

ABSTRACT
Infectious bursal disease virus (IBDV) belongs to the Birnaviridae family and is the etiological agent of a highly contagious and immunosuppressive disease (IBD) that affects domestic chickens (Gallus gallus). IBD or Gumboro disease leads to high rates of morbidity and mortality of infected animals and is responsible for major economic losses to the poultry industry worldwide. IBD is characterized by a massive loss of IgM-bearing B lymphocytes and the destruction of the bursa of Fabricius. The molecular bases of IBDV pathogenicity are still poorly understood; nonetheless, an exacerbated cytokine immune response and B cell depletion due to apoptosis are considered main factors that contribute to the severity of the disease. Here we have studied the role of type I interferon (IFN) in IBDV infection. While IFN pretreatment confers protection against subsequent IBDV infection, the addition of IFN to infected cell cultures early after infection drives massive apoptotic cell death. Downregulation of double-stranded RNA (dsRNA)-dependent protein kinase (PKR), tumor necrosis factor alpha (TNF-α), or nuclear factor κB (NF-κB) expression drastically reduces the extent of apoptosis, indicating that they are critical proteins in the apoptotic response induced by IBDV upon treatment with IFN-α. Our results indicate that IBDV genomic dsRNA is a major viral factor that contributes to the triggering of apoptosis. These findings provide novel insights into the potential mechanisms of IBDV-induced immunosuppression and pathogenesis in chickens.
IMPORTANCE IBDV infection represents an important threat to the poultry industry worldwide. IBDV-infected chickens develop severe immunosuppression, which renders them highly susceptible to secondary infections and unresponsive to vaccination against other pathogens. The early dysregulation of the innate immune response led by IBDV infection and the exacerbated apoptosis of B cells have been proposed as the main factors that contribute to virus-induced immunopathogenesis. Our work contributes for the first time to elucidating a potential mechanism driving the apoptotic death of IBDV-infected cells upon exposure to type I IFN. We provide solid evidence about the critical importance of PKR, TNF-α, and NF-κB in this phenomenon. The described mechanism could facilitate the early clearance of infected cells, thereby aiding in the amelioration of IBDV-induced pathogenesis, but it could also contribute to B cell depletion and immunosuppression. The balance between these two opposing effects might be dramatically affected by the genetic backgrounds of both the host and the infecting virus strain.
KEYWORDS: apoptosis, infectious bursal disease virus, interferon, pathogenesis
INTRODUCTION
Infectious bursal disease virus (IBDV), the best-characterized member of the Birnaviridae family, is the etiological agent of a highly contagious and immunosuppressive disease (IBD) that affects juvenile domestic chickens (Gallus gallus) (1, 2), leading to high rates of morbidity and mortality of infected animals. IBD or Gumboro disease is responsible for major economic losses to the poultry industry worldwide (3). The severity of the disease depends on several factors, i.e., the virulence of virus strain, the age of the infected animal, and the chicken breed. Two IBDV serotypes have been identified, serotype 1, which encompasses virus strains that cause disease but differ widely in their degrees of virulence, and serotype 2, which encompasses viruses that are nonpathogenic in domestic chickens (3). Chickens are highly susceptible to infection between 3 and 6 weeks after hatching. The main target cells of the virus are immature IgM-bearing B lymphocytes (4), but macrophages and monocytes are also infected (5). Virus transmission occurs through the fecal-oral route, and it is thought that ingested virus infects gut macrophages, which bring the virus to the bursa of Fabricius, the chief lymphoid organ in developing chickens and the main target of the virus (3, 6–8). IBDV infection causes a rapid depletion of the bursal B cell population and the atrophy of this lymphoid organ. Other secondary lymphoid organs, e.g., spleen, thymus, and cecal tonsils, are also affected (10). As a consequence of specific damage to immune cells, surviving chicks present severe immunosuppression, being highly susceptible to secondary infections and unresponsive to vaccination against other pathogens (10). The virus causes an acute local infection, which peaks at between 2 and 5 days postinfection (p.i.), characterized by B cell depletion in bursal follicles and the infiltration of T cells, which become activated and overexpress interferon gamma (IFN-γ) and other cytokines (10, 11). Secreted IFN-γ could in turn activate macrophages, leading to the production of proinflammatory factors, e.g., nitric oxide and interleukin 6 (IL-6), which can contribute to tissue damage (6). It has been described that an exacerbated expression of IFN-γ correlates with an aggravation of the clinical signs of the disease and the concomitant T cell immunosuppression (11).
IBDV particles are naked icosahedrons, with a diameter of ∼650 to 700 Å, containing two double-stranded RNA (dsRNA) segments of 3.2 and 2.8 kbp (segments A and B, respectively). During infection, five virus-encoded mature proteins are produced (VP1 to VP5) (12). Most of these proteins are encoded by segment A, which harbors two partially overlapping open reading frames. The first one encodes a nonstructural protein, VP5 (17 kDa), which is dispensable for virus replication in cultured cells (13) but essential for cell-to-cell transmission in vitro (14) and for viral pathogenesis in vivo (15), while the second one codes for three proteins synthesized as a polyprotein precursor (110 kDa). The viral polyprotein is cotranslationally processed by the viral protease VP4 (16, 17), giving rise to the VP2 precursor, termed pVP2 (512 residues; 54.4 kDa), which is subsequently processed to produce mature VP2, VP3 (29 kDa), and VP4 (27 kDa). Segment B contains a single open reading frame encoding VP1 (98 kDa), the RNA-dependent RNA polymerase (18).
Unlike prototypal dsRNA viruses, e.g., reoviruses, Birnaviridae family members lack the T=2 core structure. Their genome is structured into ribonucleoprotein (RNP) complexes, where the dsRNA is wrapped by the VP3 protein and complexed with the polymerase VP1. IBDV RNPs are functionally competent for RNA synthesis both in vivo and in vitro (19, 20).
The molecular bases of IBDV pathogenicity are still poorly understood. Nonetheless, there have been many reports indicating the involvement of apoptotic processes in virus-caused pathogenesis. Apoptosis of IBDV-infected cells, both in vivo, in bursal, thymic, and splenic cells from infected chickens or chicken embryos (21–24), and in vitro, in chicken peripheral blood lymphocytes (25) and in different cultured cells, including chicken embryo fibroblasts (26), chicken B lymphocytes (27), and mammalian Vero cells, has been reported (25). Moreover, apoptosis in bursal lymphocytes and thymic cells lacking detectable levels of IBDV proteins has also been documented (28–30). All these findings indicate that in vivo, IBDV induces the apoptosis of infected and probably also of uninfected bystander cells. This might contribute to the fast depletion of B lymphocytes and the destruction of the bursa of Fabricius that leads to immunosuppression. From these observations, the participation of immunological mediators in apoptosis induction has been suggested. Moreover, the viral polypeptides VP5, VP3, and VP2 have been related to the induction and/or control of the IBDV-induced apoptotic response. According to different reports, VP5 may play a dual role, preventing IBDV-induced apoptosis at the early stage of virus infection (31) and also acting as a proapoptotic polypeptide to facilitate virus dissemination (32). It has also been shown that the individual expression of VP2 activates the dsRNA-dependent protein kinase (PKR), resulting in a protein synthesis blockade and apoptotic cell death (33). It is noteworthy that this effect is efficiently blocked by the coexpression of the IBDV RNA-binding VP3 polypeptide (34). In addition, VP3 plays an important role in counteracting IBDV-induced innate antiviral host cell responses, being able to block the induction of IFN expression (34).
As mentioned above, there is growing evidence for a role of innate immunity in the pathogenesis of IBDV. IFNs are essential players in the innate immune response, being one of the primary defense systems against viral infections. IFNs are a family of cytokines exerting pleiotropic biological effects by transducing several intracellular pathways. IFNs are induced upon the recognition of pathogen-associated molecular patterns (PAMPs) by cellular sensors of the retinoic acid-inducible protein (RIG-I)-like receptor (RLR) family members RIG-I and melanoma differentiation-associated gene 5 (MDA5) or Toll-like receptors (TLRs). Upon binding to their receptors, IFNs mediate their effect by activating a signaling cascade that leads to the expression of IFN-stimulated genes (ISGs). ISGs encode a large number of proteins implicated in antiviral, immunomodulatory, and cell cycling-inhibitory effects, but also proteins involved in apoptosis. Among the proteins implicated in apoptosis are tumor necrosis factor alpha (TNF-α)-related apoptosis-inducing ligand (TRAIL), Fas-FasL, the dsRNA-activated protein kinase (PKR), and the 2′-5′A-oligoadenylate synthetase (OAS).
In addition to the well-characterized role of the IFN system for host defense against viruses, there is also a growing body of information that points to detrimental effects of type I IFNs on the outcomes of virus-induced diseases caused by certain viruses, either by inducing immunosuppression or by triggering inflammation and tissue damage (35).
Despite extensive work in the field to understand the bases for IBDV pathogenesis, the molecular mechanisms underlying the IBDV-host interaction and, specifically, the role of cytokines in IBDV pathogenicity are still unknown. Deciphering the interaction between the virus and the innate immune system and how apoptosis is triggered in infected cells will help us to understand the basis of pathogenesis and thus to design new strategies to control IBDV infection. Here we describe that exposure of IBDV-infected cells to IFN-α causes massive apoptosis. We have also identified that PKR, TNF-α, and nuclear factor κB (NF-κB) are critical proteins in the apoptotic response induced by IBDV upon treatment with IFN-α.
RESULTS
IBDV is sensitive to the antiviral action of IFN-α in HeLa cells.
To study the sensitivity of IBDV to the antiviral action of IFN-α, we used HeLa cells, which have been successfully used to study different steps of the IBDV life cycle (36). HeLa cells were pretreated with increasing amounts of human IFN-α (hIFN-α), ranging from 1 to 1,000 IU/ml, for 16 h before infection. Infections were performed by using two multiplicities of infection (MOIs), 0.05 and 2 PFU/cell, and cultures were harvested at 24 h p.i. IBDV replication was monitored by examining the expression of the VP3 protein by Western blotting. As shown in Fig. 1A, 10 and 100 IU/ml IFN were sufficient to block virus replication after infection at MOIs of 0.05 and 2, respectively, as observed by the absence of VP3 in the Western blots. In addition, extracellular virus titers from IFN-treated cultures revealed that virus yields were reduced ca. 20-fold and 65-fold in cells pretreated with 100 IU/ml of IFN and infected at MOIs of 0.05 and 2, respectively (Fig. 1B). These results show that, as previously described (37–40), IBDV does not replicate efficiently in cells already displaying the antiviral program triggered by IFN.
FIG 1.

IBDV is sensitive to the antiviral action of IFN in HeLa cells. HeLa cells were pretreated with different doses of hIFN-α (0, 1, 10, 100, and 1,000 IU/ml), as indicated, for 16 h before infection with two MOIs of IBDV, 0.05 or 2 PFU/cell, and both the culture medium and infected cells were harvested at 24 h p.i. (A) Western blot analysis of total cell extracts with anti-VP3 antibody. Detection of β-actin in the same cell extracts was used for a protein loading control. (B) Extracellular virus yields. Supernatants from infected cells were used for virus titration by a plaque assay. Striped bars, IFN-untreated cells; solid bars, infected cell samples pretreated with IFN. Bars indicate means ± standard deviations based on duplicate samples from two independent experiments. * indicates a P value of <0.05, as determined by unpaired Student's t test.
IFN-α treatment of IBDV-infected HeLa cells causes apoptosis.
To investigate whether the virus is able to counteract the antiviral activity triggered by IFN once infection has been established, we performed a different set of experiments. For this, HeLa cells were mock infected or infected with IBDV at an MOI of 2 and subsequently treated with hIFN-α (1,000 IU/ml) at 3, 6, 9, or 12 h p.i. Samples were harvested at 24 h p.i. For simplicity, henceforth samples from mock-infected cells (M) treated with IFN are denoted M+3 and M+6, etc., where the number indicates the time in hours p.i. at which IFN was added to the culture. Similarly, samples from IBDV-infected cells (I) treated with IFN are denoted I+3 and I+6, etc.
Strikingly, infected cells treated with IFN showed a strong cytopathic effect (CPE) not observed in either infected cells without IFN or mock-infected cells treated or not with IFN (Fig. 2A). CPE was more pronounced in cultures treated with hIFN-α at 3 or 6 h p.i. (I+3 or I+6) than in those treated at later times (I+9 or I+12). To discriminate between live and dead cells in these cultures, we used the Live/Dead cell imaging kit. Images were recorded at 24 h p.i. As shown in Fig. 2B, significant numbers of dead cells were observed only in IFN-treated cultures. The rate of cell death was markedly higher when IFN was added at an early time p.i. (I+3) than when it was added at a late time (I+12). Morphological changes observed in IFN-treated infected cells were reminiscent of those occurring during apoptosis. The poly(ADP-ribose) polymerase (PARP) protein, a well-known substrate for caspase 3 cleavage, is considered to be a hallmark of apoptosis (41). Thus, we examined the extent of PARP cleavage during apoptosis by Western blotting. While there was only marginal PARP cleavage at this time p.i. in IBDV-infected cells not treated with IFN, similar to what was observed for all lanes corresponding to mock-infected cells, extensive PARP cleavage was observed in all the samples of infected cells treated with IFN, although differences in the extents of cleavage were detected, being higher in the I+3 and I+6 cell samples (Fig. 2C). Similarly, when apoptosis was quantified by determining caspase 3/7 activity with the Caspase-Glo 3/7 assay kit (Fig. 2D), apoptosis was almost negligible in IBDV-infected cultures, but it was extensive in infected cultures treated with IFN, again being higher in the I+3 and I+6 cell samples than in the I+9 or I+12 ones. Moreover, we used different amounts of hIFN-α, ranging from 1 to 105 IU, and the extents of apoptotic cell death at 24 h p.i., measured with the Caspase-Glo 3/7 assay kit, were similar in samples treated with doses of >100 IU/ml (Fig. 2E), and they were also in a range similar to that for samples treated with a well-known apoptotic inducer, such as staurosporine, used as a control (Fig. 2F).
