

Abstract
In recent years, Next-Generation Sequencing (NGS) opened a new way for the study of pathogenic mechanisms and for molecular diagnosis of inherited disorders. In the present work, we focused our attention on the inherited retinal dystrophies (IRDs), a group of specific disorders of the retina, displaying a very high clinical and genetic heterogeneity, whose genetic diagnosis is not easily feasible. It represents a paradigmatic example for the integration of clinical and molecular examination toward precision medicine.
In this paper, we discuss the use of targeted NGS resequencing of selected gene panels in a cohort of patients affected by IRDs. We tested the hypothesis to apply a selective approach based on a careful clinical examination. By this approach we reached a 66% overall detection rate for pathogenic variants, with a 52% diagnostic yield. Reduction of the efforts for validation and classification of variants is a clear advantage for the management of genetic testing in a clinical setting.
Key words: multigene panels, NGS, inherited retinal dystrophies, diagnostic yield
INTRODUCTION
Inherited retinal dystrophies (IRDs) are a group of rare diseases due to a progressive degeneration of retinal photoreceptors, that can lead to vision loss [1,2]. IRDs comprise several different disorders characterized by clinical and genetic heterogeneity, often displaying a phenotypic overlap [3]. Many IRDs are characterized by progressive degeneration of both cone and rod photoreceptors, making the clinical differential diagnosis difficult, especially in the advanced stages [4].
Additionally, there are also syndromic forms in which retina is not the only affected tissue and additional organs can be involved, such as the Usher Syndrome (USH) and the Bardet-Biedl Syndrome (BBS). Furthermore, clinical symptoms can be progressive with variable onset and intra-familial variability, due to an incomplete penetrance and variable expressivity, making the clinical picture more complex [1]. All these factors often complicate or delay a precise diagnosis [1,2,5].
By a genetic point of view, IRDs displays locus and allelic heterogeneity [6], with more than 200 causative genes, that make the genetic characterization very complex. The advent of next-generation sequencing (NGS) has opened new frontiers in genetic diagnostics of IRDs, exploiting the high-throughput parallel sequencing and the simultaneous analysis of several samples. Indeed, the overall mutation detection rate for IRDs is variable [6], ranging from 36% to 60%, leaving many cases still genetically unsolved. More than 4000 pathogenic variants have been identified in causative genes, that can converge to the same phenotype [6] or can show different symptoms [1], complicating the molecular diagnosis. Lastly, since some IRDs causative genes are associated to specific inheritance traits (AR, AD, X-linked), a targeted genetic analysis could be more effective, although sometimes establishing the inheritance mode in an affected family is difficult [1].
Considering all the above, it is often complex to determine a priori which genes are to be analyzed and a “non-hypothesis-driven” approach has been applied in large NGS studies [7-9].
In the diagnostic laboratory, such an approach increases the risk to identify variants of uncertain significance, complicating the interpretation and implying a big effort in classification.
In this paper, we describe the strategy adopted by our multidisciplinary team to optimize the integration of clinical data and NGS targeted resequencing for the diagnostics of the different forms of IRDs. Our approach for the molecular diagnosis of IRDs, including genes that fit with the phenotype, allowed us to obtain a 66% overall mutation detection rate, consistent with the best rates obtained with the “non-hypothesis-driven” approach.
MATERIALS AND METHODS
Clinical diagnosis and sample collection
This investigation conformed to principles outlined in the Declaration of Helsinki.
We collected 35 unrelated affected patients with different forms of IRDs.
All patients underwent an ophthalmic evaluation at the Department of Ophthalmology of San Raffaele Hospital (Milan, Italy), including best corrected visual acuity by means of Early Treatment Diabetic Retinopathy (ETDRS) standard charts, biomicroscopy, color fundus photography, fundus autofluorescence, electrophysiological tests, and spectral-domain optical coherence tomography.
Clinical and family history details were collected during genetic counseling interview. Written informed consent for genetic analysis was obtained from all subjects. Genetic analysis was performed at Laboratory of Clinical Molecular Biology of San Raffaele Hospital (Milan, Italy).
