

SUMMARY
While gene expression dynamics have been extensively catalogued during hematopoietic differentiation in the adult, less is known about transcriptome diversity of human hematopoietic stem cells (HSCs) during development. To characterize transcriptional and post-transcriptional changes in HSCs during development, we leveraged high-throughput genomic approaches to profile miRNAs, lincRNAs, and mRNAs. Our findings indicate that HSCs manifest distinct alternative splicing patterns in key hematopoietic regulators. Detailed analysis of the splicing dynamics and function of one such regulator, HMGA2, identified an alternative isoform that escapes miRNA-mediated targeting. We further identified the splicing kinase CLK3 that, by regulating HMGA2 splicing, preserves HMGA2 function in the setting of an increase in let-7 miRNA levels, delineating how CLK3 and HMGA2 form a functional axis that influences HSC properties during development. Collectively, our study highlights molecular mechanisms by which alternative splicing and miRNA-mediated post-transcriptional regulation impact the molecular identity and stage-specific developmental features of human HSCs.
eTOC
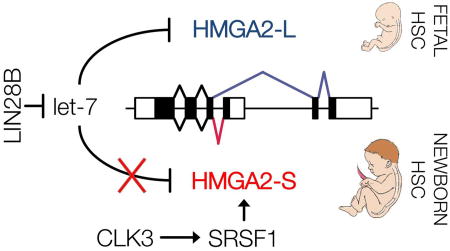
Human hematopoietic stem cells (HSCs) display substantial transcriptional diversity during development. Here, we investigated the contribution of alternative splicing on such diversity by analyzing the dynamics of a key hematopoietic regulator, HMGA2. Next, we showed that CLK3, by regulating the splicing pattern of HMGA2, reinforces an HSC-specific program.
INTRODUCTION
Hematopoiesis is the coordinated lineage commitment, differentiation, and expansion of hematopoietic stem cells (HSCs) to generate mature blood cells. Interestingly, hematopoiesis occurs at distinct anatomic sites during development, including the yolk sac and fetal liver during fetal life and bone marrow during post-natal life (Mikkola and Orkin, 2006). While HSCs from these sites are all capable of generating the full complement of mature blood cells, they differ in certain characteristics. For example, several studies have shown that HSCs from earlier developmental zones have higher regenerative capacity (Babovic and Eaves, 2014). Understanding differences in hematopoiesis along development can shed insight on processes important in HSC function and regeneration, with important clinical applications in stem cell transplantation.
Recent research has leveraged high throughput genomic profiling to characterize the hematopoietic hierarchy at a molecular level (Vedi et al., 2016). Although these studies have generated novel insights into hematopoietic lineage commitment and differentiation, our understanding of the molecular and functional differences among developmentally-distinct HSC populations remains inadequate. Murine studies have begun to address this gap (Cabezas-Wallscheid et al., 2014; McKinney-Freeman et al., 2012), but owing to species-specific differences (Doulatov et al., 2012), emerging efforts are starting to characterize the molecular diversity along the human hematopoietic hierarchy (Notta et al., 2016; Novershtern et al., 2011).
The generation and tuning of expression of alternative splicing isoforms can contribute to significant transcriptional diversity (Wang et al., 2008). However, studies are just beginning to delineate their involvement in hematopoiesis (Chen et al., 2014; Rentas et al., 2016). The discovery that core spliceosomal proteins and accessory regulatory splicing factors are frequently mutated in various hematopoietic malignancies (Sperling et al., 2016; Yoshida et al., 2011) has further spurred research into the regulation and role of alternative splicing during normal hematopoiesis (Chen et al., 2014; Crews et al., 2016a; Wong et al., 2013).
Here, we dissect the transcriptional identity of human HSCs from multiple developmental stages and establish developmental stage-specific expression signatures. Through integrative analyses, we then describe how alternative splicing and miRNA-mediated post-transcriptional regulation interplay to regulate HSC identity.
RESULTS
Transcriptional diversity in human HSCs across ontogeny
Fetal liver (FL), umbilical cord blood (CB), and bone marrow (BM) represent distinct progressive stages of hematopoiesis during development. To dissect transcriptional features of human HSC populations, we prospectively isolated immunophenotypically-defined early HSCs (CD34+ CD38− CD90+ CD45RA−) from FL, CB, and BM, as well as corresponding committed CD34+CD38+ progenitor populations (PROG). We performed RNA-sequencing (RNA-seq) and miRNA profiling (Figure 1A).
FIGURE 1. Transcriptional diversity among human HSCs along development.

A- Schematic of FACS sorting and transcriptomic analyses. Hematopoietic stem cells (HSC) and progenitor (PROG) cells were isolated according to the indicated surface markers from human Fetal Liver (FL), umbilical Cord Blood (CB) and Bone Marrow (BM) CD34+ cells. RNA-sequencing of both coding (mRNAs) and noncoding RNAs (lincRNAs) was performed along with miRNA expression quantification.
B- (left) Pairwise comparisons showing the number of differentially expressed (DE) genes at FDR<0.01. (right) DE transcripts at isoform level (FDR < 0.01), but not at gene level (FDR > 0.01). Representative genes for each category are shown on the right. The full dataset can be found in Table S1.
C- Expression heatmap of DE mRNA isoforms (top), lincRNAs (middle) and miRNAs (bottom). Representative isoforms shared among all HSC types (all-HSC) or progenitors (all-PROG), or specific to each population (as listed on left) are indicated on the right. The full dataset can be found in Table S2.
D- (top) Gene structure of the most highly expressed PROM1 isoforms (ISO) detected by RNA-seq. (bottom). Barplot showing PROM1 expression (in FPKM) in the indicated HSC samples. Reference exons numbers are listed on top (constitutive exons are not shown), with coding exons in black and UTRs in gray.
E- Violin plot representing distributions of statistically significant ΔPSI values (p<0.05) for different classes of PSI events: alternative 3’ splice site (A3), alternative 5’ splice site (A5), alternative first exon (AF), alternative last exon (AL), mutually exclusive exon (MX), retained intron (RI), and skipping exon (SE). Separate violins are shown for each pairwise comparison of HSC samples, and the number of events in each violin are shown on the right. ΔPSI values are shown for the second sample as compared to the first sample in each pair.
F- MEG3 lincRNA expression quantification by RNA-seq (in FPKM) in HSC and PROG samples.
G- Barplot showing expression of let-7 family members (red) and miR-126 (green) in HSCs. Expression is shown as the percentage of total measured miRNA counts for each HSC population. Mean +/− s.d. values are shown for D, F and G. FPKM is Fragments Per Kilobase of transcript per Million mapped reads.
H- BubbleMap visualization (Spinelli et al., 2015) of representative gene set enrichment analysis (GSEA) results between pairs of HSC samples. As indicated in the legend, for each GO category, colors (red versus blue) correspond to the sample label, shades represent statistical significance (FDR) and the area of the circle represents the enrichment (Normalized Enrichment Score, NES). Empty circles correspond to non-significant enrichments (FDR>0.05). The full dataset can be found in Table S4.
Transitions from FL to CB and from CB to BM HSCs were marked by substantial changes in gene expression (2469 and 1572 genes, respectively; FDR < 0.01) (Figure 1B, left panel). While recent studies have highlighted several hallmarks genes for HSC identity (e.g. HOXA9, LMO2, MECOM; (Ebina and Rossi, 2015), our results suggest that they are in fact highly dynamic across HSC populations, with a limited set of genes uniformly expressed across HSC populations (e.g. HLF, PRDM16 - Figure S1A and Table S1A). Additionally, our analysis highlights several factors not intrinsic to HSCs, such as genes from the niche in which HSCs develop (e.g., liver genes like KDR and FCN2 in FL-HSCs) and genes involved in blood pressure regulation (e.g., AVP in CB-HSCs, Figure S1B).
RNA processing events generate splicing isoforms that vary across cell types, contribute extensively to functional diversity (Wang et al., 2008), and have been implicated in hematopoietic aging and leukemia pathogenesis (Crews et al., 2016b). Thus, we expanded our analysis to examine the transcriptional landscape at the isoform level (Trapnell et al., 2012). We detected a large number of genes (215 in CB vs FL, 105 in CB vs BM; FDR < 0.01), including key regulators HMGA2, DNMT1, and MEIS1, that were differentially expressed among HSC populations at the isoform level, but displayed little to no differential expression at the gene level (Figure 1B, right panel and Table S1B). We also refined the isoform-level analysis by examining differential usage of 5’UTRs, 3’UTRs, coding sequences (CDS), and transcriptional start sites (TSS) (Figure S1C, related to Figure 1B).
Based on the observed transcriptional diversity, we generated a map of stage-specific mRNA and lincRNAs, isoforms, and miRNAs (Figure 1C and Table S2A-B). As an illustration, we highlight PROM1, previously implicated in stem cell biology (Miraglia et al., 1997). We detected six isoforms of PROM1, which display distinct expression patterns across HSCs. In particular, we detected isoforms with differential inclusion of exon 3, which encodes for part of the core prominin domain (Figure 1D and Figure S1D).
To further understand the alternative splicing patterns in HSC populations, we performed pairwise percent spliced-in (PSI) analyses of exons among the HSC populations (Alamancos et al., 2015). We examined different splicing events, including alternative 5’ splice site (A5), alternative 3’ splice site (A3), alternative first exon (AF), alternative last exon (AL), mutually exclusive exon (MX), retained intron (RI), and skipping exon (SE) (Figure 1E). Interestingly, there appeared to be an increase in RI events along HSC development from FL to CB and to BM-HSCs (both p<0.05).
Given the important roles for lincRNAs in stem cells (Fatica and Bozzoni, 2014), we performed de novo lincRNA discovery from the RNA-seq data. We identified 6905 lincRNAs, 76 of which were differentially-expressed among HSC and PROG populations, suggesting that lincRNAs contribute to transcriptional diversity of HSCs (Figure 1C, middle panel and Table S2A). MEG3, an HSC-specific lincRNA implicated in maintenance of LT--HSC function (Qian et al., 2016), displayed a developmentally-regulated expression pattern (Figure 1F).
Analysis of miRNA expression uncovered additional transcriptome diversity (Figure 1C, bottom panel and Table S2B). For example, let-7 family members and miR-126 are highly expressed across HSCs (together accounting for as much as 20% of the total measured miRNA content), but demonstrate a developmentally-regulated expression pattern (Figure 1G).
To understand the function of the differentially expressed genes, we applied gene set enrichment analysis (GSEA) to examine enrichment among curated gene sets (Figure 1H). FL-HSCs were enriched for “cell cycle” and “checkpoints” signatures. In contrast, CB-HSCs were enriched in “RNA metabolism” and “3’UTR mediated translational regulation” pathways. Broad expression of target genes of known transcriptional regulators (“MYC targets”, “EZH2 targets”) were also observed among different HSC populations.
Together, these analyses defined developmental-stage specific molecular signatures for each HSC population that reflect substantial transcriptional diversity at the gene and isoform levels and which is also present in noncoding RNAs (Figure 1C and Table S2A-B).
Identification of an HSC stage-specific HMGA2 isoform
Our analyses identified a number of transcripts differentially-expressed at the isoform, but not the gene level (Figure 1B, left panel and Table S1B). These include key HSC regulators whose differential expression patterns were previously undetected with gene-level analyses. Among the most differentially-expressed was an isoform of HMGA2 (ENST00000403681.2) in the transition from FL- to CB-HSCs (>5 fold decrease, FDR<0.01), but which remained relatively unchanged at the gene level (FDR>0.9) (Table S1B). HMGA2 is a component of the LIN28-let-7 axis that regulates development (Shyh-Chang and Daley, 2013). Consistent with LIN28’s role in inhibiting let-7 biogenesis (Viswanathan et al., 2008), we observed a gradual decrease of LIN28B during maturation from FL- to BM-HSCs, with a concomitant increase in levels of let-7 family members (Figure 2A). Hematopoietic progenitor cells (HPC) derived from in vitro differentiation of iPSC displayed the highest levels of LIN28B and lowest levels of let-7 members, reflecting their embryonic nature (Chadwick et al., 2003). More broadly, we observed that the top 100 in silico predicted let-7 targets (Agarwal et al., 2015) were inversely correlated with expression of let-7 (Figure 2A).