FIG 2.

IFN treatment triggers apoptosis of IBDV-infected HeLa cells. HeLa cells mock infected or infected with IBDV (MOI of 2) were treated with hIFN-α (1,000 IU/ml) at 3, 6, 9, or 12 h p.i. (samples named throughout the text M+3, M+6, M+9, and M+12 and I+3, I+6, I+9, and I+12, respectively), as indicated, or remained untreated (−) (named throughout the text M and I, respectively). Cells were analyzed at 24 h p.i. by using different assays. (A) Phase-contrast microscopy. (B) Fluorescence microscopy after incubation with the Live/Dead cell imaging reagent to discriminate live (green) from dead (red) cells. (C) Western blot analysis of cells mock infected or infected with IBDV and treated with hIFN-α at the indicated times p.i. with different antibodies: anti-PARP, anti-total PKR (t-PKR), anti-phosphorylated (Thr446) PKR (p-PKR), anti-total eIF2α (t-eIF2α), anti-phosphorylated (Ser52) eIF2α (p-eIF2α), anti-Mx, anti-ISG-56, and anti-VP3. The PARP cleavage product is denoted c-PARP. Antibodies to β-actin were used for a protein loading control. (D) Apoptosis was measured from duplicate samples by using the Caspase-Glo 3/7 assay kit, and each determination was carried out in duplicate. Caspase values from infected cell samples were normalized to those from mock-infected cells. Bars indicate means ± standard deviations based on duplicate samples from two independent experiments. Striped bars, infected cell samples not treated with IFN; solid bars, infected cell samples treated with IFN. (E) Analysis of the IFN dose-response effect on caspase 3/7 activity. Mock-infected or infected cells were treated with increasing doses (10-fold) of hIFN-α (from 1 to 105 IU/ml), and apoptosis was measured at 24 h p.i. by using the Caspase-Glo 3/7 assay kit. (F) Comparative analysis of apoptosis levels in IBDV-infected HeLa cells treated with IFN (1,000 IU/ml) at 3 h p.i. and uninfected cells treated with different doses of staurosporine (STS) (0.5, 1, and 2 μM), collected at 24 h p.i. or posttreatment, respectively. * and ** indicate P values of <0.05 and <0.01, respectively as determined by unpaired Student's t test.
From these results, we conclude that IBDV infection per se does not cause significant apoptosis in HeLa cells at 24 h p.i. Nonetheless, IFN treatment of IBDV-infected cells triggers a massive apoptotic response, and the extent of cell death is significantly greater when IFN is added early during the infection cycle. Indeed, the fact that uninfected cells treated with IFN do not undergo apoptosis indicates that a viral factor is required for triggering apoptosis.
Patterns of expression of ISGs on IBDV-infected cells after IFN-α treatment.
We next analyzed the effect of IBDV infection on the expression of several ISGs after treatment with hIFN-α by Western blotting. For this, HeLa cells infected with IBDV (MOI of 2) were treated with 1,000 IU/ml of hIFN-α at different times p.i. (3, 6, 9, or 12 h p.i.), and cell extracts were collected at 24 h p.i. As shown in Fig. 2C, we observed a general reduction in the expression of PKR in infected cells compared to mock-infected cells upon treatment with IFN, indicating that IBDV downregulates the expression of this protein. In addition, PKR phosphorylation (p-PKR) occurred only in cells infected and treated with IFN, being higher when IFN was added at an early time p.i. In concordance with the observed PKR activation, the alpha subunit of eukaryotic initiation factor 2 (eIF2α) was phosphorylated in all the samples from infected cells treated with IFN. However, the expression levels of total eIF2α (t-eIF2α) were similar in both cases, in mock-infected and infected cells, indicating that infection does not modify the expression pattern of this protein. On the other hand, the Mx and ISG-56 proteins showed similar patterns of expression in mock-infected and infected cells treated with IFN. For both proteins, there were higher levels of accumulation in the I+3 and I+6 than in the I+9 or I+12 samples. Regarding viral infection, the addition of IFN at early times p.i. resulted in a major reduction of VP3 expression (Fig. 2C). This effect was less when IFN was added at later times p.i., indicating that although IBDV is sensitive to IFN action, once infection is established, the virus expresses a factor(s) that counteracts IFN activity. In accordance with this result, while the addition of IFN at 3 h p.i. resulted in a significant drop, ca. 35-fold, in extracellular virus yields at 24 h p.i., the addition of IFN at 12 h p.i. caused a minor reduction (<2-fold) in the production of extracellular virus, which was not statistically significant (data not shown).
PKR is a crucial protein in the apoptosis pathway triggered by IFN-α in IBDV-infected HeLa cells.
Since PKR is an ISG that has been demonstrated to cause apoptosis in many instances (42), and given that PKR was activated in IBDV-infected cells treated with IFN (Fig. 2C), we decided to perform the same assays using PKR-silenced HeLa cells obtained by small interfering RNA (siRNA) technology. Control cells were generated by using a scrambled siRNA. Significantly, in PKR-silenced cells, treatment with IFN after IBDV infection did not cause the drastic morphological changes or the mortality observed in wild-type or silencing control cells (data not shown). Furthermore, PKR silencing also caused a significant reduction of the extent of PARP cleavage triggered by IFN treatment (Fig. 3A).
FIG 3.
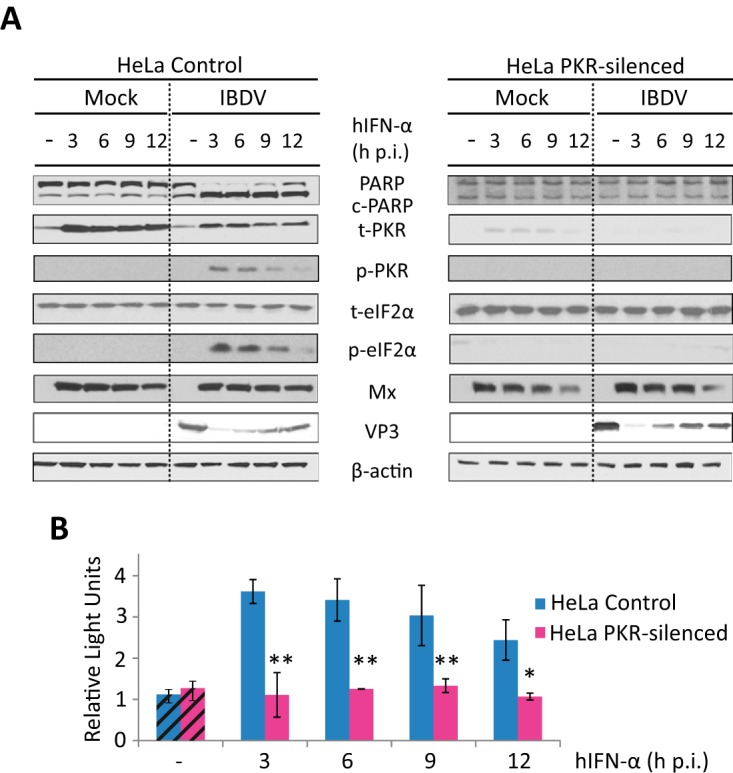
Triggering of apoptosis by IFN in IBDV-infected HeLa cells is dependent on PKR expression. HeLa cells mock infected or infected with IBDV (MOI of 2) were treated with 1,000 IU/ml of hIFN-α at 3, 6, 9, or 12 h p.i. (samples named M+3, M+6, M+9, and M+12 and I+3, I+6, I+9, and I+12, respectively, throughout the text), as indicated, or remained untreated (−) (named M and I, respectively, throughout the text), and cells were analyzed at 24 h p.i. (A) Western blot analysis of total cell extracts with different antibodies: anti-PARP, anti-t-PKR, anti-phosphorylated (Thr446) PKR, anti-t-eIF2α, anti-phosphorylated (Ser52) eIF2α, anti-Mx, and anti-VP3. Antibodies to β-actin were used for a protein loading control. The PARP cleavage product is denoted c-PARP. (B) Apoptosis was measured for duplicate samples by using the Caspase-Glo 3/7 assay kit, and each determination was carried out in duplicate. Caspase values from infected cell samples were normalized to those from mock-infected cells. Bars indicate means ± standard deviations based on duplicate samples from two independent experiments. Striped bars, infected cell samples not treated with IFN; solid bars, infected cell samples treated with IFN. * and ** indicate P values of <0.05 and <0.01, respectively, as determined by unpaired Student's t test.
When apoptosis was quantified by using the caspase 3/7 assay (Fig. 3B), we found a drastic reduction of this activity in PKR-silenced cells, obtaining values akin to those detected in infected cells not treated with IFN, independently of the time p.i. at which IFN was added.
We next analyzed the cellular response in PKR-silenced cells. While silencing control cells behaved as wild-type HeLa cells, a conspicuous absence of phosphorylated eIF2α was observed in PKR-silenced cells (Fig. 3A). These results indicate that PKR is responsible for the eIF2α phosphorylation observed in both wild-type and silencing control HeLa cells. Upon treatment with IFN, the Mx expression patterns in mock- and IBDV-infected PKR-silenced HeLa cells, as well as in silencing control cells, were similar, indicating that IFN-induced Mx upregulation is not PKR dependent and is not affected by infection. It is noteworthy that the downregulation of PKR in HeLa cells did not overcome the expression blockade of VP3 produced by IFN treatment.
PKR-dependent upregulation of IFN-β and TNF-α gene expression on IBDV-infected cells treated with IFN-α.
To further analyze the cellular response in IFN-treated IBDV-infected cells, we also studied the expression levels of several IFN response- and apoptosis-related genes. For this, M, M+3, I, and I+3 samples from control and PKR-silenced cells were harvested at 24 h p.i. for RNA extraction. Purified RNAs were then analyzed by reverse transcription-quantitative PCR (RT-qPCR). The results of this analysis are shown in Fig. 4.
FIG 4.

Upregulation of IFN-β and TNF-α gene expression in IBDV-infected HeLa cells upon IFN treatment is dependent on PKR expression. HeLa control and HeLa PKR-silenced cells were infected with IBDV (MOI of 2) and treated or not with 1,000 IU/ml of hIFN-α at 3 h p.i. Mock-infected (M and M+3) and IBDV-infected (I and I+3) cells were harvested at 24 h p.i. and subjected to RNA extraction. The expression levels of the selected genes, as indicated, were determined by SYBR green-based RT-qPCR. The expression level of each cellular target gene was normalized to the HPRT mRNA content and is presented on a log10 scale as the fold change over the level in mock-infected HeLa silencing control or HeLa PKR-silenced cells. Bars indicate means ± standard deviations based on data for triplicate samples. Dotted bars, mock-infected cell samples treated with IFN; striped bars, infected cell samples not treated with IFN; solid bars, infected cell samples treated with IFN. *, **, and *** indicate P values of <0.05, <0.01, and <0.001, respectively, as determined by unpaired Student's t test. ns, not significant.
In agreement with data from previous reports using other cell lines, IBDV infection induced the expression of the IFNB, Mx1, and ISG-56 genes in HeLa cells. However, a significantly higher level of induction (ca. 2 log units) of IFNB gene expression was observed in the I+3 samples from control cells than in either M or M+3 cell samples. Noticeably, the level of induction was significantly lower in I+3 samples from PKR-silenced cells. Moreover, the level of IFNB induction was also lower in I samples from PKR-silenced cells than in those from control cells. However, Mx, ISG-15, and ISG-56 gene expressions were induced by IFN treatment to similar levels in infected and mock-infected cells. These levels were similar in both PKR-silenced and control cells, with the only exception being ISG-15, whose expression level was highly reduced in PKR-silenced cells under all tested conditions. Intriguingly, ISG-56 and Mx expression levels were enhanced by about 10-fold in IBDV-infected PKR-silenced cells with respect to infected control cells.
To try and deciphering which factor(s) could be involved in the induction of apoptosis observed, we also analyzed the expression levels of different apoptosis-related genes, namely, BAK, FAS, TRAIL, TNFA, and BCL2. As shown in Fig. 4, only TNFA was specifically upregulated in I+3 samples from control cells. Significantly, in I+3 samples from PKR-silenced cells, TNFA expression was maintained at the same level as in the M+3 cells (Fig. 4). On the other hand, TRAIL expression was highly upregulated by IFN in both M+3 and I+3 cells, while BAK and FAS expressions were only slightly induced by IFN treatment. BCL2 gene expression remained fairly constant under all tested conditions.
Triggering of apoptosis by IFN-α in IBDV-infected HeLa cells depends on TNF-α expression.
Indeed, we were keen to assess the possible role of TNF-α in the generalized apoptosis observed in IFN-treated IBDV-infected cells. Therefore, the next step in our research was to silence TNF-α expression in HeLa cells. For this, we generated lentiviral particles expressing five short hairpin RNAs (shRNAs) targeting human TNFA. Silencing was verified by RT-qPCR at 4 and 12 days postransduction (p.t.) in cells transfected with poly(I·C) for 16 h prior to harvesting. Poly(I·C) transfection was used to induce TNF-α gene expression, since we could not detect basal levels of TNF-α mRNA in untreated HeLa cells (Fig. 5A and B, respectively). At 12 days p.t., TNFA expression levels were reduced up to 81% and 79% in cells transduced with the shRNA TNF-α 1 lentivirus (sh1) and sh4, respectively, compared with cells transduced with control shRNA (shC) lentiviral particles (Fig. 5B). A moderate reduction in TNF-α expression (ca. 50%) was observed with the other three constructs, and therefore, these constructs were discarded. Next, we carried out a biological assay by monitoring caspase 3/7 activity in cells transduced with the two successful lentiviruses, sh1 and sh4, to determine the potential role of TNF-α in the induction of apoptosis (Fig. 5C). HeLa cells transduced with sh1, sh4, or shC were mock infected or infected with IBDV (MOI of 2) at 12 days p.t. Thereafter, cells were treated with hIFN-α (1,000 IU/ml) at 3 h p.i. Samples were harvested at 24 h p.i.