Genomic DNA (gDNA) was extracted from peripheral blood using the automated extractor Maxwell16 (Promega, Milano, Italy); the concentration and gDNA quality were determined using Qubit® Fluorometer (Thermo Fisher Scientific).
Library enrichment and sequencing
Sample enrichment and paired-end libraries preparation were performed using the commercial kit TruSight One (Illumina, San Diego, CA, USA), starting from 50ng gDNA, following the manufacturer’s instructions (Document #1000000006694 v00).
TruSight One Sequencing panel includes 4,813 genes associated with known clinical phenotypes, according to the Human Gene Mutation Database_HGMD (http://www.hgmd.cf.ac.uk/ac/index.php), Online Mendelian Inheritance in Man, OMIM (www.omim.org), and GeneTests (www.genetests.org). The entire gene list is published on www.illumina.com (Pub. No. 0676-2013-016 current as of 04 January 2016). Sequencing was performed on NextSeq500 instrument (Illumina, San Diego, CA, USA) with a flow cell high output, 300 cycles PE (150 x 2).
NGS data analysis
The read alignment and variant calling were performed with BaseSpace Onsite Sequence Hub. For each case, the analysis of variants was focused on one or more gene panels based on the different clinical phenotypes. The variants were then annotated using Illumina VariantStudio data analysis software. For the identification of possible causative variants, filters were applied taking into account: 1) the quality parameter, 2) the MAF (Minor Allele Frequency) >2% in the 1000Genomes and ExAC database, 3) the localization of the variants, considering only the exonic and intronic regions at ± 20 bp from the coding regions, to identify possible splice-site variations.
In order to optimize the data analysis process and to focus on the identification of causative variants, we created panels of disease genes associated to the different forms of IRDs, as reported in Table 1. Particularly, we set panels for non-syndromic forms (Achromatopsia (ACHM); Best vitelliform macular dystrophy; Congenital stationary night blindness (CSNB); Choroideremia; Stargardt disease; Retinitis pigmentosa) and for syndromic forms (Bardet-Biedl S., Refsum disorder, Cohen S., Stickler S., Usher S.). We chose causative genes for each disease panel based on public databases, such as OMIM (http://www.ncbi.nlm.nih.gov/omim) or RetNet™ (https://sph.uth.edu/retnet/) and from the literature [4,6,10-12]. After primary analysis, the search for causative variants started by considering the panel of genes associated to the clinical suspicion. If the suspicion was less focused, more than one panel is analyzed.
Table 1.
Different panels of disease genes associated to the different forms of IRDs
| Inherited Retinal Dystrophies | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Non-Syndromic Forms | Syndromic forms | ||||||||||
| Achromatopsia | Best macular dystrophy | Congenital Stationary Night Blindness | Choroideremia | Stargardt disease-cone-rod dystrophy | Retinitis pigmentosa-rod-cone dystrophy | Bardet-Biedl Syndrome | Cohen Syndrome | Stickler Syndrome | Usher Syndrome | Refsum disease | |
| Orpha | 49382 | 1243 | 215 | 180 | 827 | 791 | 110 | 193 | 828 | 886 | 773 |
