

Abstract
Copy number variations (CNVs) comprise about 10% of reported disease-causing mutations in Mendelian disorders. Nevertheless, pathogenic CNVs may have been under-detected due to the lack or insufficient use of appropriate detection methods. In this report, on the example of the diagnostic odyssey of a patient with Marfan syndrome (MFS) harboring a hitherto unreported 32-kb FBN1 deletion, we highlight the need for and the feasibility of testing for CNVs (>1 kb) in Mendelian disorders in the current next-generation sequencing (NGS) era.
Keywords: Copy number variations (CNVs), genetic testing, Marfan syndrome (MFS), FBN1 gene, whole-genome sequencing (WGS)
Introduction
High-throughput next-generation sequencing (NGS) of targeted gene panels, the whole exome or even the whole genome is widely and increasingly used in the molecular diagnostics of Mendelian disorders. Standard NGS analysis pipelines are often restricted to the calling of single nucleotide variants (SNVs) and small insertions/deletions (indels), thereby missing copy number variations (CNVs) including deletions and duplications affecting more than 20 bp (1). However, pathogenic CNVs account for about 10% of disease-causing variants listed in the Human Gene Mutation Database (HGMD Professional 2018.1) and thus should not be missed. We believe that disease-causing CNVs tend to be under-detected in the current NGS era. In this study, we provide an example of this assumption. We investigated a family in which three members are clinically diagnosed with Marfan syndrome (MFS). Our results emphasize the need for the widespread use of NGS data to test for CNVs.
MFS is an autosomal dominant, systemic connective tissue disorder with varying clinical features, ranging from isolated traits to severe multiorgan manifestations, most frequently including the skeletal, ocular, and cardiovascular systems (2). Cardiovascular abnormalities can be life-threatening and include dilatation of the ascending aorta, which can result in dissection, mitral valve prolapse with or without regurgitation as well as tricuspid valve prolapse (3-6). MFS is caused by heterozygous pathogenic sequence variants in the FBN1 gene, which consists of 65 coding exons and encodes the protein fibrillin-1, a key component of elastic fibers (7,8). Pathogenic FBN1 sequence variants have been detected in >90% of patients fulfilling the diagnostic criteria for MFS (2). In the remaining cases, the disease-causing FBN1 mutation may have been missed by the applied genetic testing method or the clinical phenotype belongs to a non-FBN1-related syndrome. Indeed, there are MFS-related disorders with overlapping features such as Loeys-Dietz syndrome or Ehlers-Danlos syndrome vascular type, which in the absence of MFS-discriminating clinical features such as ectopia lentis can only be distinguished by genetic testing (2). As disease management and treatment guidelines differ among MFS and its related syndromes, the correct diagnosis is of clinical importance.
Clinical descriptions
In the investigated family, the index patient is a 32-year-old woman with clinically diagnosed MFS fulfilling the criteria of the revised Ghent nosology (2). She is 188 cm tall with a 195 cm arm span resulting in an arm span to height ratio of >1.05. Her joints are hypermobile and she shows positive wrist and thumb signs as well as scoliosis >20°. Other systemic manifestations include pes planus, high-arched palate with crowded teeth, atrophic striae, retrognathia, and myopia >3 diopters, resulting in a systemic score of 9. Her cardiovascular condition is also characteristic for MFS: according to echocardiography, the diameter of the ascending aorta, the sinus of Valsalva, and the aortic root were 46, 53 and 24 mm, respectively, resulting in a Z-score of 6.28 (9). In addition, mitral valve prolapse was detected, but without any hemodynamically relevant effect. According to international guidelines on the management of valvular heart disease (10), prophylactic Bentall surgery was carried out with implantation of Bio Valsalva.
Both the patient’s mother and sister show similar characteristics with a systemic score of 9 as well and conspicuous cardiovascular manifestations. The patient’s father does not fulfill the diagnostic criteria for MFS, but shares some MFS features such as reduced upper segment to lower segment and increased arm span to height ratios, tall stature, hypermobile joints, and dolichocephaly. Some phenotypic characteristics of the patient’s deceased grandparents are also known, of which the patient’s maternal grandfather showed myopia >3 diopters and a mild mitral valve prolapse. The other grandparents’ known phenotypes are not related to MFS.