FIGURE 2. HMGA2 alternative splicing in human HSCs.

A- Heatmap showing expression of LIN28B, let-7 family members, the ranked median expression of the top 100 predicted let-7 targets, and HMGA2. Previously dissected regulatory relationships of the LIN28-let-7-HMGA2 pathway (Viswanathan et al., 2008) are indicated with arrows. Scales are shown on the right as normalized z-scores.
B- Visualization of RNA-seq reads (black bars) mapping at HMGA2 (drawn to scale) in the indicated samples. A sashimi plot displaying the major splice junctions (FL-HSCs, red; CB-HSCs, blue) is superimposed. The abundance of each splicing junction is indicated and is normalized to the counts for the shared exon 2–3 splice junction (with value of 1). Arrows indicate the reads mapping to the terminal exons that distinguish the HMGA2-L and HMGA2-S isoforms.
C- Structure of the human HMGA2 locus (not drawn to scale). In the middle, locus coordinates are indicated along with coding exons (black) and UTRs (gray). Asterisk indicates location of major chromosomal rearrangements detected in malignancies (Kazmierczak et al., 1996). Red and blue dashed lines indicate how exons are spliced to result in HMGA2-L (top) and HMGA2-S (bottom). Gray boxes indicate the AT-binding hook domains. Green and black slashes indicate predicted binding sites of let-7 family members and other conserved HSC-expressed miRNAs respectively.
D- (top) Digital gel from RT-PCR electropherogram showing expression of HMGA2-L and HMGA2-S from the indicated HSC populations. Sizes of the amplicons are indicated on the right, along with the structure of each isoform (coding exons in black, UTRs in grey) and the position of the primers used for PCR co-amplification (black dots). (bottom) Digital gel from RT-PCR electropherogram showing expression of HMGA2 pre-mRNA from the indicated HSC populations. HPRT1 pre-mRNA was used as the control for both gels. Expression levels of HMGA2 isoforms are indicated in Figure S2 and Table S2A.
E- Shaded circles show HMGA2 isoform expression (z-score normalized FPKM values) across HSCs and CB hierarchy from publicly available data (Stunnenberg et al., 2016). MPP: multipotent progenitors; CMP: common myeloid progenitors; MEP: myeloid erythroid progenitors; GMP: granulocyte macrophage progenitors; CLP: common lymphoid progenitors.
HMGA2, a well-characterized target of let-7 (Lee and Dutta, 2007), was surprisingly discordant in expression with respect to let-7. HMGA2 was highly expressed in both CB- and FL-HSCs, despite let-7 levels being higher in CB as compared to FL-HSCs (Figure 2A). RNA-seq results highlighted a complex splicing pattern with at least four HMGA2 isoforms expressed in HSCs (Figure S2A–B). Quantitative visualization of the RNA-seq reads revealed that the canonical full-length isoform (ENST00000403681.2; hereafter named HMGA2-L) is highly expressed in FL-HSCs, while CB-HSCs show high expression of a shorter isoform (ENST00000393578.3; hereafter named HMGA2-S) (Figure 2B). HMGA2-L and HMGA2-S share the first three exons but differ in their terminal exon usage (including C-terminal domains and 3’UTRs) (Figure 2C, Figure S2A). Notably, the HMGA2-S 3’UTR is only one third the length of the HMGA2-L 3’UTR and is devoid of most of the conserved miRNA sites, including the seven experimentally-validated let-7 sites (Mayr et al., 2007) (Figure 2C). Semi-quantitative PCR confirmed the expression patterns of HMGA2-L and HMGA2-S isoforms (Figure 2D, top panel). Moreover, HMGA2 pre-mRNA levels were high and comparable in FL- and CB-HSCs, before decreasing in BM-HSCs, suggesting that in the transition between CB- to BM-HSCs, HMGA2 expression is likely down-regulated at the transcriptional rather than post-transcriptional level (Figure 2D, low panel). Global analyses identified several other genes that in the HSC developmental transitions display a different major isoform with a distinct 3’UTR devoid of let-7 binding sites (Table S3).
Using public data from the Blueprint Epigenome Project (Stunnenberg et al., 2016), we investigated expression of HMGA2-L and HMGA2-S in six stem and progenitor cell populations derived from the CB hierarchy (Figure 2E, bottom panel). HMGA2-S is detectable at high levels in HSCs and in multipotent progenitors. Conversely, HMGA2-L is expressed at low levels across these same six CB-derived hematopoietic populations, consistent with our data (Figure 2E, top panel).
Genome-wide mapping of HMGA2 isoform chromatin binding
HMGA2 is an architectural transcription factor, binding chromatin broadly to help recruit other transcription factors and regulate gene expression (Ozturk et al., 2014). To determine whether the proteins encoded by the HMGA2 isoforms have different functions, we evaluated HMGA2 chromatin occupancy genome-wide by performing isoform-specific chromatin immunoprecipitation-sequencing (ChIP-seq) in two hematopoietic cell types: K562 cells and conditionally-immortalized iPSC-derived hematopoietic progenitors (HPC-5F) (Doulatov et al., 2013). These cells were selected for their variable expression of endogenous HMGA2 (FPKM=0 in K562; FPKM=44 in HPC-5F) (Uhlén et al., 2015).
We used lentiviral constructs containing V5-tagged HMGA2-L (L-V5) and HMGA2-S (S-V5) open reading frame (ORF) to infect K562 and HPC-5F cells. Both V5 (Figure 3A) and HMGA2 (not shown) antibodies immunoprecipitated a large amount of DNA upon overexpression (O/E) of either construct. ChIP-seq of the immunoprecipitate from HPC-5F revealed that, unlike the H3K4me2 profile, HMGA2 binding is broad, as recently shown (Colombo et al., 2017), and that HMGA2-L and HMGA2-S proteins display comparable binding patterns (Figure 3B). Ontology analysis revealed that loci with high HMGA2-L and HMGA2-S binding are enriched for genes related to cell cycle, DNA and RNA metabolism (Figure S3A).
FIGURE 3. Protein function of HMGA2 isoforms.

A- Amount of DNA immunoprecipitated using V5 antibody in K562 and HPC-5F cells transduced with a control (C) or V5-tagged HMGA2-L (L) or HMGA2-S (S) ORF lentiviral overexpression (O/E) constructs. Mean +/− s.e.m. values are shown.
B- Visualization of ChIP-seq read mapping at a sample locus. HPC-5F cells were transduced with V5-tagged HMGA2-L or HMGA2-S ORF constructs for overexpression of the protein coding regions of the respective isoforms (L or S). ChIP-seq profiles are shown for tagged-HMGA2 (V5) or H3K4me2 (K4me2). Genes at the locus are indicated on the bottom.
C- Number of gene promoters (total n=28314) enriched for HMGA2 (V5) and H3K4me2 (K4) upon overexpression of HMGA2-L (HMGA2-L-V5) or HMGA2-S (HMGA2-S-V5) ORFs. Promoters are considered highly enriched (Hi) for V5 or K4 if they rank in the top quartile of normalized read counts or depleted (Lo) if they fall in the bottom quartile.
D- A/T content (as a percent) within immunoprecipitated promoter regions in the V5 Hi and V5 Lo categories or total promoter regions (as a background control). First and third quartiles, medians, and interquartile ranges are shown.
E- HMGA2-L and HMGA2-S binding enrichment analysis shows 8456 differentially enriched promoters over the background signal. Correlation between HMGA2-L (L) and HMGA2-S (S)-enriched promoters is shown for V5 Hi and V5 Lo fractions. Values are reported as the log2 sum of ChIP replicate signals at promoter regions. PCC is Pearson′s correlation coefficient (r^2). Enrichment of GO categories from V5 Hi promoters are shown in Figure S3A.
F- Differential expression analysis identified 1078 differentially expressed genes between HMGA2-L and HMGA2-S and the empty control. Correlation in expression between HMGA2-L (L) and HMGA2-S (S) treatments is shown. Values are reported as log2 average of FPKM replicates. PCC is Pearson′s correlation coefficient (r^2). The full pairwise comparisons are shown in Figure S3C.
G- BubbleMap visualization (Spinelli et al., 2015) of GSEA results in HPC-5F cells transduced with HMGA2-L and HMGA2-S ORFs or control (CTRL) vectors. Gene sets were derived from HSC and PROG-specific signatures from Figure 1C (see Table S2C). As indicated in the legend, colors (red versus blue) correspond to the sample label, shades represent statistical significance (FDR) and the area of the circle represents the enrichment (Normalized Enrichment Score, NES). Empty circles correspond to non-significant (NS) enrichments (FDR>0.05).
H- Human chimerism as percentage of GFP+CD45.1+ in the injected femur of xenografted mice 16 weeks after transplantation of HPC-5F cells transduced with lentivirus for HMGA2-L ORF, HMGA2-S ORF, or control (CTRL). Individual and mean values are shown. Mann-Whitney test was used individually comparing each indicated sample with respect to CTRL , p-values: <0.05 (*).
Although HMGA2 binds broadly across the genome, we identified promoter regions with the highest and the lowest level of HMGA2 binding (see Supplemental Experimental Procedures). We observed that promoters with the highest HMGA2-L or HMGA2-S binding were also marked by high H3K4me2 levels (V5 Hi - K4 Hi - Figure 3C). We also observed that highly expressed genes are skewed toward higher HMGA2 binding at their promoters (Figure S3B). Additionally, within promoters that were enriched for HMGA2 binding (V5 Hi), we observed an A/T bias (Figure 3D), consistent with in vitro observations (Winter et al., 2011). Overall, a strong correlation of binding patterns was observed for HMGA2-L and HMGA2-S for the most enriched and depleted promoter regions (r2=0.99 Figure 3E).
HMGA2 promotes expression of HSC-specific genes and enhance engraftment capacity
To examine the effects of the two isoforms on downstream gene expression, we performed RNA-seq from HPC-5F transduced with either HMGA2-L and HMGA2-S ORFs, or control vector (CTRL). A high correlation in downstream gene expression was observed between HMGA2--L and HMGA2--S treatments (r2=0.95 Figure 3F and Figure S3C). HPC-5F cells have been previously shown to partially reactivate an HSC signature and to enable short-term engraftment into immunocompromised mice (Doulatov et al., 2013). GSEA revealed that enforced expression of either HMGA2-L or HMGA2-S ORFs enhanced expression of a broad HSC-specific gene signature (Figure 3G). No significant differences were detected when comparing HMGA2-L to HMGA2-S treatment. Moreover, we observed enhanced repopulating capacity of HPC-5F cells upon O/E of either HMGA2-L or HMGA2-S ORFs in immunocompromised mice at 16 weeks (Figure 3H).
Collectively, these results revealed that HMGA2-L and HMGA2-S proteins, despite differences in their C-terminal domains, are highly comparable in their chromatin binding patterns and ability to induce transcriptional changes. The results also link HMGA2 function to activation of an HSC-specific program and enhanced self-renewal.
miRNA-mediated post-transcriptional regulation dictates differential HMGA2 isoform stability
Since HMGA2-L and HMGA2-S utilize different 3’ UTRs (Figure 2C), we hypothesized that the distinct expression patterns (Figure 2D) of the isoforms between FL- and CB-HSCs could be functional in preventing miRNA-mediated post-transcriptional regulation. To test this, we generated reporter constructs with luciferase fused to either the wild type HMGA2-L 3’UTR (RLuc-3’UTRwt_HMGA2-L), HMGA2-S 3’UTR (RLuc-3’UTRwt_HMGA2-S), or to a HMGA2-L 3’UTR mutated at all HSC-expressed miRNA binding sites (RLuc-3’UTRmt_HMGA2-L).