| ICD-10 | H53.5 | H35.5 | H53.6 | H31.2 | H35.5 | H35.5 | Q87.8 | Q87.8 | Q87.0 | H35.5 | G60.1 |
| Incidence | 1-9 /100 000 | 1-9 /100 000 | Unknown | 1-9 /100 000 | 1-5 /10 000 | 1-5 /10 000 | 1-9 /1 000 000 | Unknown | 1-9 /100 000 | 1-9 /100 000 | 1-9 /1 000 000 |
| Onset | Infancy, Neonatal | Childhood, Adolescent | Neonatal | Childhood, Adolescence, Adulthood | Childhood, Adolescence, Adulthood | Childhood, Adolescent, Adult | Prenatal, Neonatal, Childhood | Neonatal, Childhood | Childhood | Neonatal, Childhood | Infancy, Childhood, Adolescence, Adulthood |
| Inheritance mode | AR | AD | AD; AR; X-linked | X-linked | AD; AR | AD; AR; X-linked; Mitochondrial inheritance | AR | AR | AR; AD | AR | AR |
| Prevalence of mutations | 75-90% | 96%* (familial forms) 70%* (non familial forms) | 95%* | 95%* | 65-70% | 75% | 90%* | 70%* | 100%* | 80-85%* | 100%* |
| N. of genes of panel | 7 | 3 | 14 | 1 | 43 | 63 | 18 | 1 | 5 | 11 | 2 |
| Genes | ATF6 | BEST1 | CABP4 | CHM | ABCA4 | ABCA4 | ARL6 | VPS13B | COL11A1 | ADGRV1 | PEX7 |
| CNGA3 | IMPG2 | CACNA1F | ADAM9 | BBS1 | BBS1 | COL11A2 | CDH23 | PHYH | |||
| CNGB3 | PRPH2 | CACNA2D4 | AIPL1 | BBS2 | BBS10 | COL2A1 | CIB2 | ||||
| GNAT2 | GNAT1 | C2orf71 | C2orf71 | BBS12 | COL9A1 | CLRN1 | |||||
| PDE6C | GNB3 | C8orf37 | C8orf37 | BBS2 | COL9A2 | HARS | |||||
| PDE6H | GPR179 | CABP4 | BEST1 | BBS4 | MYO7A | ||||||
| RPGR | GRK1 | CACNA1F | CA4 | BBS5 | PCDH15 | ||||||
| GRM6 | CACNA2D4 | CDHR1 | BBS7 | PDZD7 | |||||||
| NYX | CDH3 | CERKL | BBS9 | USH1C | |||||||
| PDE6B | CDHR1 | CLRN1 | CEP290 | USH1G | |||||||
| RHO | CEP290 | CNGA1 | LZTFL1 | USH2A | |||||||
| SAG | CERKL | CNGB1 | MKKS | ||||||||
| SLC24A1 | CLN3 | CRB1 | MKS1 | ||||||||
| TRPM1 | CNGA3 | CRX | NPHP1 | ||||||||
| C1QTNF | CYP4V2 | SDCCAG8 | |||||||||
| CNGB3 | DHDDS | TRIM32 | |||||||||
| CNNM4 | EYS | TTC8 | |||||||||
| CRB1 | FAM161A | WDPCP | |||||||||
| CRX | FLVCR1 | ||||||||||
| CYP4V2 | FSCN2 | ||||||||||
| ELOVL4 | GUCA1B | ||||||||||
| FSCN2 | HGSNAT | ||||||||||
| GNAT2 | IDH3B | ||||||||||
| GUCA1A | IMPDH1 | ||||||||||
| GUCY2D | IMPG2 | ||||||||||
| KCNV2 | KLHL7 | ||||||||||
| PDE6C | LRAT | ||||||||||
| PDE6H | MAK | ||||||||||
| PITPNM3 | MERTK | ||||||||||
| PROM1 | NR2E3 | ||||||||||
| PRPH2 | NRL | ||||||||||
| RAB28 | PDE6A | ||||||||||
| RAX2 | PDE6B | ||||||||||
| RDH12 | PDE6G | ||||||||||
| RDH5 | PRCD | ||||||||||
| RGS9 | PROM1 | ||||||||||
| RGS9BP | PRPF3 | ||||||||||
| RIMS1 | PRPF31 | ||||||||||
| RP1L1 | PRPF6 | ||||||||||
| RPGR | PRPF8 | ||||||||||
| RPGRIP1 | PRPH2 | ||||||||||
| SEMA4A | RBP3 | ||||||||||
| TIMP3 | RBP4 | ||||||||||
| RDH12 | |||||||||||
| RGR | |||||||||||
| RHO | |||||||||||
| RLBP1 | |||||||||||
| ROM1 | |||||||||||
| RP1 | |||||||||||
| RP1L1 | |||||||||||
| RP2 | |||||||||||
| RP9 | |||||||||||
| RPE65 | |||||||||||
| RPGR | |||||||||||
| SAG | |||||||||||
| SEMA4A | |||||||||||
| SNRNP200 | |||||||||||
| SPATA7 | |||||||||||
| TOPORS | |||||||||||
| TTC8 | |||||||||||
| TULP1 | |||||||||||
| USH2A | |||||||||||
| ZNF513 | |||||||||||
Data available on Orphanet (http://www.orpha.net - Last update: August 2017) and Genereviews (https://www.ncbi.nlm.nih.gov/books/NBK1116).
