc by analyzing the concentration of C4 (nanograms per milliliter). No change was observed between baseline and month 3/ET in placebo (0.2 [–3.6, 0.8]), but there was a decrease of −6.5 (−15.8, −3.9) observed in the OCA 50 mg group (P = 0.0485; Fig. 3B). A similar reduction was observed in the OCA 10 mg group (−6.8 [−15.5, −1.7]), but this was not significant (P = 0.0508).
The Global‐PBC and UK‐PBC prognostic models were used to estimate long‐term survival before and after OCA treatment.16, 17 http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo shows the estimated survival at 5, 10, and 15 years in all treatment groups prior to receiving treatment. The baseline survival estimated by both prognostic models at 5, 10, and 15 years appears worse for the OCA 10 mg group compared to the placebo and OCA 50 mg groups. This may be due to lower platelets and elevated total bilirubin and ALT in the OCA 10 mg group at baseline, which are components of the models. After 3 months of treatment, both models predict worsening survival in patients receiving placebo and improved survival in both OCA groups (P < 0.01) (http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo).
SAFETY ASSESSMENTS
While the incidence of AEs was similar across all treatment groups, pruritus was the most common AE in all treatment groups, particularly in the OCA groups (placebo, 35%; OCA 10 mg, 70%; and OCA 50 mg, 94%) (Table 3). The incidence and severity of pruritus were dose‐dependent with OCA; 15% of patients in the OCA 10 mg and 38% of patients in the OCA 50 mg discontinued due to pruritus (http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo). At month 3/ET, there was a significant increase from baseline in the pruritus visual analogue scale score in both OCA groups compared to placebo (P < 0.05) (http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo). No differences between treatment groups were observed at month 3/ET in the total score of the 5D pruritus questionnaire (http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo). The median time to onset of pruritus was 33, 14, and 6 days in the placebo, OCA 10 mg, and OCA 50 mg groups, respectively. In the placebo, OCA 10 mg, and OCA 50 mg groups 6/6, 7/7, and 3/4 patients, respectively, experienced AEs of pruritus but received no intervention and completed the double‐blind phase of the study. Interventions for pruritus were successful in 3/3, 6/9, and 7/13 patients in the placebo, OCA 10 mg, and OCA 50 mg groups, respectively (http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo). Medications were the most frequent intervention used for pruritus, specifically the bile acid sequestrant cholestyramine (http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo). Dose frequency adjustments were typically a reduction to dosing on alternate days.
Table 3.
Incidence of Treatment‐Emergent AEs Occurring in >10% Patients in Any OCA Groupa
| Treatment Group | |||
|---|---|---|---|
|
Placebo (n = 23) |
OCA 10 mg (n = 20) |
OCA 50 mg (n = 16) |
|
| Patients (%) | Patients (%) | Patients (%) | |
| Patients with any AEs | 21 (91) | 18 (90) | 15 (94) |
| Pruritus | 8 (35) | 14 (70) | 15 (94) |
| Abdominal distension | 0 (0) | 0 (0) | 2 (13) |
| Abdominal pain | 1 (4) | 1 (5) | 2 (13) |
| Constipation | 0 (0) | 0 (0) | 2 (13) |
| Diarrhea | 1 (4) | 0 (0) | 2 (13) |
| Feces pale | 0 (0) | 0 (0) | 2 (13) |
| Hemorrhoids | 0 (0) | 0 (0) | 2 (13) |
| Nausea | 4 (17) | 0 (0) | 4 (25) |
| Nasopharyngitis | 2 (9) | 3 (15) | 1 (6) |
| Urinary tract infection | 0 (0) | 3 (15) | 1 (6) |
| Headache | 5 (22) | 4 (20) | 2 (13) |
| Insomnia | 1 (4) | 1 (5) | 2 (13) |
Any patient who received at least one dose of any treatment.