We transfected the luciferase reporter constructs into Dgcr8-knockout mouse embryonic fibroblasts (MEF K/O DGCR8), along with seven individual HSC-expressed miRNAs and a scramble control. Knockout of Dgcr8 prevents endogenous miRNA processing, which isolates the role of each miRNA in the absence endogenous miRNA activity (Han et al., 2009). Among the seven miRNAs analyzed, we found that let-7 family members had the strongest repressive effect on luciferase activity of HMGA2-L 3’UTR (Figure 4A, left panel). This effect was reversed upon mutation of binding sites for those miRNAs (Figure 4A, left panel). Additionally, other HSC-expressed miRNAs also appeared to regulate HMGA2-L. Moreover, the activity of the construct containing HMGA2-S 3’UTR was higher than that of HMGA2-L 3’UTR in the context of most miRNA treatments, indicating that HMGA2-S 3’UTR has a higher capacity to escape miRNA-mediated repression (Figure 4A, right panel).
FIGURE 4. Post-transcriptional regulation of HMGA2 isoforms.

A- (left) Normalized luciferase (Renilla) activity in Dgcr8-KO MEF of constructs carrying 3’UTR sequences of HMGA2-L (Rluc-3’UTRwt_HMGA2-L) or a mutant derivative depleted for miRNA sites (Rluc-3’UTRmt_HMGA2-L). (right) Normalized luciferase (Renilla) activity in Dgcr8-KO MEF of constructs carrying 3’UTR sequences of HMGA2-L (Rluc-3’UTRwt_HMGA2-L) or HMGA2-S (Rluc-3’UTRwt_HMGA2-S). Normalized luciferase activities were reported with respect to Rluc-3’UTRmt_HMGA2-L (left panel) or Rluc-3’UTRwt_HMGA2-S (right panel), set to 100%. Mean +/− s.e.m values are shown. Unpaired t-test was used, p-values: <0.05 (*), <0.01 (**), <0.005 (***), borderline (° = 0.055).
B- Quantification of miRNAs of interest in PC-3 and HPC-5F cells measured as by qRT-PCR. U6 snRNA was used as control.
C- Relative quantification of HMGA2 isoforms in PC-3 and HPC-5F cells transduced with lentiviral constructs carrying the HMGA2 ORFs equipped with its corresponding 3’UTRs (HMGA2-L+3’UTRwt and HMGA2-S+3’UTRwt), or a derivative HMGA2-L isoform mutated for miRNA sites (HMGA2-L+3’UTRmt). Infected cells were treated with actinomycin D (Act-D) and harvested at the indicated time points. Expression values were normalized to HPRT1 control, and then reported with respect to HMGA2-S+3’UTR-wt, set to 1. Mean +/− s.e.m values are shown. ANOVA was used, p-values: <0.05 (*), <0.01 (**), <0.005 (***).
Having demonstrated that the HMGA2-L 3’UTR is regulated by a number of miRNAs, we next evaluated whether miRNA-targeting might drive differential stability of the two HMGA2 isoforms. We used PC-3 and HPC-5F cells, which display high and low endogenous levels of the identified miRNAs, respectively (Figure 4B). Constructs carrying HMGA2-L and HMGA2-S ORFs along with their corresponding 3’UTRs were used to infect cells, which were then treated with actinomycin-D for six hours to halt transcription allowing us to isolate the effect of mRNA stability. qPCR of the HMGA2 isoforms revealed that HMGA2-L+3’UTRwt was more rapidly destabilized than HMGA2-S+3’UTRwt (Figure 4C). Conversely, HMGA2-L+3’UTRmt expression remained stable over the time points. Overall, we observed that HMGA2-L+3’UTRwt destabilization was more pronounced in cells with higher expression of the miRNAs of interest (Figure 4B, C).
To demonstrate regulation of HMGA2-L in endogenous settings, we utilized miRNA inhibitors against miRNAs highly expressed in CB-HSCs (let-7 and miR-142, Figure 5B). Consistent with reporter assay results above, inhibition of let-7 and miR-142 in CB CD34+ cells increased expression of HMGA2-L (Figure S4A).
FIGURE 5. Modulation of HMGA2 isoforms in human HSPCs.

A,B- Absolute expression of the indicated HMGA2 isoforms (in FPKM) and miRNA families (as percentage of total measured miRNA content) in the indicated HSC populations.
C- Scheme of HMGA2 isoform modulation experiments in CB and BM CD34+ hematopoietic stem and progenitor cells (HSPCs). Lentiviral constructs (pLKO.1 and pLENTI) also express GFP as a marker to allow for isolation of infected cells.
D- Phenotypic analysis of the stem cell compartment (CD133+CD34+CD38−) in CB CD34+ cells transduced with negative-control hairpin (luciferase - shLUC) or HMGA2 isoform-specific shRNAs (shHMGA2-S or shHMGA2-L) following 7 days in culture.
E- (left) Clonogenic progenitor assay of CD34+ CB cells following knockdown of HMGA2 isoforms. Cells transduced with shLUC, shHMGA2-S and shHMGA2-L hairpins were plated 3 days post-infection and CFU potential was measured 14 days post-plating. (right) CFU potential of CD34+ CB transduced with hairpins against both HMGA2 isoforms (shBOTH) was rescued upon overexpression of either the HMGA2-L or HMGA2-S ORFs.
F- Phenotypic analysis of the stem cell compartment (CD133+CD34+CD38−) in BM CD34+ cells transduced with control (CTRL), HMGA2-L+3’UTRwt, HMGA2-L+3’UTRmt, or HMGA2-S+3’UTRwt constructs after 7 days in culture.
G- Clonogenic progenitor assay of CD34+ BM cells following overexpression of HMGA2 isoforms. Cells transduced with control (CTRL) were plated 3 days after infection and CFU potential was measured 14 days post-plating. Mean +/− s.e.m. values are shown for D, E, F, G. Analysis of deviance for generalized linear models (CFU analyses) or repeated measures ANOVA (FACS analyses) were used, p-values: <0.05 (*) or <0.01 (**), non-significant (NS).
Taken together, these data demonstrate that HMGA2-L and HMGA2-S isoforms are differentially regulated at the post-transcriptional level and that miRNA levels are critical in determining their stability. These results reconcile the discordant let-7 and HMGA2 expression patterns seen in the HSC RNA-seq (Figure 2A). In FL-HSCs, where HMGA2-L is the primary isoform (Figure 5A), the cumulative levels of let-7 and other miRNAs shown to target HMGA2-L are the lowest among the HSC populations (Figure 5B). In contrast, in CB-HSCs, where HMGA2-S represents the primary isoform (Figure 5A), the expression of these miRNAs was much higher, representing more than 40% of the total measured amount of miRNA content (Figure 5B).
Modulation of HMGA2 isoforms influences human HSCs function in vitro
To determine if HMGA2 isoform dynamics impact HSC properties, we tested their role on human HSC function in vitro. We first depleted HMGA2 using isoform-specific shRNA-mediated knockdown (KD) in CB CD34+ cells (Figure 5C). We designed shRNAs that target only the long (shLONG), short (shSHORT), or both isoforms (shBOTH) (Figure S4). After one week of cytokine-driven serum-free culture, KD of either HMGA2 isoform in CB CD34+ decreased total cell output (not shown), as well as the CD34+CD133+CD38− HSC-enriched population (Figure 5D). We also found a reduction in colony-forming potential upon KD of either HMGA2-S and HMGA2-L (Figure 5E, left panel). Next, we performed a rescue experiment by co-infecting CB CD34+ cells with shBOTH as well as constructs expressing either the HMGA2-L and HMGA2-S ORFs that bear optimized sequences to escape targeting by shBOTH (see Supplemental Experimental Procedures). HMGA2 ORFs restored colony output of shBOTH-treated CB CD34+ cells (Figure 5E, right panel). Interestingly, we also observed a significant increase of E and GEMM colonies upon HMGA2 O/E, consistent with the role of HMGA2 as a positive regulator of erythropoiesis (Copley et al., 2013; Ikeda et al., 2011).
We next evaluated the effect of O/E of each isoform when under the control of its corresponding 3’UTR (Figure 5C). We performed this experiment in primary BM CD34+ cells, which have low total HMGA2 expression (Figure 5A). Cytokine-driven serum-free culture of transduced BM CD34+ cells revealed a significant increase in CD34+CD133+CD38− HSC numbers upon O/E of HMGA2-L+3’UTRmt and HMGA2-S+3’UTRwt after one week of culture (Figure 5F). Forced expression of HMGA2-L+3’UTRmt or HMGA2-S+3’UTRwt also increased the colony forming potential of transduced BM cells when compared to CTRL. The observed phenotype was more pronounced in HMGA2-S+3’UTRwt as compared to HMGA2-L+3’UTRmt, with no differences observed in HMGA2-L+3’UTRwt transduced cells (Figure 5G). These results support the functional role of HMGA2-S in regulating HSC self-renewal and clonogenic potential capacity in vitro.
CLK3 regulates HMGA2 splicing pattern through SRSF1
We next interrogated the mechanism by which HSCs are able to tune expression of the HMGA2 isoforms. To do so, we tested splicing regulators for their effect on HMGA2 isoform expression. We first filtered known splicing regulators to 23 candidates (Figure 6A) and then overexpressed each of these factors in PC-3 cells, which endogenously express both HMGA2 isoforms. Among factors tested, CLK3 had the strongest effect on HMGA2 isoform switching (Figure 6B). CLK3 belongs to the CDC-like kinases (CLK1–4) family of dual-specificity protein kinases, which regulate alternative splicing through phosphorylation of serine/arginine-rich domains on direct splicing factors (Colwill et al., 1996). To provide further evidence, we treated PC-3 cells with shRNAs against CLK3 (shCLK3) and observed a decrease of HMGA2-S with a corresponding increase of HMGA2-L expression (Figure S5A). Among HSCs profiled, CLK3 expression was the highest in CB HSCs (Figure 6C) and correlated with HMGA2-S expression across the CB hierarchy (Figure S5B). To investigate whether CLK3 affects the HMGA2 splicing pattern in human HSCs, we transduced BM CD34+ with lentiviral constructs overexpressing CLK3 or a control vector (CTRL) (Figure 6D). BM CD34+ cells were chosen for their low endogenous CLK3 and HMGA2 levels. RNA-seq analysis four days after infection of CLK3 demonstrated stimulation of expression of HMGA2-S (Figure 6E and Figure S5C). Interestingly, expression of an additional isoform bearing a short 3’UTR (HMGA2-S”) distinct from HMGA2-S and HMGA2-L was also stimulated upon CLK3 O/E; this HMGA2-S” isoform is normally expressed at very low levels in HSCs (Figure S2A–B). A similar effect of stimulating HMGA2-S expression was also observed in HPC-5F cells upon CLK3 O/E (Figure S5D).
FIGURE 6. CLK3 affects HMGA2 splicing through SRSF1.

A- Schematic representation of the filtering strategy to identify regulators of HMGA2 splicing. Genes were selected for the GO term “RNA binding” (GO:0003723), minimum expression, and ratio of expression in CB-HSC versus FL-HSC, as in the scheme.
B- Ratio of expression of HMGA2-S/HMGA2-L following overexpression of the indicated candidate splicing factors in PC-3 cells. Controls (GFP and RFP) are shaded in gray. Values were normalized to the HMGA2-S/HMGA2-L ratio upon control GFP overexpression and shown as mean +/− s.e.m.
C- CLK3 expression by RNA-seq (in FPKM) in the indicated HSC populations. Mean +/− s.d. values are shown. FDR<0.01 (**).
D- Schematic representation of CLK3 and HMGA2 modulation in BM HSPCs.
E- HMGA2 isoform quantification by RNA-seq (in FPKM) in BM-HSCs isolated upon overexpression of CLK3 or control (CTRL) four days post-infection. Mean +/− s.d. values are shown. PCR validation is shown in Figure S5E.