* Data reported on Genereview.
Interpretation of putative variants was performed using Alamut® Visual (Interactive bio-software), that integrate data from several databases, such as NCBI, UCSC, ClinVar, HGMD Professional, and in silico tools prediction, such as Polyphen, Sift, Mutation Taster. Candidate variants were classified according to the ACMG criteria in 5 categories:
class 1: benign,
class 2: likely benign,
class 3: uncertain significance (VUS),
class 4: likely pathogenic,
Analysis flow chart is reported in Figure 1.
Figure 1.
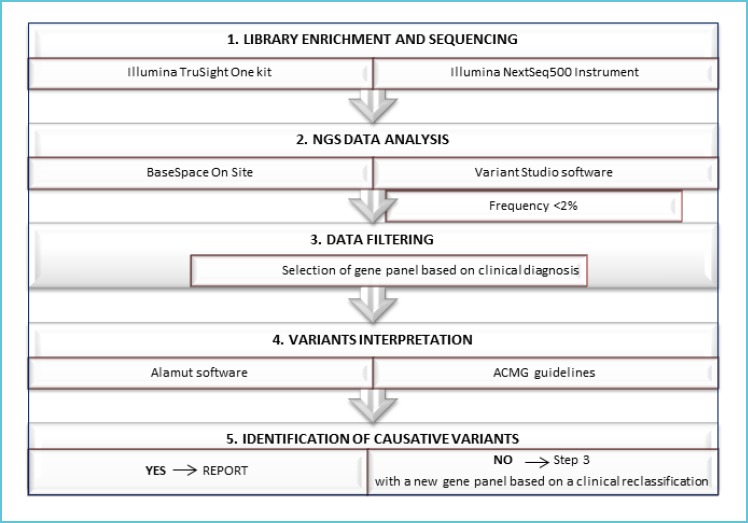
Workflow of NGS analysis
The flow chart illustrates the main steps from the sequencing to the clinical report.
Identified variants were validated using Sanger Sequencing on AB3730 sequencer (Applied Biosystem), according to the manufacturers’ protocols. (Primer and PCR conditions available on request). Moreover, in order to avoid undetected variants in regions with a low number of reads, all target regions of causative genes with a coverage <10X were analyzed by Sanger sequencing.
RESULTS AND DISCUSSION
Parameters of NGS raw data
All the 35 patients have been sequenced for 4813 genes, included in TruSight One panel (Illumina) using Illumina NextSeq500.
Runs had a mean cluster density equal to 217 k/mm2. We obtained a mean read enrichment of 59% and target aligned read of 99%. The mean coverage for the analyzed genes associated to the different forms of IRDs was 300X.
Analysis and classification of detected variants
In our cohort, excluding common variants, we detected a total of 57 variants in 29 genes; 30 were novel and 27 were already reported in dbSNP as rare variants. In three patients no variants were found (9%), while the others (91%) presented with different variants with the exception of two pathogenic variants in ABCA4 (NM_000350.2: c.5882G>A; NM_000350.2: c.5018+2T>C), identified in four different unrelated patients.
Considering all the detected variants, 66.7% (38/57) were missense, 10.5% (6/57) were stop-gain, 7% (4/57) were frameshift changes, 8.8% (5/57) may alter splice sites, 1 variant was a start-loss (1.8%), 1 was an in-frame insertion (1.8%), 1 was an in-frame deletion (1.8%) and 1 was a deletion of two whole exons (1.8%) (Figure 2). All the 57 variants were confirmed by Sanger sequencing or MLPA.