FIG 5.

Effect of TNF-α knockdown on triggering of apoptosis by IFN in IBDV-infected HeLa cells. (A and B) TNF-α silencing by shRNAs. HeLa cells were transduced with lentiviral vectors expressing an irrelevant sequence (shC) or shRNAs targeting TNF-α mRNA (sh1 to sh5). At 4 days p.t. (A) and 12 days p.t. (B), cells were transfected with poly(I·C) for 16 h to induce TNF-α gene expression, and the levels of TNF-α mRNA were determined by SYBR green-based RT-qPCR. The results of TNF-α gene expression levels, normalized against the HPRT mRNA level, are presented as fold changes relative to the level in HeLa shC cells transfected with poly(I·C). Bars indicate means ± standard deviations based on data from triplicate samples. * indicates a P value of <0.05, as determined by unpaired Student's t test. (C and D) Effect of TNF-α silencing on apoptosis. HeLa cells transduced with lentiviral vectors expressing shC, sh1, and sh4 were mock infected or infected with IBDV at an MOI of 2 at 12 days p.t. and treated (+) or not (−) with 1,000 IU/ml of hIFN-α at 3 h p.i. Cells were harvested at 24 h p.i. (C) Apoptosis was measured by using the Caspase-Glo 3/7 assay kit. Each determination was carried out in duplicate. The presented data correspond to the means ± the standard deviations of results from two independent experiments. Caspase values for infected cell samples were normalized to those for mock-infected cells. Bars indicate means ± standard deviations based on data from duplicate samples. * and ** indicate P values of <0.05 and <0.01, respectively, as determined by unpaired Student's t test. (D) PARP cleavage analyzed by Western blotting. Total cell extracts were subjected to SDS-PAGE, transferred to nitrocellulose, and immunoblotted with serum anti-PARP. The PARP cleavage product is denoted c-PARP. The Western blot corresponding to β-actin was used as a protein loading control.
Significantly, in cells where TNF-α expression was downregulated, IFN treatment applied after IBDV infection did not cause extensive apoptosis. Caspase activity was further reduced in HeLa sh4 compared to HeLa sh1 cells, and HeLa sh4 cells were then selected for subsequent studies (Fig. 5C). To further confirm this result, we analyzed the pattern of PARP cleavage in the I+3 samples from HeLa sh4 and HeLa shC cells. As shown in Fig. 5D, in contrast with the extensive PARP cleavage observed in the I+3 samples from HeLa shC cells, in HeLa sh4 cells, regardless of IFN treatment, we observed the same PARP pattern in infected and mock-infected cells. These results indicate that, like PKR, TNF-α is a critical factor in the apoptosis pathway triggered by IFN in IBDV-infected cells.
Apoptosis enhancement during IBDV infection in cells treated with IFN-α correlates with increasing levels of PKR phosphorylation and upregulation of IFN-β gene expression.
To learn about the mechanism(s) underlying the triggering of apoptosis in IFN-treated IBDV-infected cells, we carried out a detailed time course analysis to characterize different cellular and viral parameters. First of all, we analyzed the extent of apoptosis over time by examining PARP cleavage by Western blotting in M, M+3, I, and I+3 samples, from 12 h p.i. to 24 h p.i. As shown in Fig. 6A, a gradual increase in PARP cleavage was observed over time in I+3 samples, clearly being highest at 24 h p.i. Significantly, only slight PARP cleavage could be observed in I samples at late times p.i. (21 and 24 h). In mock-infected cells, either M or M+3, only a negligible band corresponding to cleaved PARP could be observed. Moreover, the gradual increase in apoptosis observed in I+3 cell samples was confirmed by real-time quantitative cell death analysis using an IncuCyte Zoom system apparatus on cultures corresponding to the different experimental conditions. In agreement with results described above, Fig. 6B shows that in I+3 cell samples, there was an increase of apoptotic cell death from 12 h p.i. until the end of the experiment, while in I samples, apoptotic cell death remained unapparent at 24 h p.i.
FIG 6.

Triggering of apoptosis during IBDV infection in cells treated with IFN correlates with PKR phosphorylation and upregulation of IFN-β gene expression. HeLa cells mock infected (M) or infected with IBDV (MOI of 2) (I) were treated or not with hIFN-α (1,000 IU/ml of at 3 h p.i.) (M+3 and I+3, respectively) and harvested at the indicated times p.i. (A) Western blot analysis of total cell extracts with anti-PARP, anti-t-PKR, anti-p-PKR, and anti-VP3 antibodies. The Western blot corresponding to β-actin was used as a protein loading control. The PARP cleavage product is denoted c-PARP. (B) Real-time quantitative cell death analysis. Mock-infected and IBDV-infected HeLa cells treated as indicated above were incubated with the IncuCyte caspase-3/7 apoptosis assay reagent as described in Materials and Methods, and cell cultures were monitored with an IncuCyte Zoom system apparatus during 24 after infection. Apoptotic cell death in duplicate cultures under each experimental condition, visualized by the appearance of green cells, was analyzed with IncuCyte Zoom software. (C to E) RT-qPCR analysis of IFN-β (C) and viral (D and E) RNAs during infection in cells treated with hIFN-α. In panel E, cells were treated or not with 7DMA (0.2 mM) after virus adsorption and then treated (I+3) (solid bars) or not (I) (striped bars) with hIFN-α at 3 h p.i. The expression levels of the selected genes, as indicated, were determined by SYBR green-based RT-qPCR. The expression level of the IFN-β target gene was normalized to the HPRT mRNA content and is presented on a log10 scale as a fold change over the level in mock-infected HeLa cells. (F) Apoptosis in samples from M, M+3, I, and I+3 cells treated or not treated with 7DMA (0.2 mM) after virus adsorption was measured at 24 h p.i. by using the Caspase-Glo 3/7 assay kit. Each determination was carried out in duplicate. The presented data correspond to the means ± the standard deviations of results from two independent experiments. The caspase values for infected cell samples were normalized to those for mock-infected cells. Bars indicate means ± standard deviations based on data from duplicate samples. * and ** indicate P values of <0.05 and <0.01, respectively, as determined by unpaired Student's t test.
Since we have determined that PKR plays a crucial role in the IFN-induced apoptotic response, we first examined PKR activation by analyzing its phosphorylation status at different times p.i. As shown in Fig. 6A, while we could not observe PKR phosphorylation in IBDV-infected cells at any of the analyzed times p.i., thus behaving in this respect as mock-infected cells, a gradual increase in PKR phosphorylation, starting at between 12 and 15 h p.i., was observed in I+3 cell samples. Although the total amount of PKR (t-PKR) was visibly larger in the M+3 cell samples, the level of PKR phosphorylation was much lower than that in I+3 cells. To monitor virus infection in these samples, we analyzed the expression of the VP3 protein by Western blotting. In IFN-untreated IBDV-infected cells, a progressive increase of VP3 protein expression from 12 to 24 h p.i. was observed (Fig. 6A). However, as described above (Fig. 2C), in the I+3 cell samples, there was a major reduction of VP3 expression at all the times analyzed (Fig. 6A).
In addition, the expression of the IFNB gene during IBDV infection was analyzed by RT-qPCR. For this, we performed a time course analysis using M, M+3, I, and I+3 cell samples. Cells were harvested at 3-h intervals from 4 to 24 h p.i. As shown in Fig. 6C, a progressive upregulation of IFNB transcription was observed in I+3 cells, reaching a ca. 42-fold increase at 24 h p.i. In contrast, as shown in Fig. 4, a modest increase in IFNB gene expression (2-fold) was detected in I and M+3 samples harvested at 24 h p.i. We also examined the kinetics of IBDV RNA accumulation (Fig. 6D). While a steady increase of the IBDV-specific RNA level, reaching a plateau at 18 h p.i., was detected in I cells, there was only an minor, although statistically significant, increase of <1 log in I+3 cells at between 4 and 12 h p.i., which reached maximum values at 24 h p.i., ca. 8-fold lower than those observed for untreated cells. Indeed, no specific IBDV RNA was detected in mock-infected samples. In agreement with these results, the production of extracellular virus was also strongly affected by IFN treatment. Thus, while we observed a progressive increase of virus titers in untreated cell samples from 12 to 24 h p.i., reaching a maximum of 3.3 × 105 PFU/ml, the production of extracellular virus was significantly reduced in IFN-treated cells, reaching a maximum level (1.1 × 104 PFU/ml) over 30-fold lower than those in untreated cultures.
These results show that in IFN-treated IBDV-infected cells, PKR phosphorylation, IFNB gene expression, and caspase activation are all induced at around 12 to 15 h p.i. Although viral RNA replication was highly reduced in the presence of IFN, there is a possibility that the limited amount of genomic dsRNA produced could be the trigger of the massive apoptotic response. To analyze this possibility, our next approach was to test the ability of a well-known polymerase inhibitor, 7-deaza-2′-C-methyladenosine (7DMA) (43), to block viral dsRNA replication. First, HeLa cells were treated with different doses of 7DMA (0.05 to 2 mM), and a toxicity assay was performed to determine the highest dose of the drug that could be used without compromising cell viability. From this assay, the 0.2 mM dose was chosen for the following experiments (data not shown). Hence, IBDV-infected cells (MOI of 2) were mock treated or treated with 7DMA after virus adsorption, and cells were harvested at 1, 4, and 24 h p.i. for RNA extraction, while one set of cells from each group, treated and not treated with 7DMA, was treated with IFN at 3 h p.i. and harvested at 24 h p.i. Quantitative analysis of viral RNA replication, performed by RT-qPCR as described above, showed that in both IFN-treated and untreated cells, treatment with 7DMA resulted in a significant reduction in the amount of viral RNA that accumulated at 24 h p.i., and the RNA levels in these samples were similar to those obtained at early times after infection, at 1 and 4 h p.i., in either the absence or presence of the inhibitor (Fig. 6E). We then treated mock-infected and IBDV-infected cells with 7DMA, cells were treated or not with IFN at 3 h p.i., and the extent of apoptosis at 24 h p.i. was analyzed by determining caspase 3/7 activity. As shown in Fig. 6F, treatment with 7DMA resulted in a significant reduction in apoptosis in the I+3 samples. A slight reduction was also observed for the samples not treated with IFN (I). Hence, these results indicate that viral RNA replication/transcription is required for massive apoptosis induction upon IFN treatment of IBDV-infected cells.
Nuclear translocation of NF-κB in infected cells treated with IFN-α.
The activation of NF-κB through the degradation of the phosphorylated NF-κB inhibitor IκB has been described to be mediated by PKR (44–47). Also, binding of TNF-α to its receptor induces apoptosis and nuclear translocation of NF-κB (48). Since these two molecules, PKR and TNF-α, seem to play a key role in the induction of apoptosis in IBDV-infected cells treated with IFN, we wished to analyze NF-κB activation at different times p.i. upon the addition of IFN-α. For this, cells were infected with IBDV and treated with IFN at 3 h p.i. Samples were harvested at 1-h intervals starting from 12 to 15 h p.i. and at 3-h intervals from 15 h to 24 h p.i. Harvested cells were split into nuclear and cytosolic fractions, and the presence of the p65 subunit of NF-κB in nuclear fractions was analyzed by Western blotting. As shown in Fig. 7, a transient activation of NF-κB was observed at late times p.i., between 15 and 22 h p.i., in the I+3 cell samples, as indicated by the presence of p65 in the nuclear fraction. However, p65 was only marginally detected in the nuclear fraction in some M and I samples. α-Tubulin and histone H2B were used as cytoplasmic and nuclear markers, respectively.
FIG 7.

NF-κB activation upon treatment with IFN in IBDV-infected HeLa cells. HeLa cells mock infected (M) or infected (I) with IBDV (MOI of 2) were treated or not with hIFN-α (1,000 IU/ml of at 3 h p.i.) (M+3 and I+3, respectively), and the nuclear translocation of the p65 subunit of NF-κB was analyzed. Shown is Western blot analysis of p65 in the nuclear and cytosolic fractions from cells harvested at the indicated times p.i. Each fraction was subjected to SDS-PAGE, transferred to nitrocellulose, and immunoblotted with serum anti-p65. The Western blots corresponding to histone H2B and α-tubulin were used as protein loading controls for nuclear and cytosolic fractions, respectively.
NF-κB activation is essential for triggering apoptosis.
