

Abstract
Ataxia telangiectasia and Rad3-related (ATR) activates checkpoint kinase 1 (CHK1) following replication fork stalling, leading to cell cycle arrest. ATR-CHK1 pathway components are considered to be promising therapeutic targets to enhance the effectiveness of replication inhibitors. The present study revealed that F-Box protein 32 (FBXO32) regulated ATR expression in pancreatic cancer PANC-1 and MIA PaCa-2 cells. Additionally, FBXO32 interacts with ATR in PANC-1 cells and ATR is a degradation substrate of E3 ubiquitin ligase FBXO32. Furthermore, FBXO32 regulated the DNA damage response induced by gemcitabine in PANC-1 cells. Taken together, the results of the present study suggested that FBXO32, as an E3 ubiquitin ligase of ATR, regulates the DNA damage response induced by gemcitabine in pancreatic cancer.
Keywords: F-Box protein 32, ataxia telangiectasia and Rad3-related, ubiquitination, degradation, pancreatic cancer, DNA damage, gemcitabine
Introduction
Pancreatic cancer is one of the most aggressive and lethal types of cancer (1). The most common type of pancreatic cancer is adenocarcinoma (accounting for 95%), which originates from the exocrine part of the pancreas and is classified as pancreatic ductal adenocarcinoma (PDAC). The prognosis of pancreatic cancer remains poor with a 5-year survival rate of only 4%, ranking the fourth leading cause of all cancer-associated mortalities in the USA (2). Chemotherapy remains an effective option, particularly for advanced pancreatic cancer (3). Unfortunately, pancreatic cancer has been considered as a relatively chemotherapy-refractory tumor. It may be difficult for patients with advanced pancreatic cancer to benefit from chemotherapy. Therefore, there is an immediate requirement to discover novel therapeutic targets for pancreatic cancer.
DNA double-strand breaks (DSB) trigger genome rearrangements, and DNA damage response (DDR) is a transduction cascade that coordinates the signaling and repair of these genomic lesions (4). The ataxia telangiectasia and Rad3-related (ATR) protein kinase is a member of the phosphoinositide 3-kinase-related kinase (PIKK) family (5). It serves an important role in DDR and activates checkpoint kinase 1 (CHK1) following replication fork stalling, leading to cell cycle arrest (6). The overexpression of a dead mutant version of ATR causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints (7). This response is an important mechanism that facilitates cancer cells in surviving anticancer treatments. Therefore, ATR may be a potential therapeutic target for the treatment of pancreatic cancer.
FBXO32 (also known as MAFbx or Atrogin-1) acts as a ubiquitin E3 ligase and has been demonstrated to target several proteins for proteasomal degradation (8,9). The present study demonstrated that FBXO32 acted as an E3 ligase of ATR in pancreatic cancer cells. These results allowed understanding of the regulatory mechanism of ATR in human pancreatic cancer and development of novel therapeutic strategies to treat pancreatic cancer.
Materials and methods
Plasmids and reagents
Myc-tagged FBXO32 (Myc-FBXO32) was cloned into the pCMV vector. To construct the Myc-FBXO32 plasmid, the full length FBXO32 gene was amplified using 293T cells via PCR amplification and cloned into the pCMV-Myc vector (Takara Bio, Inc., Otsu, Japan). Myc-FBXO32SR (shRNA-resistant FBXO32 plasmid) was generated using a KOD-Plus Mutagenesis kit (Toyobo Life Science, Osaka, Japan). Pancreatic cancer cells were transfected with varying amounts of Myc-FBXO32 plasmids (3 µg/1×106 cells; 1 µg/1×106 cells or 3 µg/1×106 cells with transfected plasmids, using Lipofectamine 2000® (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the manufacturer's protocol. All transfections occurred once with each different dose. A total of 24 h post-transfection, cells were collected for subsequent analyses. Anti-FBXO32 (dilution, 1:500) was purchased from ProteinTech Group, Inc. (Chicago, IL, USA); anti-ATR (dilution, 1:500), anti-PARP (dilution, 1:1,000) antibodies were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA); and anti-β-Tubulin (dilution, 1:5,000) was from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Gemcitabine was obtained from Eli Lilly and Company (Indianapolis, IN, USA) and dissolved in distilled water. Pancreatic cancer cells were treated with 10 µM gemcitabine and the control group was treated with equal amounts of distilled water for 24 h. MG132 was purchased from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany), and pancreatic cancer cells were treated with 20 µM MG132 for 8 h as previously described (10).