Other than pruritus, there were no trends in either OCA group with respect to AEs leading to study discontinuation. A single serious AE (rash) was reported in the double‐blind phase by a placebo‐treated patient. Results of QT interval corrected by the Fridericia formula were comparable across all treatment groups at baseline and did not change after treatment (http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo). Baseline cholesterol, high‐density lipoprotein cholesterol (HDL‐C), low‐density lipoprotein cholesterol, and triglyceride levels were comparable across treatment groups (http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo). At month 3/ET, OCA treatment resulted in decreases from baseline in total cholesterol (milligrams per deciliter) (placebo, −2.5 [–16.0, 8.7]; OCA 10 mg, −2.9 [−55.6, 7.1]; OCA 50 mg, –16.2 [−49.0, 4.6]) and HDL‐C (milligrams per deciliter) (placebo, −3.1 [−8.5, 2.9]; OCA 10 mg, −8.5 [−27.2, 0.4]; OCA 50 mg, –13.5 [−26.8, −2.5]) and increases in low‐density lipoprotein cholesterol (milligrams per deciliter) (placebo, −3.7 [−9.1, 5.6]; OCA 10 mg, 11.6 [−4.8, 14.5]; OCA 50 mg, 6.8 [−3.5, 29.3]). There were no dose‐dependent changes in triglycerides at month 3/ET (placebo, −4.4 [−14.2, 17.3]; OCA 10 mg, −3.1 [–20.4, 26.6]; OCA 50 mg, −0.4 [–45.1, 9.3]).
OPEN‐LABEL EXTENSION
Twenty‐eight (47%) patients enrolled in the OLE phase (18 patients completed through 6 years). The median (Q1, Q3) treatment duration was 6.4 (2.5, 6.7) years, and the median weighted average daily dose was 14.0 mg (10.0, 22.5). After the end of double‐blind treatment, 12 patients (43%) initiated UDCA based on the investigator's discretion. Changes in ALP, GGT, AST, ALT, and conjugated bilirubin were sustained for up to 6 years in patients who continued OCA treatment (Fig. 4). Pruritus was the most common AE reported during the OLE, experienced by 89% of patients (http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo). Twenty patients (71%) received concomitant medications for pruritus including antihistamines, bile acid sequestrants, and antibiotics (typically rifampicin). A total of 12 patients (43%) discontinued due to several causes: pruritus (3 patients, 11%), other AEs (4 patients, 14%: cirrhosis, arthralgia, colitis, lab abnormality), consent withdrawal (1 patient, 4%), or other reasons (4 patients, 14%). The safety profile in the OLE was consistent with the double‐blind phase.
Figure 4.
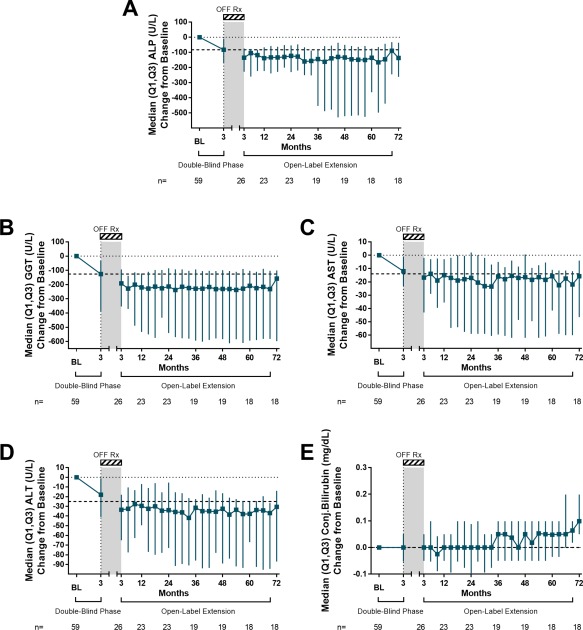
Changes in liver biochemistry over 6 years of OLE. (A) ALP change from baseline. (B) GGT change from baseline. (C) AST change from baseline. (D) ALT change from baseline. (E) Conjugated bilirubin change from baseline. For all plots, values presented are median (Q1, Q3) of all patients participating in the trial at any point regardless of dose or group. Dashed line represents the median change from baseline at the end of the double‐blind phase. Off‐Rx refers to the study period from month 3 and initiating OCA during the OLE. Abbreviation: BL, baseline.
