
Fig. 8.
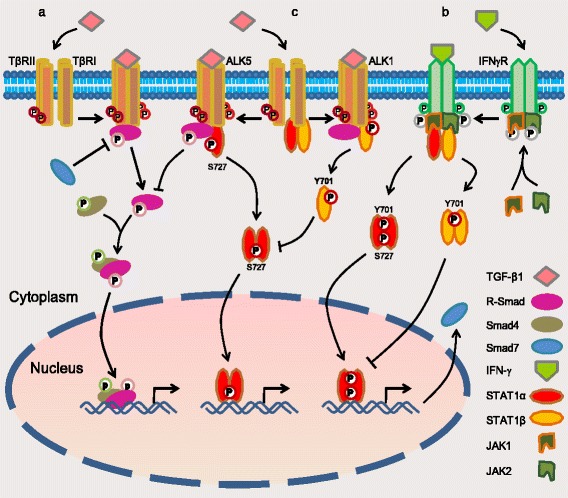
A schematic of the model of crosstalk between STAT1 and TGF-β signaling pathways. a The canonical signaling pathway of TGF-β: upon TGF-β1 binding to type II receptor (TβRII), the activated TβRII recruits and activates type I receptor (TβRI: ALK5 in most type of cells or ALK1 in endothelial cells). The activated receptor kinases then phosphorylate R-Smads such as Smad2/3 (by ALK5) and Smad1/5/8 (by ALK1). Activated R-Smads form the complex with Smad4 and translocate to the nucleus where they regulate target genes [12, 36]. b The canonical signaling pathway of STAT1: upon IFN-γ binding to its receptor, JAKs are phosphorylated and activated each other. The activated JAKs along with the intracellular tail of receptor recruit and activate STAT1 by the phosphorylation of STAT1 on Y701 and/or S727, which promotes them to dimerize and enter the nucleus where they regulate gene expression [7, 37], e.g. gene Smad7. Protein Smad7 prevents R-Smad interaction with TGF-β receptor [8]. Overexpression of STAT1β inhibits STAT1α activity to maintain the balance between two isoforms [26]. c The crosstalk of the STAT1 and TGF-β signaling pathways: STAT1 constitutively interacts with TβRII/TβRI (either ALK5 or ALK1 based on cell type). Upon TGF-β1 binding to TβRII/ALK5, the receptor-complex increases the phosphorylation of STAT1 on S727 (the active form of STAT1α). The activated STAT1 then dissociates itself from TβRII/ALK5 complex and executes its mission of transcription factor. On the other hand, STAT1 protein is increased in ovarian cancer cells and binds to TGF-β receptors. Overexpressed STAT1 suppresses TGF-β-induced Smad2 phosphorylation and blocks, at least in part, the TGF-β signaling pathway (current work)