Cell culture
Pancreatic cancer PANC-1 and MIA PaCa-2 cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and were cultured in 5% CO2 at 37°C in a 95% humidity incubator. PANC-1 and MIA PaCa-2 cells were cultured in Dulbecco's modified Eagle's medium (Thermo Fisher Scientific, Inc.), supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Inc.) and 100 U/ml penicillin and 100 µg/ml streptomycin (Thermo Fisher Scientific, Inc.).
Western blot analysis
Pancreatic cancer PANC-1 and MIA PaCa-2 cells were lysed by lysis buffer [1% Nonidet P-40, 1× phosphate-buffered saline (PBS), 0.1% sodium dodecyl sulfate and 1% protease inhibitor cocktail], followed by protein quantification using the bicinchoninic acid assay (BCA) method. Samples were diluted in loading buffer containing DTT and were boiled for 5 min. Equal amount of protein (100 µg) for each sample was separated by 10% SDS-PAGE, prior to being transferred onto nitrocellulose membranes. The membranes were blocked in 5% milk at room temperature for 1 h. Then, the membranes were immuno-blotted with anti-FBXO32 (cat. no. PA5-43915; 1:1,000; Thermo Fisher Scientific), anti-ATR (cat. no. 13934; 1:500) and anti-PARP (cat. no. 9532; 1:1,000; both Cell Signaling Technology, Inc., Danvers, MA, USA), anti-β-Tubulin (cat. no. sc-5274; 1:5,000) and anti-p53 (cat. no. sc-377567; 1:1,000; both Santa Cruz Biotechnology, Inc., Dallas, TX, USA) antibodies at 4°C overnight. Subsequently, the membrane was wash three times with 1X TBST and incubated with rabbit IgG (cat. no. MR-R100; 1:3,000) and (mouse IgG; cat. no. MR-M100; 1:3,000) horseradish peroxidase-conjugated secondary antibodies (both Shanghai MRbiotech, Co., Ltd., Shanghai, China) for 1 h at room temperature, and then visualized using SuperSignal West Pico Stable Peroxide solution (Thermo Fisher Scientific, Inc.). Blots were quantified with ImageJ software (version 1.8.0; National Institutes of Health, Bethesda, MD, USA).
Reverse transcription-quantitative polymerase chain reaction
Total RNA was extracted from PANC-1 cells using TRIzol® reagent (Life Technologies, Thermo Fisher Scientific, Inc.), as previously described (11). The cDNA was synthesized using Superscript II reverse transcriptase (Thermo Fisher Scientific). qPCR was performed using IQ SYBR Green Supermix (Bio-Rad Laboratories, Inc., Hercules, CA, USA) and an iCycleriQTX detection system (Bio-Rad Laboratories, Inc.). The thermocycling conditions were as follows: Denaturing at 95°C for 20 sec; annealing at 58°C for 30 sec; and extension at 72°C for 30 sec, for 43 cycles. All signals were normalized against β-actin and the 2-∆∆Cq method was used to quantify the fold change (12). The sequences of the primers used are as follows: FBXO32 forward, 5′-GAAGCGCTTCCTGGATGAGA-3′ and reverse, 5′-GGAATCCAGAATGGCAGTTG-3′; ATR forward, 5′-GCCGCTCCGATCGTGTAC-3′ and reverse, 5′-TTTGTATGCTCTGTGATAACCTTGTTT-3′; β-actin forward, 5′-CCCTGGCTCCTAGCACCAT-3′ and reverse, 5′-AGAGCCACCAATCCACACAGA-3′.
RNA interference
Lentivirus-based control and gene-specific shRNAs were purchased from Sigma-Aldrich; Merck KGaA. Transfections were performed using Lipofectamine 2000 (Thermo Fisher Scientific, Inc.), as described previously (13,14). A total of 2 µg gene-specific shRNA or shNT (control) were transfected into 293T cells (5×105 cells). After 48 h transfection, the cultured medium of 293T cells was collected and applied to pancreatic cancer cells. Pancreatic cancer cells were cultured in 5% CO2, at 37°C for 48 h, followed by puromycin (0.75 µg/ml; Sigma-Aldrich; Merck KGaA) selection. Cells were collected 72 h post-transfection. The knockdown efficiency was confirmed through western blotting using the aforementioned method. The shRNA sequences were as follows: shNT, 5′-CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG-3′; shFBXO32#1, 5′-CCGGCCAAGGAAAGAGCAGTATGGACTCGAGTCCATACTGCTCTTTCCTTGGTTTTTTG-3′; and shFBXO32#2, 5′-CCGGCTGCCATTCTGGATTCCAGAACTCGAGTTCTGGAATCCAGAATGGCAGTTTTTTG-3′.