Discussion
It is important to understand the safety and efficacy of OCA as a potential monotherapy, which has a distinct mechanism of action from UDCA. This double‐blind, placebo‐controlled study evaluated the efficacy and safety of OCA monotherapy in patients with PBC. The primary endpoint, percent change in ALP, was achieved with significant reductions following OCA treatment. OCA treatment improved markers of cholestasis, hepatocellular damage, and liver function, which were similarly observed in OCA‐treated patients through 6 years of open‐label treatment, suggesting a durable response without additional safety findings. OCA also increased circulating FGF‐19 and decreased C4 levels, demonstrating FXR activation. The efficacy of OCA 10 mg was comparable to OCA 50 mg, but OCA 10 mg was associated with improved tolerability and fewer discontinuations.
Evaluation of new compounds administered as monotherapy is the optimal way to define the properties of a drug in a disease regardless of whether subsequent use is in combination with other agents. Previous studies of OCA in combination with UDCA in patients with PBC have shown significant biochemical improvements.8, 15 However, this study confirms that the biochemical improvements observed with OCA occur in the absence of UDCA, further underlining the role of FXR in the regulation of bile acids and cholestatic disease.
In this study, baseline ALP was markedly elevated to ∼3.2 times ULN in the absence of UDCA. In a parallel study evaluating OCA in combination with UDCA, baseline ALP was elevated to ∼2.3 times ULN.8 Both studies used the same entry criterion for ALP, assessed OCA 10 mg and 50 mg doses, and were of identical duration. In both studies, at the end of the 3‐month double‐blind phase, ALP was ∼1.7 times ULN in OCA‐treated patients, suggesting that OCA can achieve clinically meaningful reductions in ALP in patients who either are intolerant/naive to UDCA or have an inadequate response to UDCA. Interestingly, ALP reductions observed in this OCA monotherapy study are similar to reductions reported in previous studies of UDCA (∼40%‐60%).19
UDCA has been associated with improvements in markers of cholestasis as well as various markers of hepatic damage.20, 21, 22, 23 Importantly, the mechanism by which UDCA elicits these effects is independent of FXR activation as UDCA has no affinity for FXR.24 In contrast, OCA is a selective FXR agonist, eliciting its effects through transcriptional regulation of bile acid homeostasis demonstrated by increases in FGF‐19 with concomitant reductions of bile acid synthesis.11, 12, 13 In this study, FGF‐19 increased dose‐dependently, but the reduction in C4 was similar between both OCA doses, suggesting that the effect on cholesterol 7α‐hydroxylase was saturated, resulting in nearly equal C4 reductions. In this study, reductions in serum bile acids were not significant with OCA treatment due to normal baseline levels and low patient numbers; in larger studies, OCA has been associated with reductions in serum bile acids.8, 15 Mechanistically, OCA‐driven inhibition of endogenous bile acid synthesis and choleretic properties can be effective as monotherapy and can complement UDCA therapy.8, 10
In addition to FXR‐mediated effects on bile acid homeostasis, OCA treatment has been associated with immunomodulatory and anti‐inflammatory effects.15 PBC is characterized by loss of tolerance to mitochondrial self‐antigens and frequent autoimmune comorbidities. Genome‐wide studies have shown significant associations between PBC and a range of genetic markers.25, 26 Furthermore, presence of antimitochondrial antibodies is diagnostic for PBC, and elevated IgM levels are commonly observed in patients with PBC.3, 4, 27 OCA monotherapy improved markers of immunity and inflammation including IgM and TNF‐α. In this study transforming growth factor‐β and hsCRP were not different between the placebo and OCA groups; however, in larger studies, OCA has been associated with improvements in hsCRP.8, 15
PBC is a chronic liver disease, making early prediction of long‐term prognosis critical for risk stratification and disease management. ALP28 and bilirubin,7 along with aminotransferases, have been used in previous response criteria and have served as endpoints for clinical trials.6 OCA monotherapy resulted in a greater percentage of patients achieving most published treatment response criteria compared to placebo. Furthermore, OCA monotherapy significantly increased predicted survival compared to placebo as assessed by the UK‐PBC and Global‐PBC prognostic models, which have shown improved accuracy in predicting clinical outcomes in PBC versus dichotomous response criteria.16, 17 Both prognostic models were evaluated post hoc as they were developed after this study was initiated. The improved predicted survival using both prognostic models reflects improvements in ALP together with other variables within the models. The utility of survival predictions in this study was limited by the short OCA treatment period and small patient population. Furthermore, these prognostic models were developed to predict clinical outcomes following 1 year of UDCA treatment and have not yet been validated for use with OCA. A phase 3 study evaluating clinical outcomes with OCA in patients with PBC (COBALT; http://ClinicalTrials.gov identifier NCT02308111; EudraCT no. 2014‐005012‐42) is ongoing and may be used to test the utility of these prognostic models with OCA. Nevertheless, the results are promising given that 3 months of OCA monotherapy resulted in measurable and significant improvements in predicted survival.