Figure 2.
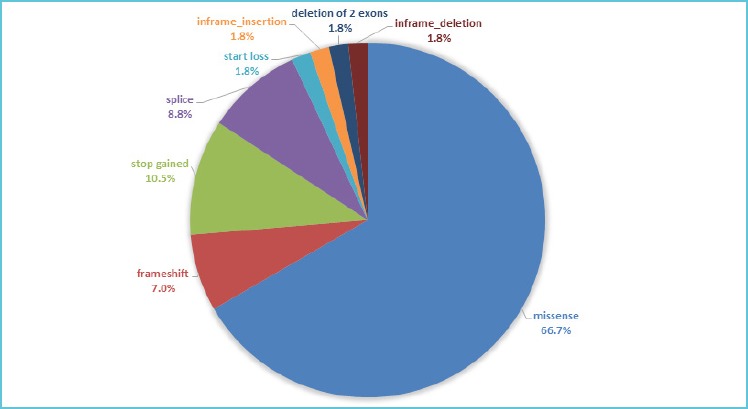
Different types of identified variants
We identified 57 variants in 29 genes in our cohort and in the pie chart the percentage of each type of detected variant is reported.
Among the 29 genes, the majority (22/29) present a single variant while seven genes are multi-variated (Figure 3).
Figure 3.
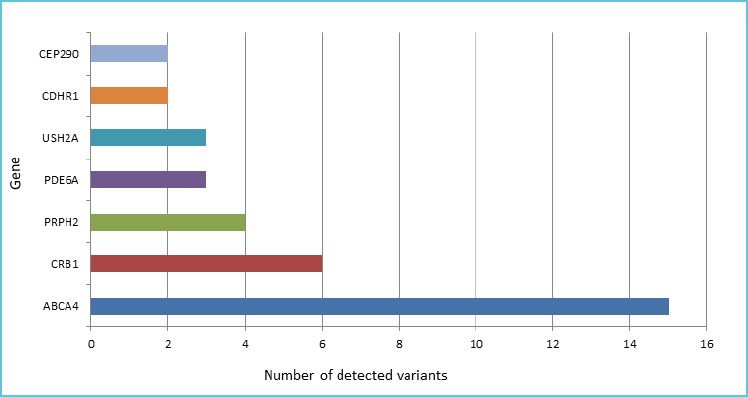
Seven genes are multivariated in our cohort
Graph represents the number of detected variants (x-axis) for each gene (y-axis).
According with the ACMG guidelines [13], 11 variants were classified as pathogenic (class 5), 19 as likely pathogenic (class 4) and the remaining 27 as variants of unknown significance (VUS, class 3).
Evaluation of the diagnostic yield and genotype-phenotype correlation
We found pathogenic or likely pathogenic variants in 23/35 (66%) patients and consistent with the subject clinical presentation. Among these, we were able to reach the genetic diagnosis in 18/35 (52%) patients while in 5/35 (14%) patients we obtained only a partial diagnosis because of the detection of only one causative recessive variant. In 9/35 (26%) patients we identified heterozygous variants with unknown significance (VUS) in disease-genes but in 5 of them the genotype did not fit to the disease inheritance manner and the genetic diagnosis remained incomplete. Finally, 3 patients were wild-type in analyzed causative genes. In these cases, a multidisciplinary re-discussion would be suggested in an attempt to define further testing or the potential for a research approach.
The majority of patients not reaching the genetic diagnosis had non-syndromic phenotypes, in particular two of the patients with no variants had a clinical diagnosis of Best, while in the case of retinal dystrophy, retinitis pigmentosa and Stargardt, a high proportion of patients had a partial or inconclusive diagnosis due to the presence of only one pathogenic variant or to the presence of VUS. In particular, for 5 patients with a partial diagnostic yield we can suspect the presence of a second pathogenic variant in a deep intronic region, as is the case for ABCA4 or the presence of a structural variant not identified by sequencing. In Table 2, we reported the obtained diagnostic yield for each disease.