Caspase-3 activity measurement
The activity of caspase-3 was measured using a Caspase-3 Colorimetric Protease Assay Sampler kit (cat. no. KHZ0022; Thermo Fisher Scientific, Inc.). The PANC-1 cells were transfected with pcDNA 3.0 or Myc-FBXO32 plasmids or transfected with shNT (control) or FBXO32-specific shRNA according to the previously described protocol. A total of 24 h post transfection, cells were counted and 3–5×106 cells were pelleted per sample. Cells were then treated with gemcitabine (10 µM) for 24 h. Cells were then lysed using 50 µl lysis buffer according to the protocol of the manufacturer (Thermo Fisher Scientific, Inc.), followed by protein quantification using the BCA method. Each cytosol extracted was diluted to a concentration of 50–200 µg protein per 50 µl cell lysis buffer (1–4 mg/ml). 2X reaction buffer (50 µl; containing 10 mM DTT) was added to each sample. The 4 mM DEVD-pNA substrate (5 µl) was added to a final concentration of 200 µM and was incubated at 37°C for 2 h in the dark. Reactions were measured in a microplate reader at a wavelength of 405 nm.
Cell proliferation assay
Cell proliferation was monitored by an MTS assay (Promega Corporation, Madison, WI, USA), according to the manufacturer's protocols. The PANC-1 cells were transfected with pcDNA 3.0 or Myc-FBXO32 plasmids or transfected with shNT (control) or FBXO32-specific shRNA, as previously described. A total of 24 h post transfection, Cells were plated onto 96-well plates at a density of 3,000 cells/well. Cells were treated with different concentrations (0, 1, 10 and 25 µM) of gemcitabine for 24 h prior to measurement. Then 20 µl CellTiter 96R AQueous One Solution Reagent (Promega Corporation) was added to each cell. A total of 50 min after incubation (37°C in a cell incubator), cell proliferation was measured using a microplate reader at a wavelength of 490 nm.
Cell cycle analysis
The PANC-1 cells were transfected with pcDNA 3.0 or Myc-FBXO32 plasmids or transfected with shNT (control) or FBXO32-specific shRNA. According to the previously described protocol. At 24 h post transfection, cells were treated with gemcitabine (10 µM) and cultured in 5% CO2 at 37°C in a 95% humidity incubator for another 24 h. Following treatment with trypsin, cells were harvested and washed with 1×PBS, prior to being fixed with 70% ethanol at 4°C overnight. The next day, the cells were washed with 1X PBS and stained with propidium iodide (10 µg/ml in 1X PBS) at room temperature for 10 min (Sigma-Aldrich; Merck KGaA). The cell cycle was analyzed by flow cytometry using a FACSCalibur system (BD Biosciences, Franklin Lakes, NJ, USA). The cell cycle fraction data were additionally analyzed using Modfit LT (Verity Software House, Inc., Topsham, ME, USA).
Statistical analysis
One-way analysis, followed by Tukey's multiple comparisons test, was performed for multiple comparisons. Student's t-test was performed for single comparisons. P<0.05 was considered to indicate a statistically significant difference.
Results
FBXO32 regulates ATR expression in pancreatic cancer cells
The present study initially examined the association between FBXO32 and ATR in pancreatic cancer cells. PANC-1 and MIA PaCa-2 cells were treated with non-specific control or FBXO32-specific shRNA. Following the effective knockdown of FBXO32, the protein expression level of ATR was increased in these cells (Fig. 1A), while knockdown of FBXO32 did not have any effect on ATR mRNA levels in PANC-1 cells (Fig. 1B). Furthermore, restored FBXO32 expression through shRNA-resistant FBXO32 plasmid reversed the effect of FBXO32 knockdown-induced ATR protein level change in PANC-1 cells. As it has been reported that c-Myc was one of the FBXO32 targets for degradation (15), the c-Myc protein levels were examined as a positive control (Fig. 1C and D). These results suggested that FBXO32 may regulate the protein level of ATR in pancreatic cancer cells.