Pruritus, a common symptom of PBC,29 was experienced by 35%, 70%, and 94% of patients receiving placebo, OCA 10 mg, and OCA 50 mg, respectively. The incidence of pruritus in the placebo and OCA 10 mg groups in this study was consistent with the incidence observed in a phase 3 study of OCA in combination with UDCA.15 Pruritus was manageable by antipruritic agents or temporary OCA interruption. Most patients who received a clinical intervention to manage pruritus completed the study regardless of OCA dose. OCA 50 mg was determined to be excessive as it resulted in 38% of patients discontinuing treatment due to pruritus without providing additional clinical benefit, as assessed by the comparable changes in ALP and other markers relative to OCA 10 mg (although the higher discontinuation rate in the OCA 50 mg group, which typically occurred shortly after initiating OCA, likely reduced observed efficacy as ET results were included with month 3 data). The phase 3 study of OCA in patients with PBC (including 7% who were receiving OCA monotherapy) investigating OCA 10 mg versus OCA 5 mg titrating to OCA 10 mg based on response and tolerability confirmed the benefit of dose titration in managing pruritus.15
Hypercholesterolemia and altered lipoprotein profiles are commonly seen in PBC, occurring in 75%‐95% of patients.30, 31 Similar to other clinical studies of OCA in patients with PBC,8, 15 OCA monotherapy resulted in a reduction in total cholesterol driven by decreased HDL‐C. The mechanism underlying the OCA‐driven reduction in HDL‐C remains to be confirmed; it is hypothesized that the reduction is due to FXR‐mediated up‐regulation of the surface HDL scavenger receptor B1, which would increase hepatic uptake of cholesterol from HDL.32
This study had several limitations, including a small patient population due to the near‐ubiquitous use of UDCA and a short double‐blind phase. The brevity of the double‐blind phase, however, is complemented by the ongoing OLE. The study did not incorporate the OLE phase into the original study design as the period between the double‐blind phase and the OLE was variable between patients. This potentially impeded recruitment of patients into the OLE, and several patients initiated UDCA between the two phases. While the OLE has been ongoing for over 6 years, the small patient population and addition of UDCA in some patients limit interpretation of data from the OLE. An additional limitation of this study is that the reason patients were not receiving UDCA at baseline was not captured.
In summary, OCA may provide an effective therapeutic option for patients with PBC in the absence of UDCA. The advent of effective second‐line therapy for PBC in the form of OCA is transforming the landscape, and it is important that we understand how to best use existing and emerging therapies. It is unclear if OCA may prove to have a role as a first‐line therapy in PBC, but the results of this study demonstrate that OCA is efficacious when given as monotherapy with a durable response with continuing treatment. This is particularly encouraging for the small but important group of PBC patients who cannot tolerate UDCA.
Supporting information
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo.
Supporting Information 1
Acknowledgment
Above all else, we thank the patients who participated in this study. Jonathan D. Roth, Ajay Malik, and Alexander Liberman provided medical writing assistance; all three were employees of Intercept Pharmaceuticals, Inc., during the writing of the manuscript.
Potential conflict of interest: Dr. Adorini consults for and owns stock in Intercept. Dr. Hirschfield consults for and is on the speakers' bureau for Intercept. He consults for GlaxoSmithKline and Novartis. Dr. Jones consults for, is on the speakers' bureau of, and received grants from Intercept. Dr. Kowdley consults for, advises, is on the speakers' bureau of, and received grants from Gilead and Intercept. He advises and received grants from Novartis. He received grants from Genfit and GlaxoSmithKline. Dr. Luketic advises GlaxoSmithKline. He received grants from Intercept, Genfit, Gilead, MSD, and Bristol‐Myers Squibb. Dr. Marschall consults and has received grants from Albiteo and Intercept. Dr. Mason consults for, is on the speakers' bureau of, and received grants from Intercept. He received grants from GlaxoSmithKline and Novartis. Dr. Parés consults for Intercept and Novartis. Dr. Pencek is employed by and owns stock in Intercept. Dr. Schramm advises Intercept. Dr. Sciacca consults for, is employed by, and owns stock in Intercept. Dr. Shapiro is employed by and owns stock in Intercept.