To study the potential contribution of NF-κB to the production of apoptosis, we used MG132, an inhibitor of the proteasome and NF-κB. MG132 blocks the proteolytic activity of the ubiquitin-proteasome system, preventing the degradation of the ubiquitinated NF-κB inhibitor IκBα and thus suppressing the activation of NF-κB (49, 50). Since proteasome inhibitors have been shown to induce the formation of reactive oxygen species, leading to a loss of cell viability (49), we performed a preliminary dose-response assay to assess the effect of MG132 on both proteasome activity and cell viability. The 2 μM dose was selected for the following experiments since it resulted in a maximal accumulation of ubiquitinated proteins and did not have a significant effect on cell viability (<5%) (data not shown). IBDV-infected and mock-infected cells were then treated with IFN (1,000 IU/ml) at 3 h p.i. in the presence of MG132 or the solvent dimethyl sulfoxide (DMSO) as a control. Cultures were harvested at 16, 18, and 20 h p.i., when the activation of NF-κB was previously detected (Fig. 7). Cells were fractionated into nuclear and cytoplasmic fractions and analyzed by Western blotting. As expected, nuclear p65 translocation was readily observed in I+3 cell samples. However, this was drastically reduced upon treatment with MG132 (Fig. 8A). Neither MG132 nor DMSO had a detectable effect on M+3 cells. On the other hand, cytoplasmic NF-κB levels remained unchanged in all cases. We then analyzed the extent of apoptosis by measuring caspase 3/7 activity. As shown in Fig. 8B, a significant reduction of caspase activity was observed in I+3 cells in the presence of MG132 compared with those treated in the presence of DMSO. These results indicate that NF-κB activation plays an important role in the triggering of apoptosis by IFN in IBDV-infected cells. However, since MG132 may have nonspecific effects, by preventing the degradation of all proteins, our next approach was to use NF-κB-specific inhibitors. For this purpose, we used the IκB kinase β (IKKβ)-specific inhibitor SC-514 (51) and the p65 translocation inhibitor JSH-23, which does not prevent IκBα degradation (52), to determine their effect on apoptosis induction. First, after assaying different doses of each compound to determine the highest dose that could be used without affecting cell viability (data not shown), we wished to confirm the effectiveness of the treatments in our HeLa cell system. For this, HeLa cells were treated with the selected doses, 50 and 25 μM SC-514 and JSH-23, respectively, or with the solvent DMSO as a control during 2 h, followed by treatment with 20 ng/ml of TNF-α during 30 min. Next, cells were collected, fractionated into nuclear and cytosolic fractions, and analyzed by Western blotting with anti-p65 and anti-IκBα antibodies. Antibodies against α-tubulin and H2B were used as cell fractionation controls. As shown in Fig. 8C, the nuclear translocation of p65 was clearly inhibited, although only partially, by both inhibitors SC-514 and JSH-23 compared with mock-treated cells. Correspondingly, the amount of p65 in the cytosolic fraction was enhanced in cells treated with both inhibitors. Additionally, as expected, in mock-treated cells and cells treated with JSH-23, IκBα was almost unapparent in the cytoplasmic fraction upon TNF-α incubation; however, IκBα degradation was prevented in cells treated with SC-514. In view of these results, IFN-treated IBDV-infected cells were treated with the same doses of SC-514 and JSH-23 as those described above at 14 h p.i., and cells were harvested at 16, 18, and 20 h p.i. Samples were analyzed by Western blotting to confirm the effectiveness of these treatments in preventing p65 nuclear translocation. As shown in Fig. 8D, under these conditions, treatment with both inhibitors also resulted in a partial inhibition of p65 nuclear translocation at 20 h p.i. (64% for SC-514 and 43% for JSH-23), compared with control cells treated with DMSO. In addition, as described above for cells incubated with TNF-α, treatment with SC-514 prevented IκBα degradation, as observed by the higher levels of accumulation of this protein in the cytosolic fractions in SC-514-treated cells than in mock- and JSH-23-treated cells at the time points analyzed (1.14-, 1.64-, and 2.45-fold more protein accumulated at 16, 18, and 20 h p.i., respectively, than in mock-treated cells). Next, we wished to determine the effect of these inhibitors on the triggering of apoptosis. For this, M, M+3, I, and I+3 cell samples were mock treated or treated with SC-514 and JSH-23 as described above, and cells were harvested at 24 h p.i. for caspase activity analysis. As shown in Fig. 8E, a significant reduction in caspase activity was observed in the I+3 samples treated with both NF-κB activation inhibitors compared with mock-treated cells. A minor reduction was also seen in the I samples treated with the inhibitors. Hence, these results confirm the involvement of the NF-κB pathway in the triggering of apoptosis by IFN in IBDV-infected cells.
FIG 8.

Inhibition of p65 nuclear translocation by treatment with the proteasome inhibitor MG132 or with NF-κB-specific inhibitors significantly affects triggering of apoptosis by IFN in IBDV-infected cells. HeLa cells mock infected or infected (I) with IBDV (MOI of 2) were treated immediately after virus adsorption with the proteasome inhibitor MG132 (2 μM) or DMSO (used as control) and incubated with hIFN-α (1,000 IU/ml) at 3 h p.i. (M+3 and I+3, respectively). Cell samples were harvested at the indicated times p.i. (A) Western blot analysis of p65 in the nuclear and cytosolic fractions. Samples were subjected to subcellular fractionation, and each fraction was subjected to SDS-PAGE, transferred to nitrocellulose, and immunoblotted with serum anti-p65. Western blots corresponding to histone H2B and α-tubulin were used as protein loading controls for nuclear and cytosolic fractions, respectively. M, untreated mock-infected cell fraction. (B) Apoptotic cell death in cell samples collected at the indicated times p.i. was measured by using the Caspase-Glo 3/7 assay kit. Each determination was carried out in duplicate. The presented data correspond to the means ± standard deviations of results from two independent experiments. Caspase values for infected cell samples were normalized to those for mock-infected cells. Bars indicate means ± standard deviations based on data from duplicate samples. (C) Analysis of p65 translocation upon TNF-α treatment in cells treated with NF-κB-specific inhibitors by Western blotting. HeLa cells were treated with the IKKβ-specific inhibitor SC-514, the p65 translocation inhibitor JSH-23 (50 and 25 μM, respectively), or DMSO as a control (−) during 2 h, followed by treatment with TNF-α (20 ng/ml) during 30 min. Nuclear and cytosolic fractions were analyzed with antibodies against p65 and IκBα. Histone H2B and α-tubulin were used as protein loading controls for nuclear and cytosolic fractions, respectively. (D) Analysis of p65 translocation in IFN-treated IBDV-infected cells upon treatment with NF-κB-specific inhibitors by Western blotting. IBDV-infected HeLa cells treated with IFN at 3 h p.i. (I+3) were treated with SC-514, JSH-23 (50 and 25 μM, respectively), or DMSO as a control (−) at 14 h p.i. Cell samples were collected at the indicated times p.i. Nuclear and cytosolic fractions were analyzed by using antibodies against p65, IκBα, and VP3. Histone H2B and α-tubulin were used as protein loading controls. (E) Apoptotic cell death in M, M+3, I, and I+3 cell samples treated with SC-514, JSH-23 (50 and 25 μM, respectively), or DMSO as a control, at 14 h p.i., was measured at 24 h p.i. as described above for panel B. * and ** indicate P values of <0.05 and <0.01, respectively, as determined by unpaired Student's t test.
Triggering of apoptosis in cells transfected with IBDV genomic dsRNA upon IFN treatment.
To further investigate the apoptosis-triggering mechanism in IBDV-infected cells treated with hIFN-α, we wished to ascertain the viral factor(s) involved in this process. For this, HeLa cells were either transfected with purified genomic IBDV dsRNA or incubated with purified IBDV virus-like particles (VLPs). After 3 h, cultures were treated with hIFN-α (1,000 IU/ml) and then harvested 21 h later. As a control, cells infected with IBDV (MOI of 2) were treated with hIFN-α at 3 h p.i. and harvested at 24 h p.i. As shown in Fig. 9, some caspase 3/7 activity was detected in cells transfected with viral dsRNA (R); however, the activity was significantly higher when cells were treated with IFN (R+3), reaching values similar to those recorded for I+3 cells. In contrast, incubation of cells with IBDV VLPs did not elicit an apoptotic response, as determined by the lack of caspase activity, in either the absence (V) or presence (V+3) of IFN treatment. These results suggest that binding to the viral receptor and the subsequent internalization of IBDV, represented here by the VLPs, are not the triggers of the apoptotic effect. On the other hand, these results suggest that genomic IBDV dsRNA might be the viral component that contributes to this effect.
FIG 9.
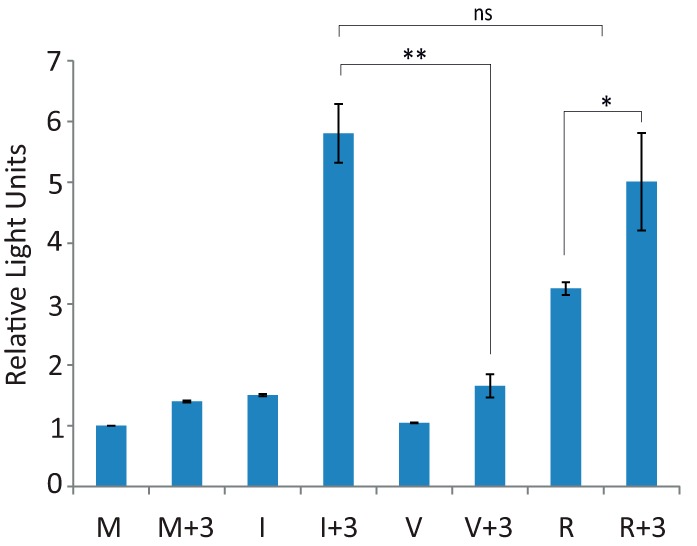
Triggering of apoptosis in cells transfected with IBDV genomic RNA, but not in cells incubated with IBDV-VLPs, upon IFN treatment. HeLa cells either mock infected (M), infected (I) with IBDV (MOI of 2), incubated with IBDV-derived VLPs (V), or transfected with genomic dsRNA (R) were treated or not with hIFN-α (1,000 IU/ml) at 3 h p.i. (M+3, I+3, V+3, and R+3, respectively). Cells were harvested at 24 h p.i., and apoptotic cell death was measured by using the Caspase-Glo 3/7 assay kit. Each determination was carried out in duplicate. The presented data correspond to the means ± the standard deviations of results from two independent experiments. Caspase values for infected cell samples were normalized to those for the corresponding mock-infected cells. Bars indicate means ± standard deviations based on data from duplicate samples. * and ** indicate P values of <0.05 and <0.01, respectively, as determined by unpaired Student's t test. ns, not significant.
IFN-α treatment of HeLa cells infected with different strains of IBDV also causes apoptosis.
Until now, all the experiments described in this report were performed with the Soroa strain of IBDV; however, it would be interesting to assess if IFN would also promote apoptosis induction upon infection with other IBDV strains. Hence, we performed a comparative analysis using Soroa, our reference strain, along with two other IBDV strains, Bursine and P2. For this, cells were infected with the different IBDV strains (MOI of 2), treated with hIFN-α (1,000 IU/ml) at 3 h p.i., and collected at 24 h p.i. for Western blot and caspase 3/7 activity analyses. Figure 10A shows that the extents of PARP cleavage in the I+3 samples were similar irrespective of the strain used for infection, and again, in all I samples, the intensity of the band corresponding to cleaved PARP was similar to that for M and M+3 samples. The expression of the VP3 protein, analyzed as a control for infection, revealed that IFN treatment affects the replication of Bursine and P2 to an extent similar to that for the Soroa strain. On the other hand, as for the Soroa strain, the level of caspase 3/7 activity was significantly higher in cells infected with either the Bursine or P2 strain followed by IFN treatment than in those not treated with the cytokine (Fig. 10B). Therefore, these results indicate that the triggering of apoptosis of IBDV-infected cells upon IFN treatment is a general phenomenon that is not restricted to the Soroa strain.
FIG 10.

IFN treatment triggers apoptosis of HeLa cells infected with different strains of IBDV. HeLa cells mock infected or infected with different strains of IBDV (MOI of 2), Soroa, P2, or Bursine, were treated with 1,000 IU/ml of hIFN-α at 3 h p.i. (M+3 and I+3, respectively), as indicated, or remained untreated (M and I, respectively). Cells were analyzed at 24 h p.i. (A) Western blot analysis of cells mock infected or infected with different strains of IBDV and treated with hIFN-α with antibodies against PARP and VP3. The PARP cleavage product is denoted c-PARP. Antibodies to β-actin were used for a protein loading control. (B) Apoptosis was measured from duplicate samples by using the Caspase-Glo 3/7 assay kit, and each determination was carried out in duplicate. Caspase values for infected cell samples were normalized to those for mock-infected cells. Bars indicate means ± standard deviations based on data for duplicate samples from two independent experiments. Yellow bars indicate mock-infected cell samples not treated (M) and treated (M+3) with IFN. * indicates a P value of <0.05, as determined by unpaired Student’s t test.
Chicken DF-1 cells infected with IBDV also undergo apoptosis upon IFN treatment.
The comprehensive analysis reported in this study was possible due to the availability of reagents for studies of human cells. However, since IBDV is an avian virus that infects only avian species, the relevance of our findings in mammalian HeLa cells to infection in chickens could be questionable. Therefore, chicken DF-1 cells were infected with IBDV (Soroa strain) at an MOI of 2, and cells were treated with chicken IFN-α (chIFN-α) (1,000 U/ml) at 3 h p.i. Microscopic inspection of infected cells incubated with the IncuCyte caspase 3/7 apoptosis assay reagent revealed that signs of infection appeared earlier than in HeLa cells, but, as for HeLa cells, treatment with IFN also exacerbated the CPE caused by IBDV and enhanced the number of apoptotic cells (Fig. 11A). Mock-infected and IBDV-infected cells treated or not with chIFN-α were harvested at 16 h p.i., and samples were analyzed to determine caspase 3/7 activity. For comparison, cultures treated with different doses of staurosporine were also included in the assay. As shown in Fig. 11B, as observed for HeLa cells, the level of caspase activity in DF-1 cells was significantly higher in the I+3 samples than in the M, M+3, or I samples. Hence, these results indicate that the apoptosis enhancement triggered by IFN in IBDV-infected cells is a general phenomenon and not an artifact resulting from the use of an avian pathogen in a mammalian cell model system.