Figure 1.

FBXO32 regulates ATR expression in pancreatic cancer cells. (A) PANC-1 and MIA PaCa-2 cells were transfected with control or one of two independent FBXO32-specific shRNAs. A total of 48 h after transfection, cells were harvested for western blot analysis. (B) PANC-1 cells were transfected with control or one of two independent FBXO32-specific shRNAs. A total of 48 h after transfection, cells were harvested for reverse transcription-quantitative polymerase chain reaction analysis of ATR and FBXO32 mRNAs. Data are mean values ± SD from three replicates (n=3). *P<0.05 vs. shControl. (C) PANC-1 cells were infected with control or one independent FBXO32-specific shRNA. A total of 24 h after transfection, cells were transfected with indicated plasmids for a further 24 h, followed by western blot analysis. (D) Protein levels of ATR in transfected cells were quantified relative to untransfected PANC-1 cells. FBXO32, F-Box protein 32; shRNA/sh, short hairpin RNA; n.s., not significant.
FBXO32 interacts with ATR in pancreatic cancer cells
It has been reported previously that FBXO32 was an E3 ubiquitin ligase that regulates the ubiquitination of substrates (15,16). Since substrate binding is a key event for E3 ligase-mediated ubiquitination and subsequent proteasome degradation, the present study examined the interaction between FBXO32 and ATR using a co-immunoprecipitation (co-IP) assay. The interaction between endogenous ATR and FBXO32 in PANC-1 cells was confirmed by reciprocal co-IP assays (Fig. 2A and B). These results suggested that FBXO32 interacts with ATR in pancreatic cancer cells and these data promoted the investigation into whether or not FBXO32 functions as an E3 ubiquitin ligase of ATR in pancreatic cancer cells.
Figure 2.

FBXO32 interacts with ATR in pancreatic cancer cells. Western blot analysis of reciprocal co-immunoprecipitation of endogenous (A) FBXO32 and (B) ATR proteins in PANC-1 cells. FBXO32, F-Box protein 32; IgG, immunoglobulin G; IP, immunoprecipitation.
ATR is a degradation substrate of the E3 ubiquitin ligase FBXO32
The present study systematically investigated whether FBXO32 degrades ATR protein in pancreatic cancer cells. ATR was co-transfected with 1 µg/1×106 cells or 3 µg/1×106 cells transfected with FBXO32 plasmids in pancreatic cancer PANC-1 cells. Ectopically expressed ATR was downregulated by co-expression of FBXO32 and this process was blocked by treatment with the proteasome inhibitor MG132 (Fig. 3A and B), which indicated that FBXO32 decreased ATR protein expression levels via the proteasome pathway. In order to determine whether FBXO32 regulates ATR polyubiquitination, endogenous FBXO32 was knocked down in PANC-1 cells, leading to decreased polyubiquitination of ATR (Fig. 3C), while restored FBXO32 expression through the shRNA-resistant FBXO32 plasmid increased the polyubiquitination of ATR. Taken together, these results suggested that ATR is a degradation substrate of E3 ubiquitin ligase FBXO32 in pancreatic cancer cells.
Figure 3.