REFERENCES
- 1. Hirschfield GM, Gershwin ME. The immunobiology and pathophysiology of primary biliary cirrhosis. Annu Rev Pathol 2013;8:303‐330. [DOI] [PubMed] [Google Scholar]
- 2. Beuers U, Gershwin ME, Gish RG, Invernizzi P, Jones DEJ, Lindor K, et al. Changing nomenclature for PBC: from “cirrhosis” to “cholangitis.” Hepatology 2015;62:1620‐1622. [DOI] [PubMed] [Google Scholar]
- 3. European Association for the Study of the Liver . EASL clinical practice guidelines: management of cholestatic liver diseases. J Hepatol 2009;51:237‐267. [DOI] [PubMed] [Google Scholar]
- 4. Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ. Primary biliary cirrhosis: AASLD practice guidelines. Hepatology 2009;50:291‐308. [DOI] [PubMed] [Google Scholar]
- 5. Lammers WJ, Kowdley KV, van Buuren HR. Predicting outcome in primary biliary cirrhosis. Ann Hepatol 2014;13:316‐326. [PubMed] [Google Scholar]
- 6. Corpechot C, Chazouilleres O, Poupon R. Early primary biliary cirrhosis: biochemical response to treatment and prediction of long‐term outcome. J Hepatol 2011;55:1361‐1367. [DOI] [PubMed] [Google Scholar]
- 7. Lammers WJ, van Buuren HR, Hirschfield GM, Janssen HL, Invernizzi P, Mason AL, et al. Levels of alkaline phosphatase and bilirubin are surrogate end points of outcomes of patients with primary biliary cirrhosis: an international follow‐up study. Gastroenterology 2014;147:1338‐1349. [DOI] [PubMed] [Google Scholar]
- 8. Hirschfield GM, Mason A, Luketic V, Lindor K, Gordon S, Mayo M, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology 2015;148:751‐761. [DOI] [PubMed] [Google Scholar]
- 9. Hempfling W, Dilger K, Beuers U. Systematic review: ursodeoxycholic acid—adverse effects and drug interactions. Aliment Pharmacol Ther 2003;18:963‐972. [DOI] [PubMed] [Google Scholar]
- 10. Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, et al. 6Alpha‐ethyl‐chenodeoxycholic acid (6‐ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem 2002;45:3569‐3572. [DOI] [PubMed] [Google Scholar]
- 11. Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev 2009;89:147‐191. [DOI] [PubMed] [Google Scholar]
- 12. Kliewer SA, Mangelsdorf DJ. Bile acids as hormones: the FXR‐FGF15/19 pathway. Dig Dis 2015;33:327‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Beuers U, Trauner M, Jansen P, Poupon R. New paradigms in the treatment of hepatic cholestasis: from UDCA to FXR, PXR and beyond. J Hepatol 2015;62(Suppl.):S25‐S37. [DOI] [PubMed] [Google Scholar]
- 14. Fiorucci S, Antonelli E, Rizzo G, Renga B, Mencarelli A, Riccardi L, et al. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology 2004;127:1497‐1512. [DOI] [PubMed] [Google Scholar]
- 15. Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A placebo‐controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med 2016;375:631‐643. [DOI] [PubMed] [Google Scholar]
- 16. Carbone M, Sharp SJ, Flack S, Paximadas D, Spiess K, Adgey C, et al. The UK‐PBC risk scores: derivation and validation of a scoring system for long‐term prediction of end‐stage liver disease in primary biliary cirrhosis. Hepatology 2015;63:930‐950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lammers WJ, Hirschfield GM, Corpechot C, Nevens F, Lindor KD, Janssen HL, et al. Development and validation of a scoring system to predict outcomes of patients with primary biliary cirrhosis receiving ursodeoxycholic acid therapy. Gastroenterology 2015;149:1804‐1812. [DOI] [PubMed] [Google Scholar]