FIG 11.

IFN treatment triggers apoptosis of IBDV-infected chicken DF-1 cells. DF-1 cells were infected with IBDV (MOI of 2) and treated or not with 1,000 U/ml of chIFN-α at 3 h p.i. Mock-infected (M and M+3) and IBDV-infected (I and I+3) cells were analyzed at 16 h p.i. (A) Cells were incubated with the IncuCyte caspase 3/7 apoptosis assay reagent, and images were recorded with an IncuCyte Zoom system apparatus at 16 h p.i. Apoptotic cells are green. (B) Apoptosis measurement. As a control for apoptosis, uninfected DF-1 cells were treated with different doses of staurosporine (0.5, 1, and 2 μM) for 16 h. Apoptosis in duplicate samples was measured by using the Caspase-Glo 3/7 assay kit, and each determination was carried out in duplicate. Caspase values for infected cell samples were normalized to those for mock-infected cells. Bars indicate means ± standard deviations based on data from duplicate samples from two independent experiments. ** indicates a P value of <0.01, as determined by unpaired Student's t test.
DISCUSSION
IBDV infection causes a strong immunosuppressive disease characterized by a massive loss of IgM-bearing B lymphocytes, the destruction of the bursa of Fabricius, and damage to secondary lymphoid organs. The molecular mechanisms underlying the pathology caused by IBDV remain largely unknown. Multiple factors may contribute to B cell depletion in chickens, but exacerbated apoptosis of B cells and the induction of the so-called “cytokine storm” or “septic shock syndrome” were recently suggested by van den Berg (53) to be potential killing mechanisms in the context of IBDV infection.
IFNs are a family of cytokines that have pleiotropic functions. Beside their well-known ability to confer protection against viral infections by eliciting the expression of a plethora of ISGs exerting antiviral activity, in both infected cells and neighboring noninfected cells, IFNs also promote the selective induction of apoptosis in cells infected with a variety of viruses, thereby limiting virus spread (35, 54, 55).
Few studies have investigated the antiviral activity exerted by type I IFN on IBDV infection both in vitro and in vivo (37–40). However, the potential contribution of IFN to IBDV pathogenesis has not yet been addressed. In agreement with data from previous studies performed with chIFN (37–40), our results demonstrate that the pretreatment of HeLa cells with IFN-α interferes with viral protein synthesis and suppresses IBDV replication in a dose-dependent manner. Similarly, infectious pancreatic necrosis virus (IPNV), another birnavirus, which causes IPN in salmonid fish, has also been shown to be sensitive to the antiviral action of IFNs (56).
IFN treatment of IBDV-infected cells triggers a PKR-dependent apoptotic response.
Interestingly, our results show that the administration of IFN shortly after the beginning of the IBDV infection process leads to extensive cell death due to caspase-mediated apoptosis. The extent of apoptosis is greater when infected cells are treated with IFN at earlier times p.i. (3 to 6 h) and is also dose dependent, although only subtle differences were observed with IFN doses above 100 IU/ml. Indeed, we also observed a significant antiviral effect of IFN under these IFN treatment conditions, as shown by the decrease of VP3 expression levels and the reduction in viral RNA levels and extracellular viral yields. In agreement with the above-mentioned observations, the antiviral effect is significantly enhanced when IFN is added soon after infection.
Although IFNs have been largely characterized for their ability to protect uninfected cells from viral infections, it has also been established that they also act as proapoptotic mediators in virus-infected cells (35, 54, 55). Although the apoptosis of infected cells can be considered a protective mechanism limiting virus spread, there are some well-documented examples evidencing that this phenomenon might also contribute to virus-induced pathogenesis (57, 58).
With these premises in mind, we sought to characterize the mechanism(s) triggering apoptosis in IBDV-infected cells following hIFN-α treatment. For this, we analyzed the expression patterns of three well-characterized ISG proteins, i.e., Mx, ISG-56, and PKR. Mx and ISG-56 were similarly expressed, in both infected and uninfected cells, upon IFN treatment. Additionally, their relative abundances were related to the length of time of IFN treatment, suggesting that they are not involved in apoptosis induction. In contrast, PKR phosphorylation was detected only in infected cells treated with IFN (Fig. 2C), even though the total amount of PKR was smaller in these cells. IFN treatment of mock-infected cells did not cause significant PKR activation, a result that is in contrast to that obtained by Panigrahi and coworkers with Huh7 cells (59). Our analysis was performed at or before 24 h after IFN treatment; therefore, since the authors of that report did not indicate the time at which the assay was performed, there is a possibility that PKR activation could require longer IFN treatment. Alternatively, differences attributable to the different cell lines used in the two studies could also account for the contrasting results. The reduction of total PKR found in infected cells treated with IFN could be due to either a specific inhibition of PKR mRNA translation or enhanced protein turnover, since the RNA levels determined by RT-qPCR were similar in IFN-treated mock-infected and IBDV-infected cells. PKR activation correlated directly with the phosphorylation of eIF2α, its best-characterized substrate, and with a significant reduction of VP3 accumulation. Significantly, the extent of PKR activation also correlated positively with the magnitude of the apoptotic response. PKR, a central player in cellular responses triggered by different stress-inducing stimuli, including viral infections (60–62), is involved in apoptosis induction by different mechanisms (42, 63), one of which is the phosphorylation of eIF2α (64). Here the role of PKR as a critical mediator of the apoptotic response in IFN-treated IBDV-infected cells was revealed by using PKR-silenced cells (Fig. 3A to C). Cells expressing a nontargeting scrambled shRNA behaved as wild-type cells. However, PKR silencing completely ablated the apoptotic response. In addition, while the level of eIF2α phosphorylation in PKR control cells increased in response to IFN treatment after IBDV infection, PKR silencing precluded eIF2α phosphorylation. Our preliminary results suggest that eIF2α phosphorylation is not required for the induction of apoptosis under the conditions described in this report. However, this possibility should be further investigated. On the other hand, as described above, the level of reduction of VP3 accumulation in IFN-treated infected cells is highest when IFN is added early after infection. Since similar VP3 accumulation patterns were found in both PKR-silenced and wild-type control cells, this reduction was not related to PKR activation. Interestingly, the amount of VP3 is inversely proportional to the levels of Mx and ISG-56, suggesting that, at least in the absence of PKR, these ISGs could be responsible for the sensitivity of IBDV to IFN when IFN is added at early times p.i. Moreover, although the mechanism remains unknown, it was previously reported that the human MxA protein inhibits IBDV replication (65).
The apoptotic response in IBDV-infected cells upon IFN treatment concurs with PKR-dependent IFNB induction that could be driven by the recognition of IBDV dsRNA by the cellular sensor machinery.
We observed a higher level of IFNB induction in infected HeLa control cells treated with IFN at 3 h p.i. than in PKR-silenced cells. This increase correlated positively with PKR activation in HeLa control cells. Concerning this observation, there is a growing body of information indicating that PKR activation contributes to the induction of type I IFN in response to some viral infections (61, 66–68). In IFN-untreated IBDV-infected cells, only a modest induction of IFNB was observed at 24 h p.i., correlating with the lack of PKR activation. Nonetheless, progressive IFNB induction was observed at later times after IBDV infection (data not shown). In this regard, the upregulation of IFNB has already been described for chicken DF-1 fibroblasts and HD11 macrophages at late times after IBDV infection (69, 70). Moreover, the recognition of naked IBDV dsRNA by the RNA cytoplasmic sensor MDA5 in DF-1 cells was found to activate chIFN-β expression (59), but it was also described that VP3 interferes with this recognition (40). This is consistent with the notion that the binding of VP3 to IBDV dsRNA shields the virus genome from host RNA sensors, thus preventing PKR-mediated apoptosis (34, 71). Regarding this, it is interesting to note that a stoichiometric analysis of IBDV structural components showed that infectious IBDV particles harbor four dsRNA genome segments associated (in the form of RNPs) with ca. 450 VP3 molecules (19). As-yet-unreported studies performed in our laboratory using surface plasmon resonance analyses indicate that, as expected for the biological role of IBDV RNPs as transcription/replication devices, VP3-dsRNA complexes are poorly stable over time and highly dependent upon the VP3 concentration. These observations suggest that although the VP3/dsRNA ratio found in IBDV virions should be enough to shield the virus genome immediately after the release of RNPs from the virus capsid, optimal protection would be achieved only upon the accumulation of higher levels of VP3 via the de novo synthesis of virus-encoded proteins. Indeed, the activation of IFN before optimal VP3-afforded protection has been reached might lead to the unleashing of the triggering of apoptosis. This hypothesis is fully consistent with data showing the progressive reduction of the effect of IFN treatments applied at late times p.i. presented here.
Our data indicate that either a viral component(s) or an intracellular signal(s) generated early during the infection process is responsible for the massive apoptotic response induced upon IFN treatment. Our results suggest that the IBDV dsRNA genome might act as a major apoptotic triggering factor. While cultures incubated with IBDV VLPs remained insensitive to IFN-induced apoptosis, those transfected with purified IBDV genomic dsRNA were highly responsive. Cooperation between dsRNA and IFN in the activation of the apoptotic pathway has been reported for both mouse embryonic cells (54) as well as primary pancreatic β cells (72). However, it could be argued that naked viral dsRNA transfected into cells would not exactly mimic the IBDV genome in the context of an infection, where the dsRNA is structured in RNPs, being complexed to VP3 and the RNA polymerase VP1. Our results from RT-qPCR analyses showed that although IBDV genome replication is highly impaired in the presence of IFN, viral RNA enhancement was detected at between 4 and 12 h p.i. Significantly, the first signs of apoptosis are detected between 12 and 15 h p.i., thus supporting the role of genomic dsRNA in the triggering of apoptosis. In addition, blocking viral RNA replication/transcription with the RNA polymerase inhibitor 7DMA drastically reduced the extent of apoptosis in infected cells treated with IFN.
Interestingly, it was recently reported that PKR participates in concert with MDA5 in recognizing foreign RNA and inducing IFN production, where MDA5 increases the virus-induced activation of PKR, probably by removing RNA-binding proteins that would otherwise interfere with PKR activation (67). Considering this hypothesis, it is tempting to speculate that the potential stripping of the VP3 polypeptide from the IBDV RNPs by MDA5 could be enhanced following IFN treatment, when MDA5 is upregulated. However, at this point, we cannot completely rule out a possible contribution of one or more newly synthesized viral proteins to the activation of the apoptotic pathway upon treatment with IFN.
TNF-α and NF-κB are essential mediators of the apoptotic response in IBDV-infected cells treated with IFN.
Our results suggest that infected HeLa cells treated with IFN could be primed to undergo apoptosis through TNF-α but not via TRAIL or Fas signaling, since an upregulation of TNFA, but not of TRAIL or FAS, was observed. Moreover, TNFA upregulation did not occur in PKR knockdown cells; TNF-α would then act downstream in the PKR-related apoptotic pathway, mediating the clearance of IBDV-infected cells. In support of this hypothesis, we observed that the downregulation of TNF-α expression in HeLa cells impairs the apoptotic response elicited by infection and IFN exposure, as measured by both caspase activation and PARP cleavage. A similar situation has been described for severe acute respiratory syndrome (SARS) infection, where FasL-Fas and TRAIL-DR5 pathways are not involved in T cell apoptosis. Moreover, a direct or indirect role for TNF-α in this process was indicated (57). Neither BAK nor BCL2, coding for proapoptotic and antiapoptotic proteins, respectively, showed a differential pattern of expression in infected cells treated with IFN with respect to IFN-treated mock-infected cells. This indicates that BAK overexpression upon IFN treatment could not by itself be responsible for the triggering of apoptosis, although we cannot discard its participation.
PKR, which is known to activate the NF-κB transcription factor required for IFN induction (68, 73), plays key roles in many important physiological responses, including cell adhesion, differentiation, apoptosis, and others (42, 60–63). In IFN-α-treated IBDV-infected cells, we detected a transient translocation of the NF-κB p65 subunit into the nucleus at late times p.i. This translocation is nearly concomitant with both PKR activation and the induction of apoptosis. The well-known feedback mechanism between TNF-α and NF-κB (74) seems to operate during the development of apoptosis in cells infected with IBDV and treated with IFN. Despite the survival role of NF-κB, its participation in apoptosis induction in HeLa as well as in B and T cells has been demonstrated (75, 76). However, the activation of NF-κB alone is not sufficient for cell killing, requiring further signals to trigger the apoptotic route (76). Here we demonstrate that the use of NF-κB inhibitors, i.e., MG132, SC-514, and JSH-23, drastically reduces apoptosis induced in infected cells treated with IFN-α, indicating an important contribution of NF-κB to the production of apoptosis under these conditions.
All the results discussed above were obtained with the mammalian HeLa cell system. However, IBDV is an avian pathogen, and although knowledge about the innate immune response in birds has only started to expand, it is already known that birds lack important components of the IFN pathway, such as RIG-I. The possibility that the observed apoptotic effect could be an artifactual phenomenon resulting from an aberrant interaction between an avian pathogen and a mammalian cell system was ruled out by showing that the IFN-induced triggering of apoptosis of IBDV-infected cells can be faithfully reproduced in chicken DF-1 cells. On the other hand, the same outcome was obtained when cells infected with different serotype 1 strains of IBDV were treated with IFN, thus showing that this is a general trait of IBDV.