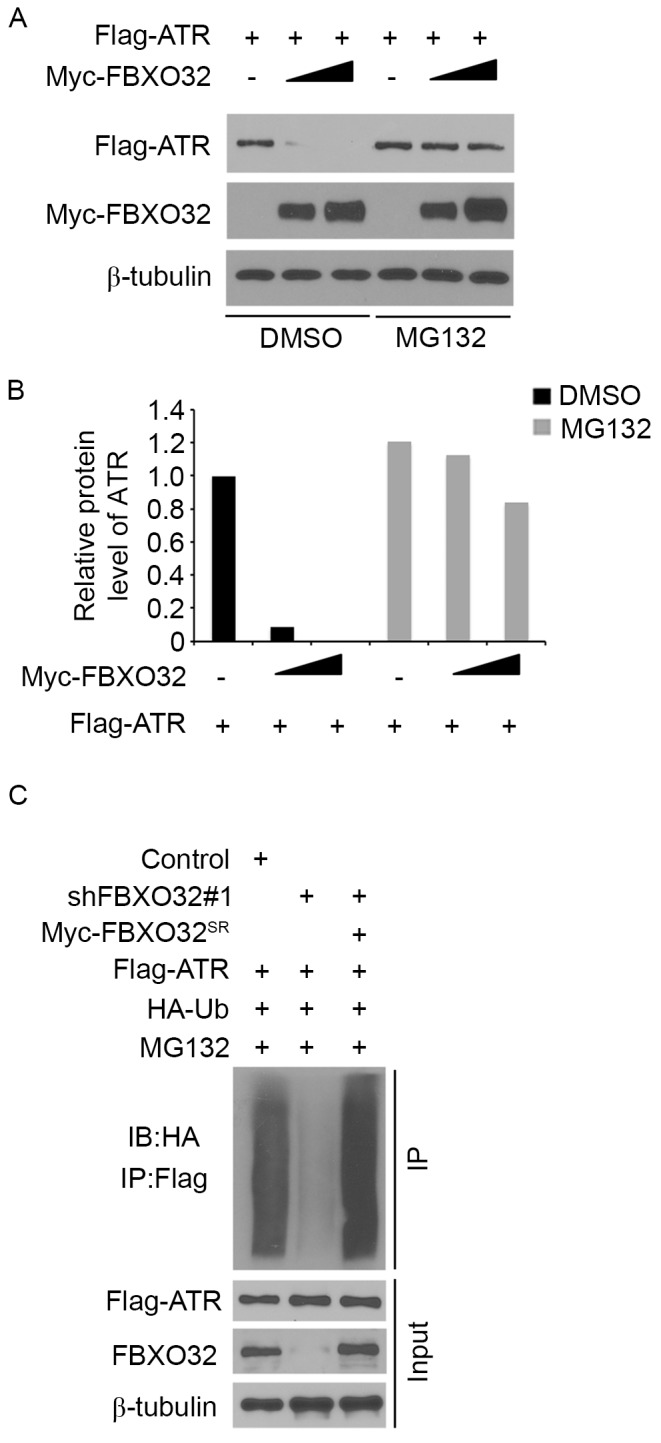
ATR is a degradation substrate of E3 ubiquitin ligase FBXO32. (A) Western blot analysis of whole cell lysates of PANC-1 cells transfected with the indicated constructs. Cells were treated with or without 20 µM of MG132 for 8 h before harvest. (B) Quantification of protein levels of ATR in PANC-1 cells. (C) PANC-1 cells were transfected with indicated plasmids for 48 h followed by treatment with 20 µM MG132 for 8 h. Immunoprecipitated Flag-ATR proteins were analyzed by western blot analysis for ubiquitination. FBXO32, F-Box protein 32; DMSO, dimethyl sulfoxide; HA, hemagglutinin; Ub, ubiquitin; IB, immunoblot; IP, immunoprecipitation.
FBXO32 regulates the DDR induced by gemcitabine in pancreatic cancer
Gemcitabine is one of the first-line therapeutic agents for pancreatic cancer (12). Gemcitabine inhibits ribonucleotide reductase and is an active chemotherapeutic agent that disrupts DNA replication (17). Gemcitabine activated checkpoint signaling pathways and acts as a strong activator of ATR (17,18). Treatment with an ATR inhibitor increased the anticancer activity of gemcitabine in pancreatic cancer cells (18). In order to investigate the role of FBXO32 in the regulation of DDR induced by gemcitabine in pancreatic cancer, pancreatic cancer cells were treated with gemcitabine alone or in combination with FBXO32 overexpression or knockdown. It was revealed that overexpression of FBXO32 led to increased expression of cleaved PARP (a pro-apoptotic protein) and P53 protein, as well as an increase in the caspase-3 activity induced by gemcitabine (Fig. 4A and B) in pancreatic cancer PANC-1 cells. Next, a cell viability assay was performed in FBXO32-overexpressing PANC-1 cells treated with different concentrations of gemcitabine. The results demonstrated that lower concentrations of gemcitabine were required to suppress the cell proliferation in FBXO32-overexpressing PANC-1 cells, compared with the normal group (Fig. 4C). The present study indicated that FBXO32 serves an important role in sensitizing pancreatic cancer cells to gemcitabine treatment. By contrast, the knockdown of FBXO32 decreased cleaved PARP and p53 expression, decreased caspase-3 activity and required a higher concentration of gemcitabine to suppress the viability of PANC-1 cells when treated with gemcitabine (Fig. 4D-F). Furthermore, Fig. 4G demonstrates that treatment of PANC-1 cells with gemcitabine resulted in S-phase cell cycle arrest and increased ratio of cells in the S phase. Overexpression of FBXO32 enhanced the effect of gemcitabine while knockdown of FBXO32 attenuated this effect in PANC-1 cells. Taken together, these results indicated that FBXO32 regulates the DDR induced by gemcitabine in pancreatic cancer.