Molecular genetic testing
The index patient underwent genetic testing for MFS and related cardiovascular diseases on genomic DNA level. First, NGS of a targeted gene panel was performed by means of PCR amplification of all coding exons and flanking intronic regions of the genes FBN1, TGFBR1, TGFBR2, SMAD3, TGFB2, TGFB3, ACTA2, COL3A1, MYH11, and SKI. Amplicons were analyzed on a MiSeq Personal Sequencer (Illumina, San Diego, CA, USA) (11). This approach revealed no pathogenic sequence variants in the analyzed amplicons. As gene panel NGS is restricted to selected amplicons/genes and limited in the detection of deletions and duplications affecting more than 20 bp (1), we subsequently carried out 60× PE150 PCR-free whole-genome sequencing (WGS) on a HiSeq X Ten platform (Illumina, San Diego, CA, USA) as previously described (12). WGS can not only identify sequence variants throughout the genome but has also the advantage of the most continuous coverage, improving both SNV and CNV detections (1).
Based on WGS, no pathogenic SNVs and small indels were detected in the ten previously analyzed genes or further genes related to the patient’s clinical features including ADAMTSL4, BGN, CBS, COL1A1, COL1A2, COL4A5, COL5A1, COL5A2, COL9A1, COL9A2, COL11A1, COL11A2, EFEMP2, ELN, FBN2, FLNA, FOXE3, GLA, JAG1, LOX, LTBP2, MAT2A, MED12, MFAP5, MYLK, NOTCH1, PLOD1, PRKG1, PTPN11, SLC2A10, SMAD2, SMAD4, and TNXB. Subsequently, the WGS data were analyzed for CNVs using the software Nexus Copy Number (BioDiscovery, El Segundo, CA, USA), revealing a 31,956-bp deletion of the FBN1 gene (NM_000138.4:c.164+13846_442+1334del). Due to the advantage of WGS, the deletion breakpoints could be identified at base-pair level. The deletion was confirmed by multiplex ligation-dependent probe amplification (MLPA, P065/P066, MRC-Holland, Amsterdam, the Netherlands) (13,14) as well as by standard PCR with a 407-bp amplicon spanning the deletion breakpoints followed by Sanger sequencing (primer_F 5’-AGGAACGATTTGAACAGATAATAGT-3’, primer_R 5’-CACCAAAGCACTTGCTATGT-3’) (Figure 1). At protein level, the deletion of coding exons 2-4 is predicted to lead to a frameshift and a premature termination codon [NP_000129.3:p.(Pro56Cysfs*3)]. Thus, nonsense mediated mRNA decay may take place resulting in functional haploinsufficiency (15). Testing of the index patient’s first-degree relatives for the detected FBN1 deletion by MLPA and Sanger sequencing revealed that the deletion is present in the patient’s sister and mother but absent in the father, as expected based on clinical features (Figure 2).
Figure 1.

Deletion characterization. Schematic representation of the 32-kb FBN1 deletion comprising coding exons 2-4 as well as an overview of the corresponding results of MLPA, WGS, and Sanger sequencing analyses. The open arrow below the gene name indicates the transcription direction. Exons are specified by bars and labeled with the corresponding number (non-coding exon 0 and the first five coding exons are indicated). The region with decreased normalized MLPA signals is indicated by a yellow box and the positions of the three MLPA probes located in exons 2-4 of FBN1 are marked by filled triangles. WGS data are displayed in the Integrative Genomics Viewer (IGV; http://software.broadinstitute.org/software/igv). Colored bars in the IGV coverage track indicate positions with ≥20% non-reference alleles; aligned reads are displayed as gray bars; red bars indicate read pairs with larger insert size than expected (due to the deletion); white and pale red bars indicate low quality reads. The region with decreased WGS read depth (i.e., the deleted region of the genomic DNA) is indicated by a gray and a brown box. Uppercase letters represent the sequence in the region of the deletion start point; lowercase letters represent the sequence in the region of the deletion end point. Due to identical sequences flanking the breakpoints, the break and re-joining could have occurred at three positions, as indicated by open triangles. The dotted lines mark the most 3’ possible breakpoints in FBN1 transcription direction (minus strand); note that the plus strand is displayed. Nucleotide positions are described in relation to the human genome reference sequence GRCh37. MLPA, multiplex ligation-dependent probe amplification; WGS, whole-genome sequencing.