It is interesting to mention that similar experiments have been carried out with IPNV, another birnavirus (77). In agreement with the results presented here, it was found that the effect of IFN on IPNV replication was moderate when IFN was added after infection (4 or 10 h p.i.) compared with IFN pretreatment. That study also showed that Mx expression induced by IFN treatment was reduced in cells previously infected with IPNV, indicating that some viral products interfere with ISG expression. Although we did not observe a similar reduction in Mx or ISG-56 expression by either Western blotting or RT-qPCR, we detected the specific inhibition of PKR expression in IBDV-infected cells treated with IFN with respect to IFN-treated mock-infected cells. However, in the report on IPNV, nothing is mentioned regarding potential cell damage after the addition of recombinant IFN-α to IPNV-infected cultures. It would be interesting to investigate whether treatment of IPNV-infected cells with IFN induces PKR and NF-κB activation, as we have shown with IBDV.
Our results conclusively show that although it efficiently halts virus replication, the addition of IFN to newly infected cells has a rather negative outcome embodied by the swift, and nearly simultaneous, destruction of all infected cells of both mammalian and chicken origins.
In our view, the findings described here recapitulate the catastrophic effect of the cytokine storm described for experimentally infected chicks, indicating that IFN overproduction during IBDV infection in vivo may be responsible for massive lymphocyte depletion by driving infected cells to undergo apoptosis, thus pointing to a potential link between IFN production and pathogenesis.
The double-edged-sword effect of IFN was previously documented for other viral infection models, where the induction of a systemic type I IFN response was clearly correlated with the induction of immunosuppression and the aggravation of clinical signs of disease. Human immunodeficiency virus (78), lymphocytic choriomeningitis virus (79, 80), classical swine fever virus (81), Newcastle disease virus (82), avian influenza virus (58), and SARS coronavirus (57) are representative examples of this direct relationship between the type I IFN response and the rigor of the clinical outcome of viral infection.
We have contributed for the first time to elucidating a potential mechanism driving the apoptotic death of IBDV-infected cells upon exposure to type I IFN. Our results also provide solid evidence about the critical importance of PKR, TNF-α, and NF-κB as key mediators of this phenomenon. The described mechanism could facilitate an early clearance of infected cells, thereby limiting viral spread and consequently ameliorating IBDV-induced pathogenesis. On the negative side, it might contribute to B cell depletion and immunosuppression. It seems likely that the balance between these two opposing effects might be dramatically affected by the genetic backgrounds of both the host and the infecting virus strain.
MATERIALS AND METHODS
Cells, viruses, and infections.
HeLa (human epithelial cervical cancer cells; ATCC CCL-2), BSC-1 (African green monkey kidney cells; ATCC CCL-26), and DF-1 (chicken embryonic fibroblasts; ATCC CRL-12203) lines were grown in Dulbecco's modified minimal essential medium (DMEM) supplemented with penicillin (100 U/ml), streptomycin (100 U/ml), gentamicin (50 μg/ml), Fungizone (12 μg/ml), nonessential amino acids, and 10% fetal calf serum (FCS) (Sigma-Aldrich). The CEC 32-511 cell line, corresponding to chicken embryonic cells that carry the luciferase reporter gene under the control of the chicken Mx promoter sequence, was kindly provided by B. Kaspers (Munich University, Germany). These cells were cultured in DMEM supplemented with 10% chicken fetal serum (Sigma-Aldrich). HeLa cells deficient in PKR (HeLa PKR silenced) and control cells (HeLa control), stably expressing an shRNA targeting the PKR gene and a scrambled shRNA, respectively, were generated as previously described (83, 84). These cells were maintained in the presence of puromycin (1 μg/ml).
IBDV infections were performed on preconfluent (<75%) cell monolayers with the Soroa strain, a cell-adapted serotype 1 virus, diluted in DMEM at an MOI of 2 PFU per cell, unless otherwise stated. After 1 h of adsorption at 37°C, the medium was removed and replaced with fresh DMEM supplemented with 2% FCS. Infected cells were incubated at 37°C until the specified times p.i. In addition, two other cell-adapted serotype 1 virus strains, Bursine and P2, both classified as classical virulent, as for the Soroa strain, were used in a set of experiments. These IBDV strains were kindly provided by N. Eterradossi (ANSES, France). The recombinant vaccinia virus VT7LacOI/POLY was used to obtain IBDV-derived virus-like particles, as previously described (85).
Treatment with IFN-α.
HeLa cells were pretreated with the indicated doses of hIFN-α (Intron-A; Merck Sharp and Dohme) 16 h before infection with IBDV, and cultures were harvested at 24 h p.i. In most of the experiments throughout this work, mock- or IBDV-infected cells were treated with 1,000 IU/ml of IFN-α at the indicated times p.i., and cultures were harvested at 24 h p.i. For simplicity, samples from mock-infected cells (M) treated with IFN are denoted M+3 and M+6, etc., where the number indicates the time in hours p.i. at which IFN was added to the culture. Similarly, samples from IBDV-infected cells (I) treated with IFN are denoted I+3 and I+6, etc. For dose-response experiments, cells were treated at 3 h p.i. with the indicated IFN-α doses, from 1 to 105 IU, and analyzed at 24 h p.i. by the determination of caspase 3/7 activation.
The expression, purification, and titration of chIFN-α will be described in detail elsewhere. Briefly, chIFN-α was produced as a recombinant protein expressed by a vaccinia virus recombinant harboring the sequence encoding IFN-α from Gallus gallus. The sequence was retrieved from the NCBI, chemically synthesized, fused to a sequence corresponding to a histidine tag, and cloned into the pVOTE transfer vector (86) to generate a vaccinia virus recombinant (rVV–chIFN-α). The virus was obtained according to well-established procedures (86). For chIFN-α production, monolayers of BSC-1 cells were infected with rVV–chIFN-α (MOI of 5). After 24 h, culture medium was collected, filtered through 0.22-μm filters to eliminate the recombinant virus, and used to purify chIFN-α by affinity chromatography (Talon metal affinity resin; Clontech). The clarified supernatant was incubated with the resin for 1 h at 4°C, and after several washes with 50 mM Tris-HCl (pH 8.0), the chIFN-α protein was eluted by incubation of the resin with elution buffer (50 mM Tris-HCl [pH 6.4] containing 150 mM NaCl and 0.5 M imidazole). After elution, the protein was dialyzed against 20 mM Tris-HCl (pH 8.0) containing 0.5 M EDTA. Purified chIFN-α was titrated according to procedures described previously by Schwarz and coworkers (87), based on a luminescence bioassay using CEC 32-511 cells that carry the luciferase gene under the control of the chicken Mx promoter sequence. For this, serial dilutions of the preparation of purified chIFN-α were added to CEC 32-511 cells seeded into 96-well plates, and luciferase activity was determined after 6 h by using a luciferase report assay kit (Promega). From the luminescence values, and by applying the Reed-Muench method (88), the chIFN-α titer in units per milliliter was determined.
Virus titration.
The supernatants from cultures infected with IBDV and treated or not treated with hIFN-α were collected and subjected to low-speed centrifugation (1,500 rpm for 5 min) to remove cell debris. The clarified cell supernatants were used to determine the extracellular virus titers by a plaque assay using a semisolid agar overlay and staining with crystal violet.
Cell viability assay.
The Live/Dead cell imaging kit (Thermo Fisher Scientific) was used to discriminate live from dead cells according to the manufacturer's instructions. Briefly, at the specified times p.i., an equal volume of the reagent was added to HeLa cell monolayers grown in 24-well plates. Cultures were incubated at room temperature for 15 min, and cells were examined under a microscope (Leica DMI6000B epifluorescence microscope; Leica Microsystems).
Real-time quantitative cell death analysis.
HeLa cell monolayers grown in 24-well plates were infected at an MOI of 2 PFU/cell. After 1 h of virus adsorption at 37°C, the medium was removed and replaced with fresh DMEM supplemented with 2% FCS and the IncuCyte caspase 3/7 apoptosis assay reagent (Essen BioScience), according to the manufacturer's instructions. Cells were then placed within an IncuCyte Zoom system apparatus. At 3 h p.i., mock-infected and infected cells were treated with 1,000 IU/ml of hIFN-α. Cultures were monitored every 30 min by using a 10× objective lens. Four randomly selected monolayer locations were imaged in each well over the entire time course (24 h). IncuCyte Zoom software was used to automatically score and quantify the number of apoptotic cells (in green). The presented data correspond to two independent infections involving the quantification of a total of 8 video recordings per sample.
Western blot analysis.
Cell monolayers were washed with phosphate-buffered saline (PBS) and lysed in Laemmli's sample buffer (62.5 mM Tris-HCl [pH 6.8], 2% sodium dodecyl sulfate [SDS], 0.01% bromophenol blue, 10% glycerol, and 5% β-mercaptoethanol). Protein samples were subjected to 10% SDS-polyacrylamide gel electrophoresis (PAGE), followed by electroblotting onto nitrocellulose membranes (Bio-Rad). Immunoblots were incubated with blocking buffer (Tris-buffered saline [TBS] containing 0.05% Tween 20 [TBST] and 5% nonfat dry milk) for 30 min at room temperature and incubated at 4°C overnight with different primary antibodies diluted in blocking buffer. Antibodies used in this study were rabbit polyclonal sera specific for IBDV VP3 (85), ISG-56 (catalog number sc-82946; Santa Cruz Biotechnology), Mx1/2/3 (catalog number sc-50509; Santa Cruz Biotechnology), PKR (catalog number sc-6282; Santa Cruz Biotechnology), p-PKR (catalog number T446; Abcam), total eIF2α (catalog number sc-11386; Santa Cruz Biotechnology), p-eIF2α (catalog number 44728G; Invitrogen), PARP (catalog number 9542; Cell Signaling), NF-κB–p65 (catalog number ab16502; Abcam), histone H2B (catalog number ab52484; Abcam), α-tubulin (catalog number 2125; Cell Signaling), ubiquitin (catalog number sc-8017; Santa Cruz Biotechnology), and β-actin (catalog number sc-47778; Santa Cruz Biotechnology). After incubation with primary antibodies, membranes were washed with TBST and incubated with either a goat anti-rabbit IgG-peroxidase conjugate (Sigma) or a goat anti-mouse IgG-peroxidase conjugate (Sigma), and immunoreactive bands were detected by an enhanced chemiluminescence (ECL) reaction (GE Healthcare).
Caspase 3/7 activity assay.
Quantification of caspase activity was carried out by using the Caspase-Glo 3/7 assay kit (Promega). For this, HeLa or DF-1 cell monolayers grown in 24-well plates were infected and treated with IFN, in duplicate, under the conditions indicated for each experiment. At the indicated times p.i., cells were harvested in medium and kept frozen until their analysis. Twenty-five microliters of the cell lysates under study per well was added to 25 μl of Caspase-Glo 3/7 reagent in a 96-well plate. Plates were shaken gently and then incubated in the dark at room temperature for 60 min before the luciferase activity was recorded by using an Appliskan luminometer (Thermo Scientific).
Quantitative PCR analysis.
Total RNA was isolated by using the High pure RNA isolation kit (Roche) according to the manufacturer's instructions. Purified RNA (500 ng) was reverse transcribed into cDNA by using a random primer and SuperScript III (Invitrogen) reverse transcriptase, according to the manufacturer's protocol. The cDNA was then subjected to qPCR using the gene-specific primers indicated in Table 1. qPCRs were performed in duplicate by using Power SYBR green PCR master mix (Thermo Fisher Scientific), according to the manufacturer's protocol, and by using a Applied Biosystems 7500 real-time PCR system instrument. Reactions were performed as follows: 2 min at 50°C; 10 min at 95°C; 40 cycles of 15 s at 95°C and 1 min at 60°C; and, finally, 15 s at 95°C, 1 min at 60°C, 30 s at 95°C, and 15 s at 60°C to build the melt curve. Gene expression levels were normalized to the hypoxanthine phosphoribosyl transferase 1 (HPRT) gene, and the results were calculated as fold changes in gene expression relative to mock-infected cells by using the delta-delta CT (threshold cycle) method of analysis.
TABLE 1.
List of primers used for RT-qPCR
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| IFNB | GTCAGAGTGGAAATCCTAAG | ACAGCATCTGCTGGTTGAAG |
| ISG-56 | GGGCAGACTGGCAGAAG | CTATAGCGGAAGGGATTTGA |
| ISG-15 | CAGCGAACTCATCTTTGCCAGTA | CCAGCATCTTCACCGTCAGG |
| Mx1 | ACAGGACCATCGGAATCTTG | CCCTTCTTCAGGTGGAACAC |
| EIF2AK2 (PKR) | GCGATACATGAGCCCAGAACAGA | CAAGAATTAGCCCCAAAGCGTAGA |
| BAK | TTGCCACCAGCCTGTTTGAGA | GAAGCCCAGAAGAGCCACCAC |
| TRAIL | TGGGACCAGAGGAAGAAGC | CAGGAATGAATGCCCACTC |
| FAS | TTGTGTGATGAAGGACATGGC | GGTCCGGGTGCAGTTTATTTC |
| TNFA | AGTGAAGTGCTGGCAACCAC | GAGGAAGGCCTAAGGTCCA |
| BCL2 | GAGAGCGTCAACCGGGAGATGTCG | GGTGTGCAGGTGCCGGTTCAGGTA |
| HPRT | TGACACTGGCAAAACAATGCA | GGTCCTTTTCACCAGCAAGCT |
| VP3 | AAGGGCAGCTACGTCGATCTAC | TGGCAACTTCGTCTATGAAAGC |
Plasmids.
For the silencing of human TNF-α, we used five shRNA expression constructs, based on plasmid pLKO.1-Puro with a U6 promoter, which were purchased from Sigma-Aldrich (sh1, TRCN0000355911; sh2, TRCN0000003757; sh3, TRCN0000003759; sh4, TRCN0000003758; sh5, TRCN0000355913). The successful 21-nucleotide human TNF-α targeting sequence was GCAGGTCTACTTTGGGATCAT (sh4). A control construct expressing a nontargeting scrambled shRNA sequence was prepared as previously described (83).