Figure 4.

FBXO32 regulates the DNA damage response induced by gemcitabine in pancreatic cancer. PANC-1 cells were transfected with indicated constructs. Cells were treated with gemcitabine (10 µM) for 24 h prior to harvest. Cells were harvested for (A) western blot analysis and (B) measurement of caspase-3 activity. Data are presented as the mean ± standard deviation of two replicate experiments. *P<0.05 for pcDNA 3.0+gemcitabine treatment vs. Myc-FBXO32+ Gemcitabine treatment. (C) PANC-1 cells were transfected with indicated constructs. Cells were treated with different concentrations (0, 1, 10 and 25 µM) of gemcitabine for 24 h prior to measurement. Cell viability was measured using an MTS assay. Data are presented as the mean ± standard deviation of three replicate experiments. PANC-1 cells were transfected with control or one of two independent FBXO32-specific shRNAs. At 24 h after transfection, cells were treated with gemcitabine (10 µM) for 24 h prior to harvest. Cells were harvested for (D) western blot analysis and (E) measurement of caspase-3 activity. Data are presented as the mean ± standard deviation of two replicate experiments. *P<0.05 for shControl+gemcitabine treatment vs. shFBXO32+gemcitabine treatment. (F) PANC-1 cells were transfected with control or one of two independent FBXO32-specific shRNAs. At 24 h after transfection, cells were treated with different concentrations (0, 1, 10 and 25 µM) of gemcitabine for 24 h prior to measurement. Cell viability was measured using an MTS assay. Data are presented as the mean ± standard deviation of three replicate experiments. (G) PANC-1 cells were transfected with indicated constructs. Cells were treated with distilled water or gemcitabine (20 µM) for 24 h. The cell cycle was analyzed after staining with PI. The cell cycle distribution was analyzed using ModFit LT software. Data from a representative experiment (from a total of two) are presented. Student's t-test was performed for single comparisons and one-way analysis of variance, followed by Tukey's multiple comparisons test was performed for multiple comparisons. *P<0.05 for comparing S phase in groups as follows: shControl vs. shControl+gemcitabine treatment, Myc-FBXO32 vs. Myc-FBXO32+gemcitabine treatment, shFBXO32 vs. shFBXO32+gemcitabine treatment, shControl+gemcitabine treatment vs. Myc-FBXO32+gemcitabine treatment, shControl+gemcitabine treatment vs. shFBXO32+gemcitabine treatment, Myc-FBXO32+gemcitabine treatment+shFBXO32+gemcitabine treatment.
Discussion
Oncogene activation has been revealed to generate replicative stress in early stages of tumor progression (19). The principle of using DNA damage to kill tumor cells has been applied for decades (20). Chemotherapeutic agents and ionizing radiation induce DNA damage, thereby activating cell cycle checkpoint pathways (21). Cell cycle checkpoint pathways are involved in regulating cell cycle progression and repairing of damaged DNA (22). This response is an important mechanism that aids cancer cells in surviving anticancer treatment (22). ATR serves an important role in DDR and activates CHK1 following replication fork stalling, leading to cell cycle arrest. As a result, ATR-CHK1 pathway components are considered to be promising therapeutic targets for enhancing the effectiveness of replication inhibitors (23). Although significant progress has been made toward understanding the function and deregulation of ATR in cancer cells, there has been little investigation into the modification of ATR after transcription. The present study provided evidence that FBXO32 interacts with ATR and regulates ATR expression via ubiquitination and degradation in pancreatic cancer cells.