- 18. Lubsen J, Kirwan BA. Combined endpoints: can we use them? Stat Med 2002;21:2959‐2970. [DOI] [PubMed] [Google Scholar]
- 19. Angulo P, Dickson ER, Therneau TM, Jorgensen RA, Smith C, DeSotel CK, et al. Comparison of three doses of ursodeoxycholic acid in the treatment of primary biliary cirrhosis: a randomized trial. J Hepatol 1999;30:830‐835. [DOI] [PubMed] [Google Scholar]
- 20. Poupon RE, Balkau B, Eschwege E, Poupon R. A multicenter, controlled trial of ursodiol for the treatment of primary biliary cirrhosis. UDCA‐PBC Study Group. N Engl J Med 1991;324:1548‐1554. [DOI] [PubMed] [Google Scholar]
- 21. Lindor KD, Dickson ER, Baldus WP, Jorgensen RA, Ludwig J, Murtaugh PA, et al. Ursodeoxycholic acid in the treatment of primary biliary cirrhosis. Gastroenterology 1994;106:1284‐1290. [DOI] [PubMed] [Google Scholar]
- 22. Heathcote EJ, Cauch‐Dudek K, Walker V, Bailey RJ, Blendis LM, Ghent CN, et al. The Canadian Multicenter Double‐blind Randomized Controlled Trial of ursodeoxycholic acid in primary biliary cirrhosis. Hepatology 1994;19:1149‐1156. [PubMed] [Google Scholar]
- 23. Pares A, Caballeria L, Rodes J, Bruguera M, Rodrigo L, Garcia‐Plaza A, et al. Long‐term effects of ursodeoxycholic acid in primary biliary cirrhosis: results of a double‐blind controlled multicentric trial. UDCA‐Cooperative Group from the Spanish Association for the Study of the Liver. J Hepatol 2000;32:561‐566. [DOI] [PubMed] [Google Scholar]
- 24. Marschall HU, Wagner M, Zollner G, Fickert P, Silbert D, Gustafsson U, et al. Combined rifampicin and ursodeoxycholic acid treatment does not amplify rifampicin effects on hepatic detoxification and transport systems in humans. Digestion 2012;86:244‐249. [DOI] [PubMed] [Google Scholar]
- 25. Liu H, Norman GL, Shums Z, Worman HJ, Krawitt EL, Bizzaro N, et al. PBC screen: an IgG/IgA dual isotype ELISA detecting multiple mitochondrial and nuclear autoantibodies specific for primary biliary cirrhosis. J Autoimmun 2010;35:436‐442. [DOI] [PubMed] [Google Scholar]
- 26. Webb GJ, Siminovitch KA, Hirschfield GM. The immunogenetics of primary biliary cirrhosis: a comprehensive review. J Autoimmun 2015;64:42‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Selmi C, Meroni PL, Gershwin ME. Primary biliary cirrhosis and Sjogren's syndrome: autoimmune epithelitis. J Autoimmun 2012;39:34‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Poupon R. Liver alkaline phosphatase: a missing link between choleresis and biliary inflammation. Hepatology 2015;61:2080‐2090. [DOI] [PubMed] [Google Scholar]
- 29. Talwalkar JA, Souto E, Jorgensen RA, Lindor KD. Natural history of pruritus in primary biliary cirrhosis. Clin Gastroenterol Hepatol 2003;1:297‐302. [PubMed] [Google Scholar]
- 30. Carey EJ, Ali AH, Lindor KD. Primary biliary cirrhosis. Lancet 2015;386:1565‐1575. [DOI] [PubMed] [Google Scholar]
- 31. Sorokin A, Brown JL, Thompson PD. Primary biliary cirrhosis, hyperlipidemia, and atherosclerotic risk: a systematic review. Atherosclerosis 2007;194:293‐299. [DOI] [PubMed] [Google Scholar]
- 32. Chao F, Gong W, Zheng Y, Li Y, Huang G, Gao M, et al. Upregulation of scavenger receptor class B type I expression by activation of FXR in hepatocyte. Atherosclerosis 2010;213:443‐448. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found at http://onlinelibrary.wiley.com/doi/10.1002/hep.29569/suppinfo.
Supporting Information 1