Figure 2.
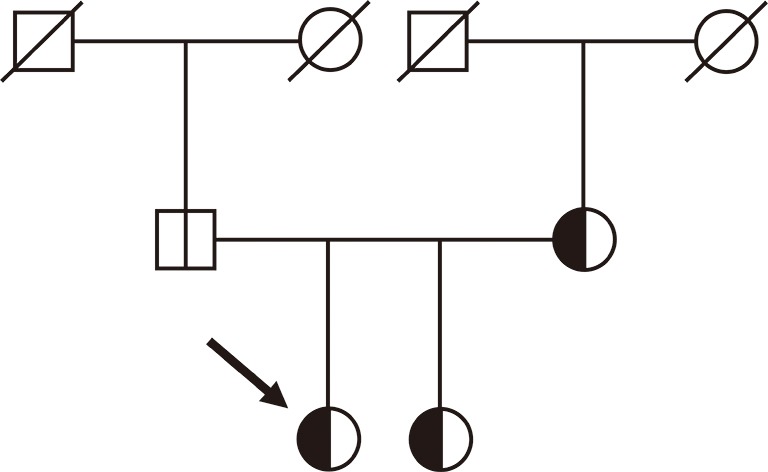
Pedigree of the examined family. Arrow indicates the index patient. The vertical line in the symbols (circle: female, square: male) denotes molecular genetic testing for the 32-kb FBN1 deletion: white halves represent normal alleles and black halves the allele carrying the deletion. A diagonal line through a symbol indicates deceased family members.
Discussion
This work exemplifies the importance of testing for CNVs in Mendelian disorders because they may be missed by standard NGS mutation screening. One of the advantages of an appropriate NGS methodology is that it allows not only the detection of SNVs and small indels but also of CNVs in a single assay. However, not all NGS applications are equally suited for CNV detection (1,16). While PCR-free WGS data facilitates CNV detection and enables CNV characterization at base-pair level due to relatively even read depth and continuous coverage (1,12), the enrichment bias and gaps of whole-exome sequencing (1,17) or targeted sequencing data introduce variable read depth and uneven coverage, hampering CNV detection (Figure 3). Consequently, CNV detection should be considered in the choice and data analysis pipeline of comprehensive NGS.
Figure 3.
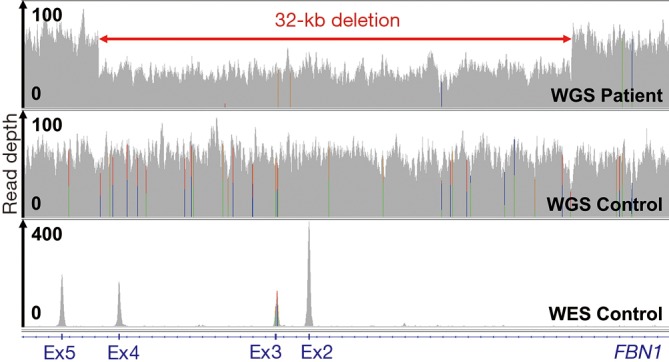
CNV detection by NGS. Read depth (coverage tracks) of 60× PE150 PCR-free WGS data of the index patient and a control as well as 100× PE100 WES data (12) of a control for the deleted and flanking genomic regions displayed in IGV. CNV, copy number variation; NGS, next-generation sequencing; WES, whole-exome sequencing; WGS, whole-genome sequencing.
Acknowledgements
Funding: This work was supported by the National Research, Development and Innovation Office of Hungary (NKFIH; NVKP-16-1-2016-0017), by the Human Resource Support Office (National Talent Program; NTP-NFTÖ-17-B-0027; to K.B.), by the “New National Excellence Program of the Ministry of Human Capacities of Hungary” (ÚNKP-17-3-I-SE-31; BÁ), and by the Foundation for People with Rare Diseases. We thank all the members of the Hungarian Marfan Foundation for enabling us to carry out this research.