FBXO32 contains the F-box domain and functions as one component of a skp1, cullin, F-box protein ubiquitin ligase complex (24,25). FBXO32 acts as a muscle-specific E3 ligase by targeting multiple substrates for ubiquitination and degradation (8,9). Furthermore, FBXO32 has been demonstrated to be involved in tumorigenesis. FBXO32 expression is decreased in ovarian cancer (26) and esophageal squamous cell carcinoma (27). Overexpression of FBXO32 in ovarian cancer cells inhibits colony formation in culture and xenograft tumor growth in mice (26). Promoter methylation of FBXO32 is responsible for the downregulation of FBXO32 in cancer cells (27). EZH2 has been reported to suppress FBXO32 expression (28). The present study demonstrated that FBXO32 acted as an E3 ligase of ATR in pancreatic cancer cells. Furthermore, FBXO32 interacts with p21 to induce p21 protein degradation and regulates DDR (16). Gemcitabine is one of the first-line therapeutic agents for pancreatic cancer. It activates checkpoint signaling pathways and acts as a strong activator of ATR (17). The results of the present study suggested that FBXO32 regulates the DDR induced by gemcitabine in pancreatic cancer, partially through inducing ATR degradation. These observations have revealed important aspects of the function of FBXO32 in tumorigenesis.
Taken together, the results of the present study demonstrated that FBXO32 acts as an E3 ubiquitin ligase of ATR and that it regulates the DDR induced by gemcitabine in pancreatic cancer, possibly through inducing ATR degradation. Therefore, we hypothesized that FBXO32 may be a potential therapeutic target for pancreatic cancer.
Acknowledgements
The present study was supported by the Scientific Research Training Program for Young Talents of Union Hospital, Tongji Medical College, Huazhong University of Science and Technology.
Funding
The present study was supported by the Chinese National Natural Science Foundation (grant no. 81702374).
Availability of data and materials
The datasets generated/analyzed in the present study are available upon reasonable request from the corresponding author.
Authors' contributions
HW and XJ conceived the study. XJ, CY and PF performed the experiments. SZ and HY performed bioinformatics and statistics analyses. XJ and HW wrote the manuscript.
Ethics statement and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.The Lancet Oncology: Pancreatic cancer in the spotlight. Lancet Oncol. 2014;15:241. doi: 10.1016/S1470-2045(14)70097-X. [DOI] [PubMed] [Google Scholar]
- 2.Blum R, Kloog Y. Metabolism addiction in pancreatic cancer. Cell Death Dis. 2014;5:e1065. doi: 10.1038/cddis.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jin X, Pan Y, Wang L, Ma T, Zhang L, Tang AH, Billadeau DD, Wu H, Huang H. Fructose-1,6-bisphosphatase inhibits ERK activation and bypasses gemcitabine resistance in pancreatic cancer by blocking IQGAP1-MAPK interaction. Cancer Res. 2017;77:4328–4341. doi: 10.1158/0008-5472.CAN-16-3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toledo LI, Murga M, Fernandez-Capetillo O. Targeting ATR and Chk1 kinases for cancer treatment: A new model for new (and old) drugs. Mol Oncol. 2011;5:368–373. doi: 10.1016/j.molonc.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cimprich KA, Shin TB, Keith CT, Schreiber SL. cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proc Natl Acad Sci USA. 1996;93:2850–2855. doi: 10.1073/pnas.93.7.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–1459. [PMC free article] [PubMed] [Google Scholar]
- 7.Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, Friend SH. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 1998;17:159–169. doi: 10.1093/emboj/17.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lagirand-Cantaloube J, Offner N, Csibi A, Leibovitch MP, Batonnet-Pichon S, Tintignac LA, Segura CT, Leibovitch SA. The initiation factor eIF3-f is a major target for atrogin1/MAFbx function in skeletal muscle atrophy. EMBO J. 2008;27:1266–1276. doi: 10.1038/emboj.2008.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xie P, Guo S, Fan Y, Zhang H, Gu D, Li H. Atrogin-1/MAFbx enhances simulated ischemia/reperfusion-induced apoptosis in cardiomyocytes through degradation of MAPK phosphatase-1 and sustained JNK activation. J Biol Chem. 2009;284:5488–5496. doi: 10.1074/jbc.M806487200. [DOI] [PubMed] [Google Scholar]