F- Violin plot representing distributions of statistically significant (p<0.05) ΔPSI values for different classes of PSI events (as in Figure 1H) in CLK3 overexpression as compared to vector control. Individual significant events are shown. The number of events of each PSI class are shown below the plot.
G- Schematic representation of HMGA2-L genomic structure (coding exons in black, UTRs in grey). SRSF1 binding motif sites are indicated by arrows in the corresponding exons. Sequence of exon 4 is indicated, and the predicted SRSF1 site (underlined in red) is shown.
H- Schematic representation of the luciferase-based splicing reporter constructs. The pcDNA3.1-Luc plasmid contains an intron-spaced Firefly luciferase coding sequence (grey boxes) that produces bioactive luciferase protein only upon proper splicing of the intervening intron. The HMGA2-L genomic region encompassing exon 4 (black box) plus 500 bp on either side of the flanking introns (wild-type [WT] or mutated [mut] by deletion of SRSF1 site) was cloned into the intron of pcDNA3.1-Luc. Its inclusion in the luciferase coding sequencing abolishes bioactive luciferase. Normalized luciferase activities were tested with or without CLK3 overexpression and reported with respect to Luc-exon-4 WT set to 100%. Mean +/− s.e.m. values are shown. Repeated measures ANOVA was used, p-value <0.05 (*).
To evaluate the impact of CLK3 O/E on the global repertoire of splicing events in BM-HSCs, we performed a ΔPSI analysis as above (see Supplemental Experimental Procedures). For most types of PSI events, approximately the same number of events were induced by CLK3 O/E as were depleted. However, CLK3 O/E significantly increased exon skipping (SE) events (Figure 6F, p=8.26×10^-30) and promoted intron retention (RI, Figure 6F, p=4.99×10^-8). These results suggest a global role of CLK3 in mediating alternative splicing.
Interestingly, exons preferentially spliced out upon CLK3 O/E were enriched for SRSF1 binding motifs (p=0.0016) (Park et al., 2016). SRSF1 is known to bind exonic enhancer sequences (ESE) and act as a barrier to prevent exon skipping (Long and Caceres, 2009). As SR proteins are regulated by CLK proteins, we searched for predicted SRSF1 binding sites in HMGA2. Motif analyses identified SRSF1 binding motifs only in the HMGA2-L mRNA sequence, specifically in exons 4 and 5, which are exclusive to HMGA2-L (Figure 6G). To test whether CLK3’s effect on HMGA2 splicing is mediated by SRSF1, we cloned exon 4 of HMGA2-L (including flanking intronic regions [500 bp]) into a luciferase splicing reporter (Luc-exon-4 WT) (Figure 6H, top panel) (Martone et al., 2016). A version of HMGA2-L exon 4 that is mutated for the SRSF1 binding motif was also generated. Upon CLK3 O/E, luciferase activity of the WT construct was enhanced, indicating that CLK3 O/E promotes skipping of the exon containing an SRSF1 binding motif. The observed effect was further enhanced when the SRSF1 site in that exon was mutated (Figure 6H, low panel). CLK3 O/E did not further increase luciferase activity of the mutated construct, supporting the direct molecular link between CLK3 and SRSF1. qPCR in PC-3 cells treated with SRSF1-specific siRNAs further confirm that depletion of SRSF1 affects HMGA2 splicing by decreasing HMGA2-L and increasing HMGA2-S expression (Figure S5E), phenocopying the effect of CLK3 modulation (Figure 6B and S5A). Collectively, these results indicate that CLK3 promotes the skipping of HMGA2-L exon in a SRSF1-dependent manner.
CLK3-HMGA2-S axis orchestrates an HSC-specific program
Flow cytometry revealed a >4-fold increase of HSCs upon CLK3 O/E four days post-infection of BM CD34+ (as compared to CTRL) (Figure 7A). A similar trend, albeit to a lesser extent, was observed upon CLK3 O/E in CD34+ CB cells, possibly due to higher endogenous CLK3 expression (Figure S6A). Next, we evaluated the global effect of CLK3 O/E on the transcriptional landscape in BM HSCs. GSEA revealed that CLK3 O/E reinforced a BM-HSC gene signature and reactivated a CB-HSC gene signature (Figure 7B). Interestingly, we observed that CLK3 O/E reactivates a broad HSC-signature in HPC-5F (Figure S6B), similar to the effect observed with forced expression of HMGA2 ORFs (Figure 3G). Furthermore, the impact of CLK3 on the HSC transcriptional landscape was phenocopied by HMGA2-S+3’UTRwt O/E in BM-HSCs (Figure 7B). On a global level, we detected an overlap of the genes modulated upon CLK3 and HMGA2-S treatment, with ~50% of the total of genes modulated upon CLK3 O/E displaying the same trend upon HMGA2-S O/E (Figure 7C). This included higher expression of key regulators of the HSC program in BM-HSCs transduced with CLK3 and HMGA2-S+3’UTRwt (Figure 7D).
FIGURE 7. _CLK3 HMGA2 axis orchestrates an HSC-specific program.

A- Phenotypic analysis of HSC content in BM CD34+ cells transduced with control (CTRL) or CLK3 lentiviral constructs 4 days post-infection. Mean +/− s.e.m. values are shown. Unpaired t-test was used, p-value <0.01 (**).
B- BubbleMap visualization (Spinelli et al., 2015) of GSEA results in BM-HSCs upon CTRL, CLK3 or HMGA2-S-wt overexpression. Gene sets were derived from HSC and PROG -specific signatures from Figure 1C (see Table S2C). As indicated in the legend, colors (red versus blue) correspond to the sample label, shades represent statistical significance (FDR) and the area of the circle represents the enrichment (Normalized Enrichment Score, NES). Empty circles correspond to non-significant (NS) enrichments (FDR>0.05). Representative GSEA plot of the boxed BubbleMap is shown on the right.
C- Venn diagrams of genes significantly DE (FDR<0.05) upon CLK3 or HMGA2-S+3’UTRwt overexpression.
D- RNA-seq based expression (in FPKM) of representative genes upon CTRL, CLK3 and HMGA2-S+3’UTRwt overexpression in BM HSCs. Mean +/− s.d. values are shown. Cufflinks FDR <0.05 in all comparisons.
E- Human chimerism as percentage of GFP+CD45.1+ in the injected femur of xenografted mice 8 weeks after transplantation of BM CD34+ HSPCs transduced with lentivirus for HMGA2-S+3’UTRwt, CLK3, or control (CTRL). Individual sample, mean +/− s.e.m. values are shown. Mann-Whitney test was used to individually compare each indicated sample with respect to CTRL , p-values: <0.05 (*).
To further support our observation that CLK3-mediated promotion of stemness potential is mediated by HMGA2-S, we performed a rescue experiment by co-infecting CB CD34+ cells with shCLK3 and an HMGA2-S O/E construct. CFU analysis revealed that shCLK3 treatment of CB CD34+significantly reduced E and GEMM colonies (Figure S6C), paralleling the effect of HMGA2-S KD (Figure 5E), In contrast, co-overexpression of HMGA2-S rescued output of E and GEMM colonies (Figure S6C).
To evaluate whether elevation of HSC-specific gene expression enhances HSC function in vivo, we transduced human BM CD34+ cells with HMGA2-S+3’UTRwt, CLK3 or CTRL vectors and transplanted cells into immunodeficient mice. Human BM CD34+ cells display significantly reduced proliferative potential in vivo compared to CB and FL cells, corresponding to an age-related decline in HSC function (Bernitz et al., 2016). Consistent with this, CTRL-treated BM cells displayed only ~1% engraftment at 8 weeks post-transplant. In contrast, induction of HMGA2-S+3’UTRwt or CLK3 significantly enhanced the human chimerism (Figure 7E). Collectively, our findings demonstrate that CLK3, at least in part by regulating the splicing pattern of HMGA2, reinforces an HSC-specific program in vitro and in vivo.
DISCUSSION
Recent studies have leveraged high-throughput genetic, epigenetic, and transcriptomic data to better understand the underlying mechanisms of hematopoiesis (Notta et al., 2016). Here, we have comprehensively characterized the transcriptional landscape of HSCs along development (FL, CB, and BM), including gene and isoform-level expression of coding genes, as well as expression of non-coding RNAs. Building on prior work (Chen et al., 2014), our analyses highlighted extensive alternative splicing among HSC populations, including a stage-specific alternative splicing pattern for HMGA2. Comprehensive functional experiments further reveal that interplay between alternative splicing and miRNA-mediated regulation profoundly impacts regulation and expression of HMGA2, with consequences for the molecular identity and behavior of human HSCs.
HMGA2 is an important downstream effector of the LIN28/let-7 pathway (Viswanathan et al., 2008), and its expression is tightly regulated at its 3’UTR by let-7 miRNAs (Lee and Dutta, 2007). Expression of aberrant HMGA2 transcripts is frequently seen in human malignancies (Schoenmakers et al., 1995) as the result of chromosomal rearrangements, and in gene therapy trials, as a consequence of insertional mutagenesis that dissociate the HMGA2 3’UTR from its protein-coding region (Cavazzana-Calvo et al., 2010). Loss of let-7 sites has been proposed as the major driver of HMGA2-mediated oncogenic transformation (Fedele et al., 1998; Mayr et al., 2007) and promotion of hematopoietic cell proliferation (Ikeda et al., 2011). Here we report that CB-HSCs express an alternative HMGA2 isoform bearing a 3’UTR devoid of miRNA sites (HMGA2-S) and that expression of HMGA2-S allows for preserved expression and function of HMGA2 in spite of physiologically high levels of let-7 and other miRNAs present in CB-HSCs. Independent reports have implicated HMGA2 in promoting cell proliferation and stem cell properties in different contexts (Copley et al., 2013; Ikeda et al., 2011; Li et al., 2007; Nishino et al., 2008). Our work expands the role of HMGA2 in HSCs and provides evidence for similar functions of the proteins encoded by the two HMGA2 isoforms.
Our work also delineated the molecular mechanism for differential HMGA2 splicing observed across HSCs. Here, we describe the role of a splicing kinase, CLK3, in dictating HSC development by promoting expression of an HMGA2 isoform insensitive to miRNA--mediated targeting. Indeed, we observed a substantial overlap between genes modulated by either CLK3 or HMGA2-S. We also demonstrate that enforced expression of CLK3 and HMGA2-S can induce a more proliferative phenotype in BM-HSCs by reactivating a CB-specific transcriptional signature and promoting engraftment of human BM HSPCs, which normally display low repopulating capacity.
Furthermore, we demonstrated that CLK3 stimulates exon skipping, including at HMGA2. A dynamic cycle of phosphorylation and dephosphorylation of SR proteins is essential for splicing (Mermoud et al., 1994). Among splicing kinases, CLK proteins have broad roles in this phosphorylation process to regulate SR proteins (Ngo et al., 2005, Aubol et al., 2016). In line with previous observations that high levels of CLK inhibit the activity of SR proteins in promoting splicing (Prasad et al., 1999), we show that i) exons preferentially spliced out upon CLK3 O/E are enriched for SRSF1 binding sites; ii) only HMGA2-L (and not HMGA2-S) contains SRSF1 binding sites that prevent the proper splicing of HMGA2-L isoform upon CLK3 O/E. Thus, in the context of HMGA2, we have demonstrated that CLK3 acts through SRSF1 to shift the balance of HMGA2-L versus HMGA2-S splicing.
In summary, we show marked differences among HSC populations at different developmental stages and implicate alternative splicing as a mechanism that contributes to the transcriptional diversity. Our comprehensive map of the transcriptome of HSCs represents a valuable tool for understanding the mechanisms of hematopoiesis that can be applied to complement and improve cell fate conversion and HSC expansion approaches.