Ethical Statement: Ethical approval for this study was obtained from the Scientific and Research Ethical Committee of the Medical Research Council of Hungary (ETT-TUKEB, 12751-3/2017-EKU). Written informed consent was obtained from the patient for publication of this manuscript and any accompanying images.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Caspar SM, Dubacher N, Kopps AM, et al. Clinical sequencing: From raw data to diagnosis with lifetime value. Clin Genet 2018;93:508-19. 10.1111/cge.13190 [DOI] [PubMed] [Google Scholar]
- 2.Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010;47:476-85. 10.1136/jmg.2009.072785 [DOI] [PubMed] [Google Scholar]
- 3.Benke K, Ágg B, Szilveszter B, et al. The role of transforming growth factor-beta in Marfan syndrome. Cardiol J 2013;20:227-34. 10.5603/CJ.2013.0066 [DOI] [PubMed] [Google Scholar]
- 4.Benke K, Agg B, Matyas G, et al. Gene polymorphisms as risk factors for predicting the cardiovascular manifestations in Marfan syndrome. Role of folic acid metabolism enzyme gene polymorphisms in Marfan syndrome. Thromb Haemost 2015;114:748-56. 10.1160/TH15-02-0096 [DOI] [PubMed] [Google Scholar]
- 5.Benke K, Agg B, Polos M, et al. The effects of acute and elective cardiac surgery on the anxiety traits of patients with Marfan syndrome. BMC Psychiatry 2017;17:253. 10.1186/s12888-017-1417-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benke K, Ágg B, Szabó L, et al. Bentall procedure: quarter century of clinical experiences of a single surgeon. J Cardiothorac Surg 2016;11:19. 10.1186/s13019-016-0418-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agg B, Benke K, Szilveszter B, et al. Possible extracardiac predictors of aortic dissection in Marfan syndrome. BMC Cardiovasc Disord 2014;14:47. 10.1186/1471-2261-14-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dietz HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature 1991;352:337-9. 10.1038/352337a0 [DOI] [PubMed] [Google Scholar]
- 9.The Marfan Foundation. Available online: https://www.marfan.org/dx/zscore
- 10.Vahanian A, Alfieri O, Andreotti F, et al. Guidelines on the management of valvular heart disease (version 2012): the Joint Task Force on the Management of Valvular Heart Disease of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS). Eur J Cardiothorac Surg 2012;42:S1-44. 10.1093/ejcts/ezs455 [DOI] [PubMed] [Google Scholar]
- 11.Campens L, Callewaert B, Muiño Mosquera L, et al. Gene panel sequencing in heritable thoracic aortic disorders and related entities - results of comprehensive testing in a cohort of 264 patients. Orphanet J Rare Dis 2015;10:9. 10.1186/s13023-014-0221-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meienberg J, Bruggmann R, Oexle K, et al. Clinical sequencing: is WGS the better WES? Hum Genet 2016;135:359-62. 10.1007/s00439-015-1631-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schouten JP, McElgunn CJ, Waaijer R, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002;30:e57. 10.1093/nar/gnf056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mátyás G, Alonso S, Patrignani A, et al. Large genomic fibrillin-1 (FBN1) gene deletions provide evidence for true haploinsufficiency in Marfan syndrome. Hum Genet 2007;122:23-32. 10.1007/s00439-007-0371-x [DOI] [PubMed] [Google Scholar]
- 15.Magyar I, Colman D, Arnold E, et al. Quantitative sequence analysis of FBN1 premature termination codons provides evidence for incomplete NMD in leukocytes. Hum Mutat 2009;30:1355-64. 10.1002/humu.21058 [DOI] [PubMed] [Google Scholar]
- 16.Tattini L, D'Aurizio R, Magi A. Detection of Genomic Structural Variants from Next-Generation Sequencing Data. Front Bioeng Biotechnol 2015;3:92. 10.3389/fbioe.2015.00092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meienberg J, Zerjavic K, Keller I, et al. New insights into the performance of human whole-exome capture platforms. Nucleic Acids Res 2015;43:e76. 10.1093/nar/gkv216 [DOI] [PMC free article] [PubMed] [Google Scholar]