Table 2.
The percentance of complete, partial and total diagnostic yield obtained using our multigene panel approach for each disease
| Total patients = 35 | Clinical phenotype | Patients (n) | Complete diagnostic yield % (n) | Partial diagnostic yield % (n) | Total diagnostic yield % |
|---|---|---|---|---|---|
| Overall diagnostic yield (%) = 51 | |||||
| Disease | Pattern dystrophy | 1 | 100(1) | 100 | |
| Bardet-Biedl S. | 1 | 100(1) | 100 | ||
| Best Disease | 5 | 60(3) | 60 | ||
| Complex phenotype; retinal dystrophy (rod-cone or cone-rod) | 11 | 36(4) | 18(2) | 54 | |
| Retinitis Pigmentosa | 4 | 75(3) | 75 | ||
| Stargardt disease | 11 | 36(4) | 27(3) | 63 | |
| Stickler S. | 1 | 100(1) | 100 | ||
| Usher S. | 1 | 100(1) | 100 |
In Table 3 are listed all the genes with variants identified in the present work in association with different diseases. It is possible to appreciate that the larger genetic overlap is between the retinal dystrophies and RP phenotypes (Table 3, the shaded lines).
Table 3.
The genetic overlapping between retinal dystrophies and RP phenotypes
| Pattern dystrophy | Bardet-Biedl Syndrome | Best Disease | Complex Phenotype; retinal dystrophy (rod-cone or cone-rod) | Retinitis Pigmentosa | Stargardt disease | Stickler Syndrome | Usher Syndrome |
|---|---|---|---|---|---|---|---|
| PRPH2 | BBS4 | BEST1 | ABCA4 | ABCA4 | ABCA4 | COL2A1 | USH2A |
| IMPG2 | CDH23 | C2ORF71 | ATF6 | ||||
| PRPH2 | CDHR1 | CDHR1 | CEP290 | ||||
| CEP290 | CRB1 | CRX | |||||
| CNGA3 | USH2A | GNAT2 | |||||
| CRB1 | GPR98 | ||||||
| FSCN2 | PCDH15 | ||||||
| IMPDH1 | TOPORS | ||||||
| KCNV2 | |||||||
| PDE6A | |||||||
| PDE6B | |||||||
| PITPNM3 | |||||||
| PRPH2 | |||||||
| RIMS1 | |||||||
| RP1 | |||||||
| RP1L1 | |||||||
| RPGRIP1 |
The coloured cells indicate the genes mutated in different clinical phenotypes.
In the present work, we applied a targeted NGS resequencing for genetic testing of IRDs; selection of gene panels was done based on the clinical suspicion (Table 1) allowing us to reduce the number of genes tested. We reached a diagnosis in a proportion of patients that was consistent with the results from other studies, where wider panels were used. Based on these findings, this approach, reducing the efforts needed for classification and validation of variants, seems to be more suited in the diagnostic field.
CONCLUSION AND GENERAL REMARKS
Thanks to NGS, genetic testing costs are reducing rapidly with the potential for a broader access in the frame of health care systems. As NGS allows parallel analysis, it currently realizes a real improvement for personalized medicine, shortening the time needed to reach a diagnosis, nevertheless we still have to face a number of criticisms [15]. This report, showing an overall mutation detection rate for IRDs of approximately 60%, addresses the challenges ahead, which include: a better understanding of the clinical significance of variants in disease genes; improvement of variant calling, especially for deep-intronic regions, regulatory sequences, promoters and structural variants (i.e.: extension of captured regions and improvement of tools for CNV detection); improvement of geno-type-phenotype correlations and comprehension of more complex or not yet understood genetic mechanisms of diseases.
Correspondingly, the simultaneous sequencing of a large number of genes has resulted in increased detection of variants of unknown significance, which require interpretation for clinical purposes. The development of databases such as ClinVar and WES (Whole Exome Sequencing) variant allele frequency by ExAC Browser are gradually improving variant interpretation.