Our work also revises the canonical LIN28/let-7 pathway, where LIN28 proteins inhibit let-7 biogenesis, which in turn repress expression of target genes, including HMGA2 (Lee and Dutta, 2007). Our results highlight a physiologic isoform of HMGA2 that escapes regulation by the upstream LIN28/let-7 pathway. CLK3 appears to function as a tuner that, by regulating the balance of HMGA2 isoforms, impacts the developmental identity of HSCs. Collectively, our findings open up new directions of investigation into the mechanisms of altered HMGA2 splicing that might contribute to developmental regulation and malignancies.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, George Q. Daley (George.Daley@childrens.harvard.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary hematopoietic cell source and FACS analyses
Human CD34+ cells (Lonza and AllCells) from all sources were obtained as viable frozen states. The age of the individuals used for the profiling and treatments are as follows: FL-CD34+ (17–20 weeks gestation); CB-CD34+ (newborns); BM-CD34+ (24–36 years old). For the generation of HSC/PROG transcriptional profiles, at least three distinct lot numbers, each corresponding to independent individuals or pools of distinct individuals (of random male or female samples), were utilized to attain maximal representation. For each biological replicate, HSC and PROG populations were sorted from the same pool of cells. Cells were stained and sorted using a BD FACS Aria II cell sorter for panels of cell surface markers and dyes as indicated below.
HSC panel: CD34 PE-Cy7 (8G12; BD), CD38 PE-Cy5 (HIT2; BD), CD90 PE (5E10; BD), CD45RA-FITC (HI100; BioLegend), DAPI
PROG panel: CD34 PE-Cy7 (8G12; BD), CD38 PE-Cy5 (HIT2; BD), DAPI
Stem Cell panel: CD34 PE-Cy7 (8G12; BD), CD38 PE-Cy5 (HIT2; BD), CD90 PE (5E10; BD), CD45RA-V450 (H100; BD), CD133/1-APC (AC133; Miltenyi Biotec)
Cell culture and viral transduction
All cell culture incubations were performed at 37 C with 5%CO2.
cDNA sequences for overexpression were cloned into a pSMAL-GFP or pSMAL-BFP (Doulatov et al., 2013) or pLVX-PURO (Clontech) lentiviral backbones, using Infusion HD Cloning Kit (Clontech). HMGA2 cDNAs were cloned as full ORF sequences including wild-type or mutated 3’UTRs (see below), as ORFs only, or as RNAi-resistant ORFs (by degenerating the shRNA target sequences). For RNA interference, shRNA sequences were cloned in the pLKO.1_hPGK-Puro-CMV-tGFP lentiviral backbone (Sigma-Aldrich). Lentiviral particle productions and quantifications were performed using standard procedures.
CB and BM CD34+ cells were thawed and plated in X-VIVO media (Lonza) supplemented with 20% BIT 9500 (StemCell Technologies), 2 mM L-glutamine, and 100 U/ml penicillin/streptomycin, and incubated for 4–6 hours before transduction. Cells were then seeded on retronectin-coated (10 µg/cm2) 96 well plates (Clontech) at a density of 0.5–1×105 cells per well, and viral particles were added at a multiplicity of infection (MOI) of 30–50 (for the knockdown treatments) or 100 (for the overexpression treatments) in a final volume of 150 ul. The following cytokines were used along with protamine sulfate at 8 µg/ml (Sigma-Aldrich): stem cell factor (SCF) 100 ng/ml, Flt3 ligand (FLT3-L) 100 ng/ml, thrombopoietin (TPO) 50 ng/ml, and interleukin 6 (IL-6) 20 ng/ml (all PeproTech). Cells were then spininfected for 1 hr at 2500 rpm at room temperature (RT) and subsequently incubated for a minimum of 24 hrs before changing media. Cells were then expanded in StemSpan SFEM (StemCell Technologies), supplemented with half the concentration of the indicated cytokines. Infected cells were sorted 3 days post-transduction for use in downstream assays.
HPC-5F were obtained according to (Doulatov et al., 2013). Infection was performed in StemSpan SFEM (StemCell Technologies) with 50 ng/ml SCF, 50 ng/ml FLT3, 50 ng/ml TPO, 50 ng/ml IL6, 10 ng/ml IL3 (all R&D Systems) and an MOI of 5–10 was used for the indicated constructs. Media was changed 24 hrs post-infection and supplemented with doxycycline at 2 µg/ml (Sigma-Aldrich) to allow for the expression of the 5 factors. Prostate cancer cells (PC-3) and K562 cells were obtained from ATCC and cultured according to the suggested specifications. Cells were transduced with an MOI of 4–8 and media was changed 24 hrs after infection. Cell were treated with Actinomycin D at 1ug/ml (Sigma-Aldrich) for the indicated amount of time and then harvested.
Colony forming unit assays
1×10^3 cells were plated into 3 ml of complete MethoCult (H4434; StemCell Technologies) and supplemented with 10 ng/ml FLT3, 10 ng/ml IL6, and 50 ng/ml TPO (all PeproTech). Colonies were scored manually after 14 days of incubation.
Mouse transplantation and assessment of human cell engraftment
NOD/LtSz-scidIL2Rgnull (NSG) (Jackson Labs) mice were bred and housed at the Boston Children’s Hospital animal care facility, and experiments were performed in accordance to institutional guidelines approved by the BCH animal care committee. Briefly, 6–10 week old female mice were irradiated (275 rads) 24 hrs before transplant. BM-transduced cells were GFP+ sorted 3 days post-infection.7.5×10^4 cells were transplanted per mouse in a 25 µL volume using a 28.5g insulin needle after temporarily sedating the animals with isoflurane. For HPC-5F transplantation experiments, cells were cultured for 14 days before injection, and 8×10^6 cells were transplanted per mouse. For all the transplantation experiments, Sulfatrim was administered in drinking water to prevent infections after irradiation. Mice were sacrificed at the indicated time points.njected femur, uninjected femur, and tibiae were collected and single cell suspensions were prepared using standard flushing and cell dissociation techniques. Samples were stained with a panel of human markers: CD19 PE (4G7; BD), CD45 PE-Cy5 (Immu19.2; Coulter), CD45 APC-Cy7 (2D1; BD),CD33 APC (P67.6; BD) and DAPI. Un-injected mouse bone marrow was used as a control for non-specific staining and BM mononuclear cells (Lonza) were used as a positive control for antibody staining and proper compensation. All acquisitions were performed on a BD Fortessa cytometer.
Luciferase reporter assays
For the HMGA2-3’UTR reporter assay, 3’UTR sequences for the HMGA2-L (Rluc-3’UTRwt_HMGA2-L) and HMGA2-S (Rluc-3’UTRwt_HMGA2-S) isoforms were cloned into psiCHECK2 plasmid (which contains both Renilla [RLuc] and Firefly [FLuc] luciferases from Promega) downstream the Renilla luciferase (RLuc) ORF. A mutant derivative of HMGA2-L 3’UTR mutated for all the miRNA binding sites of interest (Rluc-3’UTRmt_HMGA2-L) was synthesized (GeneScript). Dgcr8 K/O MEFs (Novus Biologicals) were transfected along with miRIDIAN microRNA mimics using the DharmaFECT Duo reagent (Dharmacon). RLuc and Fluc activities were measured by Dual Luciferase assay (Promega) 72 hrs after transfection. Ratios between Rluc and Fluc were calculated, and outliers of biological triplicates removed. To report HMGA2-L 3’UTR stability upon deletion of the miRNA sites, Rluc-3’UTRwt_HMGA2-L values were normalized to those of Rluc-3’UTRmt_HMGA2-L (set to the value of 100%), within the same miRNA treatment. To show which HMGA2 3’UTR isoform was more stable upon miRNA treatments, Rluc-3’UTRwt_HMGA2-L values were normalized to those of Rluc-3’UTRwt_HMGA2-S (set to the value of 100%), within the same miRNA treatment.
For the splicing reporter assay, the genomic region of HMGA2-L exon 4 along with 500bp on either side (Luc-exon-4-WT) was cloned in between the splicing acceptor and donor sequence of a spliced FLuc ORF reporter plasmid (pcDNA3.1-Luc (Martone et al., 2016)). The mutant derivative of the SRSF1 site within exon 4 was generated by deleting its consensus motif.
Transfection of miRNA hairpin inhibitors
CD34+ cells isolated from Cord Blood were seeded at a density of 1.6 × 10^5/well in a 12-well plate and pre-stim for 4 hours in StemSpan SFEM (StemCell Technologies) supplemented with FLT3, SCF, IL-6 and TPO, as indicated. miRIDIAN miRNA hairpin inhibitors (Dharmacon) were transfected using DOTAP Liposomal Transfection Reagent (Sigma-Aldrich). After 24 hours, the media was replaced with fresh media and cells were harvested at 72 hours post-transfection.
METHOD DETAILS
RNA and DNA sequencing libraries
RNA was extracted from cells using the miRNeasy kit (Qiagen). Transcriptional profiling of HSC and PROG populations and HPC-5F cells transduced with CLK3/HMGA2 constructs was generated from 100 ng of total RNA for each biological replicate using the Truseq RNA Library Preparation Kit v2 (Illumina). Transcriptional profiles of BM-HSCs transduced with CLK3 and HMGA2-S+3’UTRwt constructs was performed from 1–5ng of total RNA using the SMART-Seq v4 Ultra Low Input RNA Kit (Clontech) in combination with the Nextera XT DNA Library Preparation Kit from 150pg of cDNA (Illumina). ChIP-seq libraries were performed as previously described (Cacchiarelli et al., 2015) after immunoprecipitation with antibodies for V5 (MBL Int., Lot #5) and H3K4me2 (Diagenode). Libraries were sequenced on an Illumina HiSeq 2000 or 2500 according to protocol specifications.
miRNA profiling and analysis
For each biological replicate, 100 ng of RNA was utilized for miRNA profiling using the nCounter Human v2 miRNA Expression Assay (NanoString Technologies). Several steps were then undertaken to normalize the Nanostring data. First, Nanostring miRNA counts underwent QC and normalization according to manufacturer’s specifications. The normalized counts were subsequently grouped and summed by their respective miRNA family. Then, for each sample, the expression of a given miRNA family was calculated as a percentage of the total counts for that sample (i.e., total measured miRNA content) in the Nanostring data. A two-sided unpaired t-test was applied to compare of expression of each miRNA family across samples.
RNA-seq alignment and transcript assembly
RNA sequences were aligned using TopHat v2.0.14 (Trapnell et al., 2012) using default parameters. Sequences were aligned to the hg19 reference genome. For the HSC/PROG samples, Gencode v17 transcript annotation was used as the transcriptome index. For all other RNA-seq samples, Gencode v19 transcript annotations were used. For HPC-5F cells transduced with CLK3/HMGA2 and for BM-HSCs transduced with CLK3/HMGA2-S+3’UTRwt, no novel junctions were considered (using the parameter “—no-novel-juncs”). Visualizations and Sashimi plots of RNA-seq alignments were performed using IGV v2.3 (Robinson et al., 2011).
Downstream transcript assembly and differential expression analysis were performed using Cufflinks v2.2.1 (Trapnell et al., 2012). For HSC/PROG samples, Cufflinks-assembled transcripts were merged with the Gencode v17 annotations using the Cuffcompare and Cuffmerge functions to generate a “Gencode v17 + Cufflinks” annotation file. Novel lincRNA discovery and annotation for Cufflinks-assembled transcripts was then performed according to Cabilli et al (Cabili et al., 2011).
For the HSC/PROG transcriptome profiling, aligned sequences were quantified using the Gencode v17 + Cufflinks annotation file using Cuffquant with default parameters. For the CLK3 and HMGA2 overexpression in BM-HSCs and HPC-5F cells, aligned sequences were quantified using Cuffquant with the Gencode v19 annotation file. Lastly, the Cuffnorm function was used to calculate expression in Fragments Per Kilobase of transcript per Million mapped reads (FPKM). A mask file was used during quantification for all datasets; this mask file was comprised of all rRNA, scRNA, snoRNA, snRNA, miRNA, “misc_RNA”, and tRNA sequences as annotated in Gencode v17 or v19. Unless otherwise noted, all Cufflinks default parameters were used.