The following plasmids required to generate vesicular stomatitis virus G protein (VSV-G)-pseudotyped lentiviral particles were acquired from Addgene: pMDLg/pRRE (Addgene plasmid 12251), pMD2.G (Addgene plasmid 12259), and pRSV-Rev (Addgene plasmid 12253).
Lentiviral particle production and HeLa cell transduction.
Lentiviral particles were produced in HEK-293T cells by the cotransfection of plasmids pMDLg/pRRE, pMD2.G, and pRSV-Rev, together with the pLKO.1-Pure-TNF-α shRNA vector, as described previously (89). Supernatants were collected at 36 h posttransfection, filtered through a 0.45-μm filter, and used to inoculate HeLa cells. Since the shRNA-expressing lentiviral vectors used confer puromycin resistance, we determined the smallest amount of the supernatant that was sufficient to confer resistance to puromycin (1 μg/ml) to 100% of the cells and used this concentration for cell transduction. Silencing was verified by RT-qPCR at days 4 and 12 postransduction.
Purification of IBDV-derived virus-like particles.
Cultures of BSC-1 cells were infected with VT7LacOI/POLY at an MOI of 10 PFU/cell and maintained in the presence of isopropyl-β-d-thiogalactopyranoside (IPTG). At 20 h p.i., cells were collected and used to purify VLPs by ultracentrifugation on sucrose gradients, as previously described (85). Purified VLPs were visualized by electron microscopy after negative staining and by Western blotting using an anti-VP3 serum along with a virus stock to determine the amount of VLPs required to mimic infection at an MOI of 2 PFU/cell.
Purification of genomic IBDV dsRNA.
Genomic IBDV dsRNA was extracted from preparations of sucrose gradient-purified IBDV as previously described (20). Isolated dsRNA samples were treated with 50 U/mg of RNase T1 (Roche), specifically digesting single-stranded RNA (ssRNA), at 37°C for 30 min to eliminate contaminating ssRNA traces and then subjected to a second round of purification using silica-based minispin columns (Roche). dsRNA concentrations were determined by using a NanoDrop spectrophotometer (Thermo Scientific).
Transfection with purified IBDV dsRNA.
HeLa cell monolayers grown in 24-well plates were transfected with 100 ng of IBDV RNase T1-treated purified dsRNA using Lipofectamine 2000 reagent (Invitrogen) at a 1:1 ratio (RNA/Lipofectamine 2000), according to a previously described procedure (85).
Preparation of cytosolic and nuclear extracts.
Nuclear and cytosolic extracts were prepared from HeLa cells grown in 24-well plates and treated under different experimental conditions by using NE-PER nuclear and cytoplasmic extraction reagents (Thermo Scientific), according to the manufacturer's instructions.
Statistics.
GraphPad Prism version 5.03 software (GraphPad Software, La Jolla, CA) was used to determine statistical significance, using the Student unpaired two-tailed t test.
ACKNOWLEDGMENTS
We are grateful for the excellent technical assistance provided by Antonio Varas, the work of Silvia Gutiérrez-Erlandsson and Ana M. Oña from the CNB Confocal Laser Scanning Microscopy scientific service, and the work of Cristina Patiño and Javier Bueno Chamorro from the CNB Electron Microscopy service. We are also grateful to Nicolas Eterradossi and Sébastien Soubies for lending us the IBDV Bursine and P2 strains and Bernd Kaspers for the CEC 32-511 cell line. In addition, we thank Ramón Rodríguez for critical reading of the manuscript.
This work was supported by grant AGL2014-60095-P (AEI/FEDER, UE) to D.R. and J.F.R. L.L.C.-G. was supported by a predoctoral research contract from the International Fellowship Program of the Caixa Foundation. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Mahgoub HA, Bailey M, Kaiser P. 2012. An overview of infectious bursal disease. Arch Virol 157:2047–2057. doi: 10.1007/s00705-012-1377-9. [DOI] [PubMed] [Google Scholar]
- 2.Ingrao F, Rauw F, Lambrecht B, van den Berg T. 2013. Infectious bursal disease: a complex host-pathogen interaction. Dev Comp Immunol 41:429–438. doi: 10.1016/j.dci.2013.03.017. [DOI] [PubMed] [Google Scholar]
- 3.van den Berg TP, Eterradossi N, Toquin D, Meulemans G. 2000. Infectious bursal disease (Gumboro disease). Rev Sci Tech 19:509–543. doi: 10.20506/rst.19.2.1227. [DOI] [PubMed] [Google Scholar]
- 4.Rodenberg J, Sharma JM, Belzer SW, Nordgren RM, Naqi S. 1994. Flow cytometric analysis of B cell and T cell subpopulations in specific-pathogen-free chickens infected with infectious bursal disease virus. Avian Dis 38:16–21. doi: 10.2307/1591831. [DOI] [PubMed] [Google Scholar]
- 5.Burkhardt E, Muller H. 1987. Susceptibility of chicken blood lymphoblasts and monocytes to infectious bursal disease virus (IBDV). Arch Virol 94:297–303. doi: 10.1007/BF01310722. [DOI] [PubMed] [Google Scholar]
- 6.Kim IJ, Karaca K, Pertile TL, Erickson SA, Sharma JM. 1998. Enhanced expression of cytokine genes in spleen macrophages during acute infection with infectious bursal disease virus in chickens. Vet Immunol Immunopathol 61:331–341. doi: 10.1016/S0165-2427(97)00135-9. [DOI] [PubMed] [Google Scholar]
- 7.Lam KM. 1998. Alteration of chicken heterophil and macrophage functions by the infectious bursal disease virus. Microb Pathog 25:147–155. doi: 10.1006/mpat.1998.0224. [DOI] [PubMed] [Google Scholar]
- 8.Khatri M, Sharma JM. 2006. Infectious bursal disease virus infection induces macrophage activation via p38 MAPK and NF-kappaB pathways. Virus Res 118:70–77. doi: 10.1016/j.virusres.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 9.Reference deleted.
- 10.Sharma JM, Kim IJ, Rautenschlein S, Yeh HY. 2000. Infectious bursal disease virus of chickens: pathogenesis and immunosuppression. Dev Comp Immunol 24:223–235. doi: 10.1016/S0145-305X(99)00074-9. [DOI] [PubMed] [Google Scholar]
- 11.Rauw F, Lambrecht B, van den Berg T. 2007. Pivotal role of ChIFNgamma in the pathogenesis and immunosuppression of infectious bursal disease. Avian Pathol 36:367–374. doi: 10.1080/03079450701589159. [DOI] [PubMed] [Google Scholar]
- 12.Dobos P, Hill BJ, Hallett R, Kells DT, Becht H, Teninges D. 1979. Biophysical and biochemical characterization of five animal viruses with bisegmented double-stranded RNA genomes. J Virol 32:593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mundt E, Kollner B, Kretzschmar D. 1997. VP5 of infectious bursal disease virus is not essential for viral replication in cell culture. J Virol 71:5647–5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mendez F, de Garay T, Rodriguez D, Rodriguez JF. 2015. Infectious bursal disease virus VP5 polypeptide: a phosphoinositide-binding protein required for efficient cell-to-cell virus dissemination. PLoS One 10:e0123470. doi: 10.1371/journal.pone.0123470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yao K, Goodwin MA, Vakharia VN. 1998. Generation of a mutant infectious bursal disease virus that does not cause bursal lesions. J Virol 72:2647–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feldman AR, Lee J, Delmas B, Paetzel M. 2006. Crystal structure of a novel viral protease with a serine/lysine catalytic dyad mechanism. J Mol Biol 358:1378–1389. doi: 10.1016/j.jmb.2006.02.045. [DOI] [PubMed] [Google Scholar]
- 17.Birghan C, Mundt E, Gorbalenya AE. 2000. A non-canonical lon proteinase lacking the ATPase domain employs the ser-Lys catalytic dyad to exercise broad control over the life cycle of a double-stranded RNA virus. EMBO J 19:114–123. doi: 10.1093/emboj/19.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.von Einem UI, Gorbalenya AE, Schirrmeier H, Behrens SE, Letzel T, Mundt E. 2004. VP1 of infectious bursal disease virus is an RNA-dependent RNA polymerase. J Gen Virol 85:2221–2229. doi: 10.1099/vir.0.19772-0. [DOI] [PubMed] [Google Scholar]
- 19.Luque D, Saugar I, Rejas MT, Carrascosa JL, Rodriguez JF, Caston JR. 2009. Infectious bursal disease virus:ribonucleoprotein complexes of a double-stranded RNA virus. J Mol Biol 386:891–901. doi: 10.1016/j.jmb.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dalton RM, Rodriguez JF. 2014. Rescue of infectious birnavirus from recombinant ribonucleoprotein complexes. PLoS One 9:e87790. doi: 10.1371/journal.pone.0087790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lam KM. 1997. Morphological evidence of apoptosis in chickens infected with infectious bursal disease virus. J Comp Pathol 116:367–377. doi: 10.1016/S0021-9975(97)80053-9. [DOI] [PubMed] [Google Scholar]
- 22.Vasconcelos AC, Lam KM. 1995. Apoptosis in chicken embryos induced by the infectious bursal disease virus. J Comp Pathol 112:327–338. doi: 10.1016/S0021-9975(05)80014-3. [DOI] [PubMed] [Google Scholar]
- 23.Ojeda F, Skardova I, Guarda MI, Ulloa J, Folch H. 1997. Proliferation and apoptosis in infection with infectious bursal disease virus: a flow cytometric study. Avian Dis 41:312–316. doi: 10.2307/1592183. [DOI] [PubMed] [Google Scholar]
- 24.Nieper H, Teifke JP, Jungmann A, Lohr CV, Muller H. 1999. Infected and apoptotic cells in the IBDV-infected bursa of Fabricius, studied by double-labelling techniques. Avian Pathol 28:279–285. doi: 10.1080/03079459994777. [DOI] [PubMed] [Google Scholar]
- 25.Vasconcelos AC, Lam KM. 1994. Apoptosis induced by infectious bursal disease virus. J Gen Virol 75(Part 7):1803–1806. doi: 10.1099/0022-1317-75-7-1803. [DOI] [PubMed] [Google Scholar]
- 26.Shahsavandi S, Ebrahimi MM, Sadeghi K, Mahravani H. 2014. Apoptotic response of chicken embryonic fibroblast cells to infectious bursal disease virus infections reflects viral pathogenicity. In Vitro Cell Dev Biol Anim 50:858–864. doi: 10.1007/s11626-014-9783-9. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez-Lecompte JC, Nino-Fong R, Lopez A, Frederick Markham RJ, Kibenge FS. 2005. Infectious bursal disease virus (IBDV) induces apoptosis in chicken B cells. Comp Immunol Microbiol Infect Dis 28:321–337. doi: 10.1016/j.cimid.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 28.Sharma JM, Dohms J, Walser M, Snyder DB. 1993. Presence of lesions without virus replication in the thymus of chickens exposed to infectious bursal disease virus. Avian Dis 37:741–748. doi: 10.2307/1592023. [DOI] [PubMed] [Google Scholar]
- 29.Jungmann A, Nieper H, Muller H. 2001. Apoptosis is induced by infectious bursal disease virus replication in productively infected cells as well as in antigen-negative cells in their vicinity. J Gen Virol 82:1107–1115. doi: 10.1099/0022-1317-82-5-1107. [DOI] [PubMed] [Google Scholar]
- 30.Inoue M, Fukuda M, Miyano K. 1994. Thymic lesions in chicken infected with infectious bursal disease virus. Avian Dis 38:839–846. doi: 10.2307/1592122. [DOI] [PubMed] [Google Scholar]
- 31.Liu M, Vakharia VN. 2006. Nonstructural protein of infectious bursal disease virus inhibits apoptosis at the early stage of virus infection. J Virol 80:3369–3377. doi: 10.1128/JVI.80.7.3369-3377.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Z, Wang Y, Xue Y, Li X, Cao H, Zheng SJ. 2012. Critical role for voltage-dependent anion channel 2 in infectious bursal disease virus-induced apoptosis in host cells via interaction with VP5. J Virol 86:1328–1338. doi: 10.1128/JVI.06104-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fernandez-Arias A, Martinez S, Rodriguez JF. 1997. The major antigenic protein of infectious bursal disease virus, VP2, is an apoptotic inducer. J Virol 71:8014–8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Busnadiego I, Maestre AM, Rodriguez D, Rodriguez JF. 2012. The infectious bursal disease virus RNA-binding VP3 polypeptide inhibits PKR-mediated apoptosis. PLoS One 7:e46768. doi: 10.1371/journal.pone.0046768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A. 2015. Type I interferons in infectious disease. Nat Rev Immunol 15:87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delgui LR, Rodriguez JF, Colombo MI. 2013. The endosomal pathway and the Golgi complex are involved in the infectious bursal disease virus life cycle. J Virol 87:8993–9007. doi: 10.1128/JVI.03152-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mo CW, Cao YC, Lim BL. 2001. The in vivo and in vitro effects of chicken interferon alpha on infectious bursal disease virus and Newcastle disease virus infection. Avian Dis 45:389–399. doi: 10.2307/1592978. [DOI] [PubMed] [Google Scholar]
- 38.O'Neill AM, Livant EJ, Ewald SJ. 2010. Interferon alpha-induced inhibition of infectious bursal disease virus in chicken embryo fibroblast cultures differing in Mx genotype. Avian Dis 54:802–806. doi: 10.1637/9001-072309-Reg.1. [DOI] [PubMed] [Google Scholar]