Similarly, programs such as SIFT, PolyPhen-2, and NNSPLICE are now widely used to predict the influence of a variant on protein localization, structure, and/or function. However, in silico predictions are not always consistent with functional studies and, despite recent advances, pathogenicity assessment remains challenging, particularly for hypomorphic, synonymous and non-coding variants. Ultimately, better tools are required, as well as improved knowledge of the genome and genome function.
Acknowledgements
We acknowledge all the clinicians of the Department of Ophthalmology and the personnel of the Clinical Molecular Biology Laboratory at IRCCS San Raffaele Hospital, Milan, Italy.
REFERENCES
- 1.Chiang JP-W, Lamey T, McLaren T, Thompson JA, Montgomery H, De Roach J. Progress and prospects of next-generation sequencing testing for inherited retinal dystrophy. Expert Rev Mol Diagn. 2015;15: 1269–1275. doi:10.1586/14737159.2015.1081057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bravo-Gil N, Méndez-Vidal C, Romero-Pérez L, González-del Pozo M, Rodríguez-de la Rúa E, Dopazo J, et al. Improving the management of Inherited Retinal Dystrophies by targeted sequencing of a population-specific gene panel. Sci Rep. 2016;6: 23910. doi:10.1038/srep23910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368: 1795–1809. doi:10.1016/S0140-6736(06)69740-7 [DOI] [PubMed] [Google Scholar]
- 4.den Hollander AI, Black A, Bennett J, Cremers FPM. Lighting a candle in the dark: advances in genetics and gene therapy of recessive retinal dystrophies. J Clin Invest. 2010;120: 3042–3053. doi:10.1172/JCI42258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Werdich XQ, Place EM, Pierce EA. Systemic diseases associated with retinal dystrophies. Semin Ophthalmol. 2014;29: 319–328. doi:10.3109/08820538.2014.959202 [DOI] [PubMed] [Google Scholar]
- 6.Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res. 2010;29: 335–375. doi:10.1016/j.preteyeres.2010.03.004 [DOI] [PubMed] [Google Scholar]
- 7.Audo I, Bujakowska KM, Léveillard T, Mohand-Saïd S, Lancelot M-E, Germain A, et al. Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J Rare Dis. 2012;7: 8. doi:10.1186/1750-1172-7-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Neveling K, Collin RWJ, Gilissen C, van Huet RAC, Visser L, Kwint MP, et al. Next-generation genetic testing for retinitis pigmentosa. Hum Mutat. 2012;33: 963–972. doi:10.1002/humu.22045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang F, Wang H, Tuan H-F, Nguyen DH, Sun V, Keser V, et al. Next generation sequencing-based molecular diagnosis of retinitis pigmentosa: identification of a novel genotype-phenotype correlation and clinical refinements. Hum Genet. 2014;133: 331–345. doi:10.1007/s00439-013-1381-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Broadgate S, Yu J, Downes SM, Halford S. Unravelling the genetics of inherited retinal dystrophies: Past, present and future. Prog Retin Eye Res. 2017;59: 53–96. doi:10.1016/j.preteyeres.2017.03.003 [DOI] [PubMed] [Google Scholar]
- 11.Hohman TC. Hereditary Retinal Dystrophy. Handbook of experimental pharmacology. 2016. pp. 337–367. doi:10.1007/164_2016_91 [DOI] [PubMed] [Google Scholar]
- 12.Ferrari S, Di Iorio E, Barbaro V, Ponzin D, Sorrentino FS, Parmeggiani F. Retinitis pigmentosa: genes and disease mechanisms. Curr Genomics. 2011;12: 238–249. doi:10.2174/138920211795860107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17: 405–423. doi:10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Q, Wang K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am J Hum Genet. Elsevier; 2017;100: 267–280. doi:10.1016/j.ajhg.2017.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kricka LJ, Di Resta C. Translating genes into health. Nat Genet. 2013;45: 4–5. doi:10.1038/ng.2510 [DOI] [PubMed] [Google Scholar]