Processed RNA-seq gene and isoform-level expression profiles for human hematopoietic cells from the cord blood lineage (n=63 samples) were downloaded from the Blueprint Epigenome dataset (http://www.blueprint-epigenome.eu, downloaded on May 4, 2016).
We note that Gencode v17 and v19 annotate the HMGA2-S isoform (ENST00000393578) as being 316 bp in length. Newer versions of Gencode (e.g., v27) annotate it as 911 bp, which is consistent with the observed sequencing alignments and RT-PCR. As there are no competing exons near the HMGA2-S 3’UTR, using Gencode v17 or v19 does not substantially impact our quantifications or differential expression results.
Differential expression analyses, RT-PCR and qPCR
Differentially expressed genes and isoforms were identified using the cuffdiff function within Cufflinks. All default parameters were used, and a mask file as described above was applied. For the HSC/PROG data, the Gencode v17 + Cufflinks annotation was used. For the CLK3 and HMGA2 overexpression in BM-HSCs and HPC-5F cells, the Gencode v19 annotation was used.
For the HSC and PROG data, specific filtering criteria were applied to generate a subset of differentially expressed genes/isoforms. For each pairwise comparison, significantly differentially expressed (FDR<0.01) genes were identified. To define isoform-exclusive events, we identified significantly differentially expressed (FDR<0.01) isoforms that are non-significant (FDR>0.01) for their corresponding gene-level differential expression.
For the CLK3/HMGA2 overexpression data in BM-HSCs, significantly differentially expressed genes were defined as showing absolute fold change >2 relative to control, FPKM>1 in either the overexpression or control sample, and FDR<0.05.
FPKM values were plotted as the mean and standard deviation of the FPKM replicate mean values and when indicated, statistical significance was reported as the p-value output by the Cuffdiff function.
Semi-quantitative PCR was performed to validate isoform splicing and usage by direct amplification of the relevant exon-intron structures. To quantify full HMGA2 mRNAs, PCR were performed on RNA-seq libraries (performed using polyA+ capture), amplifying the full coding sequence. To quantify HMGA2 pre-mRNA, PCR weas performed on cDNA reverse transcribed with random hexamers on polyA- RNA, amplifying a 5’ portion of pre-mRNA. All PCR products were purified and run on a Bioanalyzer or Tapestation (Agilent Technologies) to obtain digital gels from electropherograms. In each sample, HPRT1 was used as an endogenous housekeeping control. Virtual run traces are shown at global scale visualization adjusting brightness/contrast for best representation.
Quantitative PCR (qPCR) of mRNAs and miRNAs was performed using the miScript system (Qiagen). Outliers of technical replicates were removed, and ΔCt or ΔΔCt analyses were performed using HPRT1 and U6 as endogenous controls.
Definition of HSC transcriptional signatures
For HSC/PROG RNA-seq samples, significant isoforms and lincRNAs were defined as having expression of FPKM > 5 in at least one sample, an absolute fold change > 2, and FDR<0.05 in at least one pairwise comparison. For HSC/PROG miRNA expression, normalized Nanostring expression data was used (see above). The mean values across sample replicates were used. miRNAs families were filtered for having an absolute fold change > 2 absolute change and a p-value < 0.05 in at least one pairwise comparison and representing > 0.1% of total measured miRNA content.
To classify these significant isoforms, lincRNAs, and miRNAs into groups, the Jensen–Shannon divergence was calculated using the csSpecificity function implemented in CummeRbund (Trapnell et al., 2012). A specificity score > 0.25 was used to classify isoforms, and lincRNAs, and miRNAs as being enriched in a given sample and to define transcriptional signatures for each sample.
To generate a heatmap (Figure 1C), the expression values were then z-score normalized, and the plotting order of isoforms, lncRNAs, or miRNAs were based on the specificity classifications. The heatmap.2 function within the gplots package in R v3.1.1 was used to generate the heatmap.
Gene set enrichment analysis (GSEA)
GSEA were performed as previously described using curated gene sets available in MSigdb (Subramanian et al., 2005), or gene sets generated from HSC and PROG gene signatures (see above). Visualization of GSEA results was performed using BubbleGum (Spinelli et al., 2015).
ChIP-Seq alignment and promoter analysis
Reads were aligned to the hg19 human genome using Bowtie2 (Langmead and Salzberg, 2012) with default parameters. Enrichment at genomic 1kb tiles and promoters (defined as 1 kb up- and downstream from RefSeq transcription start sites) was computed by using the Bedtools “coverage” command to count the number of reads in tile or promoter region (Quinlan and Hall, 2010). Visualizations of ChIP-seq alignments were performed using IGV v2.3 (Robinson, 2011). In order to control for tiles or promoter regions with an over- or under- enrichment of reads due to technical artifacts such as low sequence complexity, the top 15% of regions with the most number of reads and the bottom 15% of regions with the fewest reads from the whole cell extract samples were discarded. The number of reads from sample replicates were summed to yield sample totals for each region.
Differentially-bound promoters were defined as those in which the number of both H3K4me2 and V5 reads were less than the 25th or greater than the 75th percentile in either the short or long experiments. Differentially-expressed genes from the same samples were defined as those genes that were expressed at >5 FPKM in any sample and showed at least an absolute 2-fold change in a comparison between two samples. RNA-seq and ChIP-seq results were then combined to study the relationship between HMGA2 occupancy on expression. The median expression and median occupancy levels were used as cutoffs to separate the data into four quadrants.
let-7 target analysis
Data from TargetScan human version 7 was used to identify predicted conserved miRNA binding sites (www.targetscan.org). Predicted miRNA binding scores for all isoforms was kindly generated by George Bell (Whitehead Institute). The top 50% of let-7 target isoforms were determined by having the lowest (i.e., strongest) weighted context++ scores. For target isoforms with more than one let-7 binding site, the binding site with the strongest weighted context++ score was used. The mean expression of the let-7 target isoforms for each sample (FL-HSC, CB-HSC, BM-HSC, FL-PROG, CB-PROG, BM-PROG, and HPC) was then calculated. The mean expressions were then z-score normalized for plotting.
PSI analysis
Percent-spliced-in (PSI) analysis was performed using SUPPA (Alamancos et al., 2015). Splicing events (alternative 5’ splice site [A5], alternative 3’ splice site [A3], alternative first exon [AF], alternative last exon [AL], mutually exclusive exon [MX], retained intron [RI], and skipping exon [SE]) were generated from the Gencode v19 annotation file or Gencode v17 + Cufflinks annotation file using default parameters. Input RNA-seq quantifications were based on the RNA-seq quantifications as described above, except FPKM expression values were converted to transcripts per kilobase million (TPM) as recommended by the software developers. Events were filtered for TPM > 1. PSI calculations were then performed using the “psiPerEvent” tool. Differential splicing analysis was performed using the “diffSplice” tool. Default parameters were used, and the “empirical” method was used to calculate significance. A p-value threshold of 0.05 was used for declaring significance. Of note, the direction of dPSI for SE events was flipped from the software default to reflect actual splicing changes.
Splicing factor screen
To identify candidate splicing factors for HMGA2 splicing, we applied several filters. We selected genes from the GO category “RNA binding” (GO:0003723). We then filtered for candidate genes that display an absolute fold change >2 in FL-HSCs relative to CB-HSCs and expressed at FPKM >10 in either those samples. Among the filtered candidates genes, 23 genes available as lentiviral constructs from the Broad Institute Genome Perturbation Platform (GPP) were subsequently tested through overexpression in PC-3 cells.
Identification of SRSF1 binding motif
A search for SRSF1 binding sites weas performed using the scanMotifGenomeWide function in HOMER (http://homer.ucsd.edu/homer/motif/genomeWideMotifScan.html) usign the hg19 reference genome (Heinz et al., 2010). All software default parameters were used. The SRSF1 binding position-weighted matrix from (Wang et al., 2011) was used. A p-value threshold of 1 × 10-8 was applied and only “+” strand hits were kept.
RNA binding protein enrichment
The PSI data from above was used to generate exons skipped (SE events) upon CLK3 overexpression in BM-HSCs as compared to control. A p-value threshold from the PSI analysis of 0.05 was used to identify exons that are preferentially increased or decreased upon CLK3 overexpression. Background control exons were identified using exons with p-value > 0.10. The rMAPS v1.0.6 online software (http://rmaps.cecsresearch.org) was used to perform the enrichment analysis (Park et al., 2016). All default parameters were used in the enrichment analysis, including the binding motifs provided by the software.
QUANTIFICATION AND STATISTICAL ANALYSIS
No statistical methods were used to predetermine sample size. In general, as descriptive statistics, we reported mean +/− s.e.m. values with the exception of RNA-seq data where we reported mean +/− s.d. values. If not stated otherwise, t-test (for two groups) or ANOVA (more than two groups) where the standard statistical tests applied. Pairing, repeated measurements or other test corrections were applied as needed.
To assess statistical significance for CFU discrete measurements, we applied analysis of deviance for generalized linear models (Chambers, 1991). For in vivo engraftment capacity, the Mann-Whitney test was applied individually by comparing each treated sample with respect to the control sample.
DATA AND SOFTWARE AVAILABILITY
Sequencing data can be found at NCBI Geo DataSets under the Superseries GSE109093.