- 10.Jin X, Pan Y, Wang L, Zhang L, Ravichandran R, Potts PR, Jiang J, Wu H, Huang H. MAGE-TRIM28 complex promotes the Warburg effect and hepatocellular carcinoma progression by targeting FBP1 for degradation. Oncogenesis. 2017;6:e312. doi: 10.1038/oncsis.2017.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tian S, Li P, Sheng S, Jin X. Upregulation of pyruvate kinase M2 expression by fatty acid synthase contributes to gemcitabine resistance in pancreatic cancer. Oncol Lett. 2018;15:2211–2217. doi: 10.3892/ol.2017.7598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 13.Jin X, Yang C, Fan P, Xiao J, Zhang W, Zhan S, Liu T, Wang D, Wu H. CDK5/FBW7-dependent ubiquitination and degradation of EZH2 inhibits pancreatic cancer cell migration and invasion. J Biol Chem. 2017;292:6269–6280. doi: 10.1074/jbc.M116.764407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jin X, Tian S, Li P. Histone acetyltransferase 1 promotes cell proliferation and induces cisplatin resistance in hepatocellular carcinoma. Oncol Res. 2017;25:939–946. doi: 10.3727/096504016X14809827856524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mei Z, Zhang D, Hu B, Wang J, Shen X, Xiao W. FBXO32 targets c-Myc for proteasomal degradation and inhibits c-Myc activity. J Biol Chem. 2015;290:16202–16214. doi: 10.1074/jbc.M115.645978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu Z, Lee ST, Qiao Y, Li Z, Lee PL, Lee YJ, Jiang X, Tan J, Aau M, Lim CZ, Yu Q. Polycomb protein EZH2 regulates cancer cell fate decision in response to DNA damage. Cell Death Differ. 2011;18:1771–1779. doi: 10.1038/cdd.2011.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karnitz LM, Flatten KS, Wagner JM, Loegering D, Hackbarth JS, Arlander SJ, Vroman BT, Thomas MB, Baek YU, Hopkins KM, et al. Gemcitabine-induced activation of checkpoint signaling pathways that affect tumor cell survival. Mol Pharmacol. 2005;68:1636–1644. doi: 10.1124/mol.105.012716. [DOI] [PubMed] [Google Scholar]
- 18.Woods D, Turchi JJ. Chemotherapy induced DNA damage response: Convergence of drugs and pathways. Cancer Biol Ther. 2013;14:379–389. doi: 10.4161/cbt.23761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, Venere M, Ditullio RA, Jr, Kastrinakis NG, Levy B, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 20.Matt S, Hofmann TG. The DNA damage-induced cell death response: A roadmap to kill cancer cells. Cell Mol Life Sci. 2016;73:2829–2850. doi: 10.1007/s00018-016-2130-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wagner JM, Kaufmann SH. Prospects for the use of ATR inhibitors to treat cancer. Pharmaceuticals (Basel) 2010;3:1311–1334. doi: 10.3390/ph3051311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sclafani RA, Holzen TM. Cell cycle regulation of DNA replication. Annu Rev Genet. 2007;41:237–280. doi: 10.1146/annurev.genet.41.110306.130308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tao Y, Leteur C, Yang C, Zhang P, Castedo M, Pierré A, Golsteyn RM, Bourhis J, Kroemer G, Deutsch E. Radiosensitization by Chir-124, a selective CHK1 inhibitor: Effects of p53 and cell cycle checkpoints. Cell Cycle. 2009;8:1196–1205. doi: 10.4161/cc.8.8.8203. [DOI] [PubMed] [Google Scholar]
- 24.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science. 2001;294:1704–1708. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 25.Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA. 2001;98:14440–14445. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chou JL, Su HY, Chen LY, Liao YP, Hartman-Frey C, Lai YH, Yang HW, Deatherage DE, Kuo CT, Huang YW, et al. Promoter hypermethylation of FBXO32, a novel TGF-beta/SMAD4 target gene and tumor suppressor, is associated with poor prognosis in human ovarian cancer. Lab Invest. 2010;90:414–425. doi: 10.1038/labinvest.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo W, Zhang M, Shen S, Guo Y, Kuang G, Yang Z, Dong Z. Aberrant methylation and decreased expression of the TGF-β/Smad target gene FBXO32 in esophageal squamous cell carcinoma. Cancer. 2014;120:2412–2423. doi: 10.1002/cncr.28764. [DOI] [PubMed] [Google Scholar]
- 28.Ciarapica R, De Salvo M, Carcarino E, Bracaglia G, Adesso L, Leoncini PP, Dall'Agnese A, Walters ZS, Verginelli F, De Sio L, et al. The Polycomb group (PcG) protein EZH2 supports the survival of PAX3-FOXO1 alveolar rhabdomyosarcoma by repressing FBXO32 (Atrogin1/MAFbx) Oncogene. 2014;33:4173–4184. doi: 10.1038/onc.2013.471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated/analyzed in the present study are available upon reasonable request from the corresponding author.