- 39.Cai M, Zhu F, Shen P. 2012. Expression and purification of chicken beta interferon and its antivirus immunological activity. Protein Expr Purif 84:123–129. doi: 10.1016/j.pep.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 40.Ye C, Jia L, Sun Y, Hu B, Wang L, Lu X, Zhou J. 2014. Inhibition of antiviral innate immunity by birnavirus VP3 protein via blockage of viral double-stranded RNA binding to the host cytoplasmic RNA detector MDA5. J Virol 88:11154–11165. doi: 10.1128/JVI.01115-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.O'Brien MA, Moravec RA, Riss TL. 2001. Poly(ADP-ribose) polymerase cleavage monitored in situ in apoptotic cells. Biotechniques 30:886–891. [DOI] [PubMed] [Google Scholar]
- 42.Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, Esteban M. 2006. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev 70:1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zmurko J, Marques RE, Schols D, Verbeken E, Kaptein SJ, Neyts J. 2016. The viral polymerase inhibitor 7-deaza-2′-C-methyladenosine is a potent inhibitor of in vitro Zika virus replication and delays disease progression in a robust mouse infection model. PLoS Negl Trop Dis 10:e0004695. doi: 10.1371/journal.pntd.0004695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zamanian-Daryoush M, Mogensen TH, DiDonato JA, Williams BR. 2000. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol Cell Biol 20:1278–1290. doi: 10.1128/MCB.20.4.1278-1290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gil J, Alcami J, Esteban M. 2000. Activation of NF-kappa B by the dsRNA-dependent protein kinase, PKR involves the I kappa B kinase complex. Oncogene 19:1369–1378. doi: 10.1038/sj.onc.1203448. [DOI] [PubMed] [Google Scholar]
- 46.Gil J, Garcia MA, Gomez-Puertas P, Guerra S, Rullas J, Nakano H, Alcami J, Esteban M. 2004. TRAF family proteins link PKR with NF-kappa B activation. Mol Cell Biol 24:4502–4512. doi: 10.1128/MCB.24.10.4502-4512.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Takada Y, Ichikawa H, Pataer A, Swisher S, Aggarwal BB. 2007. Genetic deletion of PKR abrogates TNF-induced activation of IkappaBalpha kinase, JNK, Akt and cell proliferation but potentiates p44/p42 MAPK and p38 MAPK activation. Oncogene 26:1201–1212. doi: 10.1038/sj.onc.1209906. [DOI] [PubMed] [Google Scholar]
- 48.Hayden MS, Ghosh S. 2014. Regulation of NF-κB by TNF family cytokines. Semin Immunol 26:253–266. doi: 10.1016/j.smim.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Han YH, Moon HJ, You BR, Park WH. 2009. The effect of MG132, a proteasome inhibitor on HeLa cells in relation to cell growth, reactive oxygen species and GSH. Oncol Rep 22:215–221. [PubMed] [Google Scholar]
- 50.Gilmore TD, Herscovitch M. 2006. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene 25:6887–6899. doi: 10.1038/sj.onc.1209982. [DOI] [PubMed] [Google Scholar]
- 51.Lee KH, Jeong J, Yoo CG. 2013. Long-term incubation with proteasome inhibitors (PIs) induces IkappaBalpha degradation via the lysosomal pathway in an IkappaB kinase (IKK)-dependent and IKK-independent manner. J Biol Chem 288:32777–32786. doi: 10.1074/jbc.M113.480921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shin HM, Kim MH, Kim BH, Jung SH, Kim YS, Park HJ, Hong JT, Min KR, Kim Y. 2004. Inhibitory action of novel aromatic diamine compound on lipopolysaccharide-induced nuclear translocation of NF-kappaB without affecting IkappaB degradation. FEBS Lett 571:50–54. doi: 10.1016/j.febslet.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 53.van den Berg TP. 2000. Acute infectious bursal disease in poultry: a review. Avian Pathol 29:175–194. doi: 10.1080/03079450050045431. [DOI] [PubMed] [Google Scholar]
- 54.Tanaka N, Sato M, Lamphier MS, Nozawa H, Oda E, Noguchi S, Schreiber RD, Tsujimoto Y, Taniguchi T. 1998. Type I interferons are essential mediators of apoptotic death in virally infected cells. Genes Cells 3:29–37. doi: 10.1046/j.1365-2443.1998.00164.x. [DOI] [PubMed] [Google Scholar]
- 55.Chawla-Sarkar M, Lindner DJ, Liu Y-F, Williams BR, Sen GC, Silverman RH, Borden EC. 2003. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 8:237–249. doi: 10.1023/A:1023668705040. [DOI] [PubMed] [Google Scholar]
- 56.Robertsen B, Bergan V, Rokenes T, Larsen R, Albuquerque A. 2003. Atlantic salmon interferon genes: cloning, sequence analysis, expression, and biological activity. J Interferon Cytokine Res 23:601–612. doi: 10.1089/107999003322485107. [DOI] [PubMed] [Google Scholar]
- 57.Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, Perlman S. 2016. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe 19:181–193. doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moulin HR, Liniger M, Python S, Guzylack-Piriou L, Ocana-Macchi M, Ruggli N, Summerfield A. 2011. High interferon type I responses in the lung, plasma and spleen during highly pathogenic H5N1 infection of chicken. Vet Res 42:6. doi: 10.1186/1297-9716-42-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Panigrahi R, Hazari S, Chandra S, Chandra PK, Datta S, Kurt R, Cameron CE, Huang Z, Zhang H, Garry RF, Balart LA, Dash S. 2013. Interferon and ribavirin combination treatment synergistically inhibit HCV internal ribosome entry site mediated translation at the level of polyribosome formation. PLoS One 8:e72791. doi: 10.1371/journal.pone.0072791. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 60.Dabo S, Meurs EF. 2012. dsRNA-dependent protein kinase PKR and its role in stress, signaling and HCV infection. Viruses 4:2598–2635. doi: 10.3390/v4112598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Munir M, Berg M. 2013. The multiple faces of proteinkinase R in antiviral defense. Virulence 4:85–89. doi: 10.4161/viru.23134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marchal JA, Lopez GJ, Peran M, Comino A, Delgado JR, Garcia-Garcia JA, Conde V, Aranda FM, Rivas C, Esteban M, Garcia MA. 2014. The impact of PKR activation: from neurodegeneration to cancer. FASEB J 28:1965–1974. doi: 10.1096/fj.13-248294. [DOI] [PubMed] [Google Scholar]
- 63.Garcia MA, Meurs EF, Esteban M. 2007. The dsRNA protein kinase PKR: virus and cell control. Biochimie 89:799–811. doi: 10.1016/j.biochi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 64.Kaufman RJ. 1999. Double-stranded RNA-activated protein kinase mediates virus-induced apoptosis: a new role for an old actor. Proc Natl Acad Sci U S A 96:11693–11695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mundt E. 2007. Human MxA protein confers resistance to double-stranded RNA viruses of two virus families. J Gen Virol 88:1319–1323. doi: 10.1099/vir.0.82526-0. [DOI] [PubMed] [Google Scholar]
- 66.McAllister CS, Samuel CE. 2009. The RNA-activated protein kinase enhances the induction of interferon-β and apoptosis mediated by cytoplasmic RNA sensors. J Biol Chem 284:1644–1651. doi: 10.1074/jbc.M807888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pham AM, Santa Maria FG, Lahiri T, Friedman E, Marie IJ, Levy DE. 2016. PKR transduces MDA5-dependent signals for type I IFN induction. PLoS Pathog 12:e1005489. doi: 10.1371/journal.ppat.1005489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McAllister CS, Toth AM, Zhang P, Devaux P, Cattaneo R, Samuel CE. 2010. Mechanisms of protein kinase PKR-mediated amplification of beta interferon induction by C protein-deficient measles virus. J Virol 84:380–386. doi: 10.1128/JVI.02630-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lee CC, Wu CC, Lin TL. 2014. Chicken melanoma differentiation-associated gene 5 (MDA5) recognizes infectious bursal disease virus infection and triggers MDA5-related innate immunity. Arch Virol 159:1671–1686. doi: 10.1007/s00705-014-1983-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee CC, Wu CC, Lin TL. 2015. Role of chicken melanoma differentiation-associated gene 5 in induction and activation of innate and adaptive immune responses to infectious bursal disease virus in cultured macrophages. Arch Virol 160:3021–3035. doi: 10.1007/s00705-015-2612-y. [DOI] [PubMed] [Google Scholar]
- 71.Valli A, Busnadiego I, Maliogka V, Ferrero D, Caston JR, Rodriguez JF, Garcia JA. 2012. The VP3 factor from viruses of Birnaviridae family suppresses RNA silencing by binding both long and small RNA duplexes. PLoS One 7:e45957. doi: 10.1371/journal.pone.0045957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rasschaert J, Ladriere L, Urbain M, Dogusan Z, Katabua B, Sato S, Akira S, Gysemans C, Mathieu C, Eizirik DL. 2005. Toll-like receptor 3 and STAT-1 contribute to double-stranded RNA+ interferon-gamma-induced apoptosis in primary pancreatic beta-cells. J Biol Chem 280:33984–33991. doi: 10.1074/jbc.M502213200. [DOI] [PubMed] [Google Scholar]
- 73.Ghosh S, Dass JF. 2016. Study of pathway cross-talk interactions with NF-kappaB leading to its activation via ubiquitination or phosphorylation: a brief review. Gene 584:97–109. doi: 10.1016/j.gene.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 74.Baud V, Karin M. 2001. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol 11:372–377. doi: 10.1016/S0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- 75.Jing H, Lee S. 2014. NF-κB in cellular senescence and cancer treatment. Mol Cells 37:189–195. doi: 10.14348/molcells.2014.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kaltschmidt B, Kaltschmidt C, Hofmann TG, Hehner SP, Droge W, Schmitz ML. 2000. The pro- or anti-apoptotic function of NF-kappaB is determined by the nature of the apoptotic stimulus. Eur J Biochem 267:3828–3835. doi: 10.1046/j.1432-1327.2000.01421.x. [DOI] [PubMed] [Google Scholar]
- 77.Skjesol A, Aamo T, Hegseth MN, Robertsen B, Jorgensen JB. 2009. The interplay between infectious pancreatic necrosis virus (IPNV) and the IFN system: IFN signaling is inhibited by IPNV infection. Virus Res 143:53–60. doi: 10.1016/j.virusres.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fernandez S, Tanaskovic S, Helbig K, Rajasuriar R, Kramski M, Murray JM, Beard M, Purcell D, Lewin SR, Price P, French MA. 2011. CD4+ T-cell deficiency in HIV patients responding to antiretroviral therapy is associated with increased expression of interferon-stimulated genes in CD4+ T cells. J Infect Dis 204:1927–1935. doi: 10.1093/infdis/jir659. [DOI] [PubMed] [Google Scholar]
- 79.Teijaro JR, Ng C, Lee AM, Sullivan BM, Sheehan KC, Welch M, Schreiber RD, de la Torre JC, Oldstone MB. 2013. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340:207–211. doi: 10.1126/science.1235214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wilson EB, Yamada DH, Elsaesser H, Herskovitz J, Deng J, Cheng G, Aronow BJ, Karp CL, Brooks DG. 2013. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340:202–207. doi: 10.1126/science.1235208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Summerfield A, Alves M, Ruggli N, de Bruin MG, McCullough KC. 2006. High IFN-alpha responses associated with depletion of lymphocytes and natural IFN-producing cells during classical swine fever. J Interferon Cytokine Res 26:248–255. doi: 10.1089/jir.2006.26.248. [DOI] [PubMed] [Google Scholar]
- 82.Rue CA, Susta L, Cornax I, Brown CC, Kapczynski DR, Suarez DL, King DJ, Miller PJ, Afonso CL. 2011. Virulent Newcastle disease virus elicits a strong innate immune response in chickens. J Gen Virol 92:931–939. doi: 10.1099/vir.0.025486-0. [DOI] [PubMed] [Google Scholar]
- 83.Zhang P, Samuel CE. 2007. Protein kinase PKR plays a stimulus- and virus-dependent role in apoptotic death and virus multiplication in human cells. J Virol 81:8192–8200. doi: 10.1128/JVI.00426-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fernandez-Escobar M, Najera JL, Baldanta S, Rodriguez D, Way M, Esteban M, Guerra S. 2015. Suppression of NYVAC infection in HeLa cells requires RNase L but is independent of protein kinase R activity. J Virol 90:2135–2141. doi: 10.1128/JVI.02576-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Fernandez-Arias A, Risco C, Martinez S, Albar JP, Rodriguez JF. 1998. Expression of ORF A1 of infectious bursal disease virus results in the formation of virus-like particles. J Gen Virol 79(Part 5):1047–1054. doi: 10.1099/0022-1317-79-5-1047. [DOI] [PubMed] [Google Scholar]
- 86.Ward GA, Stover CK, Moss B, Fuerst TR. 1995. Stringent chemical and thermal regulation of recombinant gene expression by vaccinia virus vectors in mammalian cells. Proc Natl Acad Sci U S A 92:6773–6777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schwarz H, Harlin O, Ohnemus A, Kaspers B, Staeheli P. 2004. Synthesis of IFN-beta by virus-infected chicken embryo cells demonstrated with specific antisera and a new bioassay. J Interferon Cytokine Res 24:179–184. doi: 10.1089/107999004322917025. [DOI] [PubMed] [Google Scholar]
- 88.Ramakrishnan MA. 2016. Determination of 50% endpoint titer using a simple formula. World J Virol 5:85–86. doi: 10.5501/wjv.v5.i2.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gastaminza P, Cheng G, Wieland S, Zhong J, Liao W, Chisari FV. 2008. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J Virol 82:2120–2129. doi: 10.1128/JVI.02053-07. [DOI] [PMC free article] [PubMed] [Google Scholar]