Supplementary Material
Document S1. Figures S1-S6
Table S1. Differentially expressed transcripts at gene and isoform level, related to Figure 1
Table S2. Quantification of mRNAs and non coding RNAs, related to Figure 1
Table S3. Summary of isoform switching events and effect on 3’UTR usage, related to Figure 2
Table S4. GSEA results, related to Figure 1
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD34 PE-Cy7 | BD Biosciences | Cat. 348791; CLONE 8G12 |
| CD38 PE-Cy5 | BD Biosciences | Cat. 555461; CLONE HIT2 |
| CD90 PE | BD Biosciences | Cat. 555596; CLONE 5E10 |
| CD45RA FITC | BioLegend | Cat. 304105; CLONE HI100 |
| DAPI solution | BD Biosciences | Cat. 564907 |
| CD45RA V450 | BD Biosciences | Cat. 560362; CLONE HI100 |
| CD133/1 APC | Miltenyi Biotec | Cat. 130-090-826; CLONE AC133 |
| CD19 PE | BD Biosciences | Cat. 349209; CLONE 4G7 |
| CD45 PE-Cy5 | Coulter | CLONE Immu19.2 |
| CD33 APC | BD Biosciences | Cat. 340474; CLONE P67.6 |
| CD45 APC-Cy7 | BD Biosciences | Cat. 561863; CLONE 2D1 |
| V5 | MBL | Cat. M167-3 |
| H3K4me2 | Diagenode | Cat. 035-050 |
| Biological Samples | ||
| Cord Blood CD34+ Cells | Lonza | Cat. 2C-101 |
| Cord Blood CD34+ Cells | AllCells | Cat. CB008F |
| Fetal Liver CD34+ Cells | AllCells | Cat. FL-CD34-002F |
| Bone Marrow CD34+ Cells | AllCells | Cat. ABM017F |
| Bone Marrow CD34+ Cells | Lonza | Cat. 2M-101C |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Actinomycin D | Sigma-Aldrich | Cat. A1410 |
| Doxycycline Hyclate | Sigma-Aldrich | Cat. D9891 |
| Protamine Sulfate | Sigma-Aldrich | Cat. P4020 |
| Recombinant Human SCF | Peprotech | Cat. 300-07 |
| Recombinant Human FLT3L | Peprotech | Cat. 300-19 |
| Recombinant Human TPO | Peprotech | Cat. 300-18 |
| Recombinant Human IL6 | Peprotech | Cat. 200-06 |
| Retronectin | Clontech | Cat. T100A |
| DOTAP Liposomal Transfection Reagent | Sigma-Aldrich | Cat. 11202375001 |
| DharmaFECT Duo Transfection Reagent | Dharmacon | Cat. T-2010-03 |
| Critical Commercial Assays | ||
| Dual-Luciferase Reporter Assay System | Promega | Cat. E1910 |
| TruSeq RNA Library Prep Kit v2 | Illumina | Cat. RS-122-2001 |
| SMART-Seq v4 Ultra Low Input RNA Kit | Clontech | Cat. 634888 |
| Nextera XT DNA Library Preparation Kit | Illumina | Cat. FC-131-1024 |
| nCounter Human v2 miRNA Expression Assay | NanoString Technologies | Cat. GXA-MIR2-24 |
| ChiP-Seq Assay | Broad Institute Epigenomics Program | n/a |
| Experimental Models: Cell Lines | ||
| PC-3 | ATCC | Cat. CRL-1435 |
| K562 | ATCC | Cat. CCL-243 |
| HPC-5F | Doulatov et al. 2013 | n/a |
| DGCR8 knockout MEF | Novus Biologicals | Cat. NBP2-25171 |
| Experimental Models: Organisms/Strains | ||
| NOD/LtSz-scidIL2Rgnull (NSG) | Jackson Laboratory | n/a |
| Recombinant DNA | ||
| pLVX_HMGA2-L_ORF_V5_PURO | In this paper | n/a |
| pLVX_HMGA2-S_ORF_V5_PURO | In this paper | n/a |
| pLVX_CTRL_V5_PURO | In this paper | n/a |
| pSMAL_HMGA2-L_ORF_BFP (RNAi resistant) | In this paper | n/a |
| pSMAL_HMGA2-S_ORF_BFP (RNAi resistant) | In this paper | n/a |
| pSMAL_CTRL_BFP | In this paper | n/a |
| pSMAL_HMGA2-L_GFP | In this paper | n/a |
| pSMAL_HMGA2-S_GFP | In this paper | n/a |
| pSMAL_HMGA2-L+3′UTRwt_GFP | In this paper | n/a |
| pSMAL_HMGA2-L+3′UTRmt_GFP | In this paper | n/a |
| pSMAL_HMGA2-S+3′UTRwt_GFP | In this paper | n/a |
| pSMAL_CLK3_GFP | In this paper | n/a |
| pSMAL_CTRL_GFP | In this paper | n/a |
| pLKO.1_shRNA_HMGA2-L_1 | In this paper | n/a |
| pLKO.1_shRNA_HMGA2-L_2 | In this paper | n/a |
| pLKO.1_shRNA_HMGA2-S_1 | In this paper | n/a |
| pLKO.1_shRNA_HMGA2-S_2 | In this paper | n/a |
| pLKO.1_shRNA_BOTH | In this paper | n/a |
| pLKO.1_CLK3_1 | Sigma-Aldrich | TRCN0000196926 |
| pLKO.1_CLK3_2 | Sigma-Aldrich | TRCN0000000749 |
| pLKO.1_shRNA_LUC | In this paper | n/a |
| pLX_TRC304/317_splicing_regulators_ORFs | Broad Institute GPP | n/a |
| psiCHECK2_Rluc-3’UTRwt_HMGA2-L | In this paper | n/a |
| psiCHECK2_Rluc-3’UTRmt_HMGA2-L | In this paper | n/a |
| psiCHECK2_Rluc-3’UTRwt_HMGA2-S | In this paper | n/a |
| pcDNA3.1_Luc-exon-4-WT (HMGA2-L) | In this paper | n/a |
| pcDNA3.1_Luc-exon-4-mut (HMGA2-L) | In this paper | n/a |
| Oligonucleotides | ||
| HMGA2 detection primers (full ORF cDNA) | ||
| HMGA2-common_FW | AGCGCCTCAGAAGAGAGGAC | n/a |
| HMGA2-L_RV | TGAGGATGTCTCTTCAGTTTCC | n/a |
| HMGA2-S_RV | TGGAAGAAAGGCTTCTAAGCTG | n/a |
| HMGA2-RI_RV | AGGCTCCTGTAGTCAGTCATTG | n/a |
| HMGA2-S”_RV | TGAAGACACTTTCCTTGGATCC | n/a |
| HMGA2 detection primers (partial ORF cDNA) | ||
| HMGA2-L-ORF_FW | AGACCTAGGAAATGGCCACA | n/a |
| HMGA2-L-ORF_RV | GTCCTCTTCGGCAGACTCTT | n/a |
| HMGA2-S-ORF_FW | AGTCCCTCTAAAGCAGCTCA | n/a |
| HMGA2-S-ORF_RV | TGAACACCACATGACACCAA | n/a |
| HMGA2-ex2-ORF_FW | GAACCAACCGGTGAGCCCT | n/a |
| HMGA2-ex2-ORF_RV | CTTTTGAGCTGCTTTAGAGGG | n/a |
| PROM1 detection primers | ||
| CAGAAGGCATATGAATCCAAA | n/a | |
| PROM1_FW | A | |
| PROM1_RV | GGTGCATTTCTCCACCACAT | n/a |
| Commercial detection primers | ||
| QuantiTect Primer Assay Hs_CLK3_1_SG | Qiagen | Cat. QT00197428 Prod. 249900 |
| QuantiTect Primer Assay Hs_SRSF1_1_SG | Qiagen | Cat. QT00203056 Prod. 249900 |
| QuantiTect Primer Assay Hs_HPRT1_1_SG | Qiagen | Cat. QT00059066 Prod. 249900 |
| miScript Primer Assay (miRNAs in this paper) | Qiagen | Cat. MS000XXXXX Prod. 218300 |
| miScript Primer Assay Hs_RNU6-2_11 | Qiagen | Cat. MS00033740 Prod.218300 |
| siRNA, miRNA inhibitors and mimics | ||
| ON-TARGETplus Human SRSF1 (6426) siRNA pool | Dharmacon | Cat. L-018672-01-0005 |
| ON-TARGETplus Non-targeting pool | Dharmacon | Cat. D-001810-10-05 |
| miRIDIAN Hairpin Inhibitor (miRNAs in this paper) | Dharmacon | Cat. IH-HMR-XX-0002 |
| miRIDIAN Hairpin Inhibitor Negative Control #1 | Dharmacon | Cat. IN-001005-01-05 |
| miRIDIAN Hairpin Inhibitor Negative Control #2 | Dharmacon | Cat. IN-002005-01-05 |
| miRIDIAN Mimic (miRNAs in this paper) | Dharmacon | Cat. C-HMR-XX-0002 |
| miRIDIAN Mimic Negative Control #1 | Dharmacon | Cat. CN-001000-01-05 |
| Deposited Data | ||
| RNA-seq, ChIP-seq, miRNA nanostring data | In this paper | NCBI GEO: GSE109093 |
| TargetScan | Agarwal et al., 2015 | v7 |
| MSigDb | Subramanian et al., 2005 | v6.1 |
| Blueprint Epigenome dataset | Stunnenberg et al., 2016 | 7th data release |
| Gencode gene annotations | Harrow et al., 2012 | v17, v19 |
| Human reference genome, hg19 | UCSC Genome Browser | hg19 |
| SRSF1 motifs | Wang et al., 2011 | n/a |
| Software and Algorithms | ||
| BEDTools | Quinlan and Hall. 2010 | v2.2.6 |
| Cufflinks | Trapnell et al., 2012 | v2.2.1 |
| TopHat | Trapnell et al., 2012 | v2.0.14 |
| cummeRbund | Trapnell et al., 2012 | v2.20.0 |
| Bowtie2 | Langmead and Salzberg. 2012 | v2.2.1 |
| GSEA | Subramanian et al., 2015 | v2.0 |
| BubbleGUM | Spinelli et al., 2015 | v1.3.19 |
| IGV | Robinson et al., 2011 | v2.3 |
| SUPPA | Alamancos et al., 2015 | v2.2.1 |
| rMAPS | Park et al., 2016 | v1.0.6 |
| HOMER | Heinz et al., 2010. | v4.9 |
| R | The R Project | v3.1 |
| Python | Python | v2.7 |
n/a: does not apply
HIGHLIGHTS.
-
-
Substantial diversity of human HSC transcriptomes at distinct developmental stages;
-
-
HMGA2 alternative 3’UTR usage is functional to avoid miRNA-mediated inhibition;
-
-
CLK3 affects HMGA2 splicing pattern by promoting exon skipping;
Acknowledgments
We are grateful to Trista North for critical review of the manuscript; Tarjei Mikkelsen, Michael Ziller and Areum Han for technical assistance and for helpful discussion; Ronald Mathieu and Mahnaz Paktinat at the BCH Flow Cytometry core for sorting; Annamaria Carissimo for assistance with statistical analyses; Julie Martone and Irene Bozzoni for providing the plasmid for the splicing assay. This work is supported by grants to G.Q.D. from the NIH NIDDK (R24-DK092760, R24-DK49216), NHLBI Progenitor Cell Biology Consortium (UO1-HL100001), NHLBI R01HL04880, NIH R24OD017870-01, to J.N.H. from NIH grant R01DK075787, to A.M. from New York Stem Cell Foundation, to A.C. from NCI Outstanding Investigator Award (R35 CA197745) and AC, BS, and FG from Centers for Cancer Systems Biology (U54 CA209997), to DC from Fondazione Telethon Core Grant, Armenise-Harvard Foundation career development award, European Research Council (grant agreement 759154, CellKarma) and the Rita-Levi Montalcini program from MIUR. M.C is a Leukemia and Lymphoma Society Fellow, L.T.V. is supported by the NSF Graduate Research Fellowship. A.M. is a New York Stem Cell Foundation Robertson Investigator, K.M.T. was supported by the HHMI International Student Research Fellowship and the Herchel Smith Graduate Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions. M.C. and G.Q.D. conceived the experimental plan. M.C., M.H.G., and D.C. wrote the manuscript. M.C, L.W., B.T.V., D.C., K.M.T., A.B. performed experimental work. S.D., L.T.V. and L.W. performed intra-femoral injections and provided HPC-5F cells. P.M.S. and J.B. assisted with mouse work. M.H.G, C.T., K.C., P.C. processed and analyzed data. D.C. supervised computational analyses. G.Q.D., J.N.H., A.M., J.L.R., A.C. provided mentoring and assisted with data interpretation.
Competing financial interests.
GQD holds equity interest in True North Therapeutics and 28/7 Therapeutics.
References
- Agarwal V, Bell GW, Nam J-W, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015:4. doi: 10.7554/eLife.05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alamancos GP, Pagès A, Trincado JL, Bellora N, Eyras E. Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA. 2015;21:1521–1531. doi: 10.1261/rna.051557.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubol BE, Wu G, Keshwani MM, Movassat M, Fattet L, Hertel KJ, Fu X-D, Adams JA. Release of SR Proteins from CLK1 by SRPK1: A Symbiotic Kinase System for Phosphorylation Control of Pre-mRNA Splicing. Mol. Cell. 2016;63:218–228. doi: 10.1016/j.molcel.2016.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babovic S, Eaves CJ. Hierarchical organization of fetal and adult hematopoietic stem cells. Exp. Cell Res. 2014;329:185–191. doi: 10.1016/j.yexcr.2014.08.005. [DOI] [PubMed] [Google Scholar]
- Bernitz JM, Kim HS, MacArthur B, Sieburg H, Moore K. Hematopoietic Stem Cells Count and Remember Self-Renewal Divisions. Cell. 2016;167:1296–1309. doi: 10.1016/j.cell.2016.10.022. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezas-Wallscheid N, Klimmeck D, Hansson J, Lipka DB, Reyes A, Wang Q, Weichenhan D, Lier A, von Paleske L, Renders S, et al. Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell. 2014;15:507–522. doi: 10.1016/j.stem.2014.07.005. [DOI] [PubMed] [Google Scholar]
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacchiarelli D, Trapnell C, Ziller MJ, Soumillon M, Cesana M, Karnik R, Donaghey J, Smith ZD, Ratanasirintrawoot S, Zhang X, et al. Integrative Analyses of Human Reprogramming Reveal Dynamic Nature of Induced Pluripotency. Cell. 2015;162:412–424. doi: 10.1016/j.cell.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, Down J, Denaro M, Brady T, Westerman K, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick K, Wang L, Li L, Menendez P, Murdoch B, Rouleau A, Bhatia M. Cytokines and BMP-4 promote hematopoietic differentiation of human embryonic stem cells. Blood. 2003;102:906–915. doi: 10.1182/blood-2003-03-0832. [DOI] [PubMed] [Google Scholar]
- Chambers JM. Statistical Models in S. Boca Raton, FL, USA: CRC Press, Inc.; 1991. [Google Scholar]
- Chen L, Kostadima M, Martens JHA, Canu G, Garcia SP, Turro E, Downes K, Macaulay IC, Bielczyk-Maczynska E, Coe S, et al. Transcriptional diversity during lineage commitment of human blood progenitors. Science. 2014;345:1251033. doi: 10.1126/science.1251033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo DF, Burger L, Baubec T, Schübeler D. Binding of high mobility group A proteins to the mammalian genome occurs as a function of AT-content. PLoS Genet. 2017;13:e1007102. doi: 10.1371/journal.pgen.1007102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colwill K, Pawson T, Andrews B, Prasad J, Manley JL, Bell JC, Duncan PI. The Clk/Sty protein kinase phosphorylates SR splicing factors and regulates their intranuclear distribution. EMBO J. 1996;15:265–275. [PMC free article] [PubMed] [Google Scholar]
- Copley MR, Babovic S, Benz C, David JH, Beer PA, Kent DG, Wohrer S, Treloar DQ, Day C, Rowe K, et al. The Lin28b–let-7-Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat. Cell Biol. 2013;15:916–925. doi: 10.1038/ncb2783. [DOI] [PubMed] [Google Scholar]
- Crews LA, Balaian L, Delos Santos NP, Leu HS, Court AC, Lazzari E, Sadarangani A, Zipeto MA, La Clair JJ, Villa R, et al. RNA Splicing Modulation Selectively Impairs Leukemia Stem Cell Maintenance in Secondary Human AML. Cell Stem Cell. 2016a;19:599–612. doi: 10.1016/j.stem.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews LA, Balaian L, Delos Santos NP, Leu HS, Court AC, Lazzari E, Sadarangani A, Zipeto MA, La Clair JJ, Villa R, et al. RNA Splicing Modulation Selectively Impairs Leukemia Stem Cell Maintenance in Secondary Human AML. Cell Stem Cell. 2016b;19:599–612. doi: 10.1016/j.stem.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulatov S, Notta F, Laurenti E, Dick JE. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10:120–136. doi: 10.1016/j.stem.2012.01.006. [DOI] [PubMed] [Google Scholar]
- Doulatov S, Vo LT, Chou SS, Kim PG, Arora N, Li H, Hadland BK, Bernstein ID, Collins JJ, Zon LI, et al. Induction of multipotential hematopoietic progenitors from human pluripotent stem cells via respecification of lineage-restricted precursors. Cell Stem Cell. 2013;13:459–470. doi: 10.1016/j.stem.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebina W, Rossi DJ. Transcription factor-mediated reprogramming toward hematopoietic stem cells. EMBO J. 2015;34:694–709. doi: 10.15252/embj.201490804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat. Rev. Genet. 2014;15:7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- Fedele M, Berlingieri MT, Scala S, Chiariotti L, Viglietto G, Rippel V, Bullerdiek J, Santoro M, Fusco A. Truncated and chimeric HMGI-C genes induce neoplastic transformation of NIH3T3 murine fibroblasts. Oncogene. 1998;17:413–418. doi: 10.1038/sj.onc.1201952. [DOI] [PubMed] [Google Scholar]
- Han J, Pedersen JS, Kwon SC, Belair CD, Kim Y-K, Yeom K-H, Yang W-Y, Haussler D, Blelloch R, Kim VN. Posttranscriptional crossregulation between Drosha and DGCR8. Cell. 2009;136:75–84. doi: 10.1016/j.cell.2008.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Mason PJ, Bessler M. 3’ UTR-truncated Hmga2 cDNA causes MPN-like hematopoiesis by conferring a clonal growth advantage at the level of HSC in mice. Blood. 2011;117:5860–5869. doi: 10.1182/blood-2011-02-334425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak B, Rosigkeit J, Wanschura S, Meyer-Bolte K, Van de Ven WJ, Kayser K, Krieghoff B, Kastendiek H, Bartnitzke S, Bullerdiek J. HMGI-C rearrangements as the molecular basis for the majority of pulmonary chondroid hamartomas: a survey of 30 tumors. Oncogene. 1996;12:515–521. [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes Dev. 2007;21:1025–1030. doi: 10.1101/gad.1540407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li O, Li J, Dröge P. DNA architectural factor and proto-oncogene HMGA2 regulates key developmental genes in pluripotent human embryonic stem cells. FEBS Lett. 2007;581:3533–3537. doi: 10.1016/j.febslet.2007.06.072. [DOI] [PubMed] [Google Scholar]
- Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem. J. 2009;417:15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- Martone J, Briganti F, Legnini I, Morlando M, Picillo E, Sthandier O, Politano L, Bozzoni I. The lack of the Celf2a splicing factor converts a Duchenne genotype into a Becker phenotype. Nat. Commun. 2016;7:10488. doi: 10.1038/ncomms10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315:1576–1579. doi: 10.1126/science.1137999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney-Freeman S, Cahan P, Li H, Lacadie SA, Huang H-T, Curran M, Loewer S, Naveiras O, Kathrein KL, Konantz M, et al. The transcriptional landscape of hematopoietic stem cell ontogeny. Cell Stem Cell. 2012;11:701–714. doi: 10.1016/j.stem.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermoud JE, Cohen PT, Lamond AI. Regulation of mammalian spliceosome assembly by a protein phosphorylation mechanism. EMBO J. 1994;13:5679–5688. doi: 10.1002/j.1460-2075.1994.tb06906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkola HKA, Orkin SH. The journey of developing hematopoietic stem cells. Development. 2006;133:3733–3744. doi: 10.1242/dev.02568. [DOI] [PubMed] [Google Scholar]
- Miraglia S, Godfrey W, Yin AH, Atkins K, Warnke R, Holden JT, Bray RA, Waller EK, Buck DW. A novel five-transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood. 1997;90:5013–5021. [PubMed] [Google Scholar]
- Ngo JCK, Chakrabarti S, Ding J-H, Velazquez-Dones A, Nolen B, Aubol BE, Adams JA, Fu X-D, Ghosh G. Interplay between SRPK and Clk/Sty kinases in phosphorylation of the splicing factor ASF/SF2 is regulated by a docking motif in ASF/SF2. Mol. Cell. 2005;20:77–89. doi: 10.1016/j.molcel.2005.08.025. [DOI] [PubMed] [Google Scholar]
- Nishino J, Kim I, Chada K, Morrison SJ. Hmga2 promotes neural stem cell self-renewal in young but not old mice by reducing p16Ink4a and p19Arf Expression. Cell. 2008;135:227–239. doi: 10.1016/j.cell.2008.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notta F, Zandi S, Takayama N, Dobson S, Gan OI, Wilson G, Kaufmann KB, McLeod J, Laurenti E, Dunant CF, et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science. 2016;351:aab2116. doi: 10.1126/science.aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novershtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, McConkey ME, Habib N, Yosef N, Chang CY, Shay T, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144:296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozturk N, Singh I, Mehta A, Braun T, Barreto G. HMGA proteins as modulators of chromatin structure during transcriptional activation. Front Cell Dev Biol. 2014;2:5. doi: 10.3389/fcell.2014.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Jung S, Rouchka EC, Tseng Y-T, Xing Y. rMAPS: RNA map analysis and plotting server for alternative exon regulation. Nucleic Acids Res. 2016;44:W333–W338. doi: 10.1093/nar/gkw410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad J, Colwill K, Pawson T, Manley JL. The protein kinase Clk/Sty directly modulates SR protein activity: both hyper- and hypophosphorylation inhibit splicing. Mol. Cell. Biol. 1999;19:6991–7000. doi: 10.1128/mcb.19.10.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian P, He XC, Paulson A, Li Z, Tao F, Perry JM, Guo F, Zhao M, Zhi L, Venkatraman A, et al. The Dlk1-Gtl2 Locus Preserves LT-HSC Function by Inhibiting the PI3K–mTOR Pathway to Restrict Mitochondrial Metabolism. Cell Stem Cell. 2016;18:214–228. doi: 10.1016/j.stem.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentas S, Holzapfel NT, Belew MS, Pratt GA, Voisin V, Wilhelm BT, Bader GD, Yeo GW, Hope KJ. Musashi-2 attenuates AHR signalling to expand human haematopoietic stem cells. Nature. 2016;532:508–511. doi: 10.1038/nature17665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenmakers EF, Wanschura S, Mols R, Bullerdiek J, Van den Berghe H, Van de Ven WJ. Recurrent rearrangements in the high mobility group protein gene, HMGI-C, in benign mesenchymal tumours. Nat. Genet. 1995;10:436–444. doi: 10.1038/ng0895-436. [DOI] [PubMed] [Google Scholar]
- Shyh-Chang N, Daley GQ. Lin28: primal regulator of growth and metabolism in stem cells. Cell Stem Cell. 2013;12:395–406. doi: 10.1016/j.stem.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat. Rev. Cancer. 2016 doi: 10.1038/nrc.2016.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli L, Carpentier S, Montañana Sanchis F, Dalod M, Vu Manh T-P. BubbleGUM: automatic extraction of phenotype molecular signatures and comprehensive visualization of multiple Gene Set Enrichment Analyses. BMC Genomics. 2015;16:814. doi: 10.1186/s12864-015-2012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stunnenberg HG International Human Epigenome Consortium, and Hirst, M. The International Human Epigenome Consortium: A Blueprint for Scientific Collaboration and Discovery. Cell. 2016;167:1897. doi: 10.1016/j.cell.2016.12.002. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Vedi A, Santoro A, Dunant CF, Dick JE, Laurenti E. Molecular landscapes of human hematopoietic stem cells in health and leukemia. Ann. N. Y. Acad. Sci. 2016;1370:5–14. doi: 10.1111/nyas.12981. [DOI] [PubMed] [Google Scholar]
- Viswanathan SR, Daley GQ, Gregory RI. Selective blockade of microRNA processing by Lin28. Science. 2008;320:97–100. doi: 10.1126/science.1154040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Juan L, Lv J, Wang K, Sanford JR, Liu Y. Predicting sequence and structural specificities of RNA binding regions recognized by splicing factor SRSF1. BMC Genomics. 2011;12(Suppl 5):S8. doi: 10.1186/1471-2164-12-S5-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter N, Nimzyk R, Bösche C, Meyer A, Bullerdiek J. Chromatin immunoprecipitation to analyze DNA binding sites of HMGA2. PLoS One. 2011;6:e18837. doi: 10.1371/journal.pone.0018837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong JJ-L, Ritchie W, Ebner OA, Selbach M, Wong JWH, Huang Y, Gao D, Pinello N, Gonzalez M, Baidya K, et al. Orchestrated intron retention regulates normal granulocyte differentiation. Cell. 2013;154:583–595. doi: 10.1016/j.cell.2013.06.052. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Figures S1-S6
Table S1. Differentially expressed transcripts at gene and isoform level, related to Figure 1
Table S2. Quantification of mRNAs and non coding RNAs, related to Figure 1
Table S3. Summary of isoform switching events and effect on 3’UTR usage, related to Figure 2
Table S4. GSEA results, related to Figure 1