

Abstract
The large surface area, good vascularization, immense capacity for solute exchange and ultra-thinness of the alveolar epithelium are unique features of the lung that can facilitate systemic delivery via pulmonary administration of peptides and proteins. Physical and biochemical barriers, lack of optimal dosage forms and delivery devices limit the systemic delivery of biotherapeutic agents by inhalation. Current efforts to overcome these difficulties in order to deliver metabolic hormones (insulin, calcitonin, thyroid-stimulating hormone [TSH], follicle-stimulating hormone [FSH] and growth hormones) systemically, to induce systemic responses (immunoglobulins, cyclosporin A [CsA], recombinant-methionyl human granulocyte colony-stimulating factor [r-huG-CSF], pancreatic islet autoantigen) and to modulate other biological processes via the lung are reviewed. Safety aspects of pulmonary peptide and protein administration are also discussed.
Keywords: lung, peptides, proteins, pulmonary absorption, systemic delivery
Introduction
The techniques of recombinant DNA technology have been well refined during the past 20 years such that it is now possible to produce, under good manufacturing practice conditions, commercial quantities of therapeutic peptides and proteins. It is expected that, during the next decade, an even greater number of molecular targets will be identified for treatment of various diseases. These are exciting developments, not only for scientists, but also for patients, because such biotherapeutic agents are very specific in their actions, and thus will greatly improve the quality of life for the majority of patients.
Hundreds of bioengineered proteins and peptides are either already on the market or are undergoing clinical investigation; these include growth factors, hormones, monoclonal antibodies, cytokines and anti-infective agents, among others. However, these compounds have unusual characteristics that present considerable challenges to formulation scientists. The combination of their large molecular size, hydrophilicity and lability (both chemical and enzymatic) virtually exclude their formulation in traditional dosage forms such as tablets and capsules. Consequently, most proteins and peptides currently on the market are injectable. This route of drug administration is generally not preferable to patients, in particular because the indication for the use of these agents is usually treatment of a chronic condition. This leads to low patient compliance and an increase in the cost of therapy.
Formulation scientists have generally approached this challenge from two directions (Fig. 1): controlled release injections or drug administration via alternative routes. Unlike the limited surface area available for drug absorption (approximately 180 cm2) in the nasal cavity, the lung offers a large surface area for drug absorption (approximately 75 m2) [1]. In addition, the alveolar epithelium is very thin (approximately 0.1-0.5 μm thick) [2], thereby permitting rapid drug absorption. The alveoli can be effectively targeted for drug absorption by delivering the drug as an aerosol, with mass median aerodynamic diameter (MMAD) less than 5 μm. Also, the first-pass metabolism of the gastrointestinal tract is avoided. Although metabolic enzymes can be found in the lungs, the metabolic activities and pathways may be different from those observed in the gastrointestinal tract [3], which makes pulmonary administration of many peptides and proteins very promising. In addition to the challenges of dosage form, those posed by the delivery device should also be considered.
Figure 1.
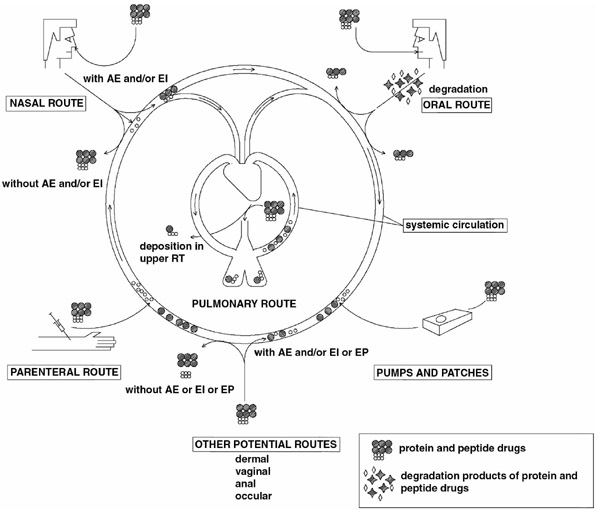
Clinical and potential routes of administration for therapeutic peptides and proteins. Note that small peptides may be absorbed in limited amounts without absorption enhancers (AE) and/or enzyme inhibitors (EI) via some routes (eg nasal). EP, electroporation/iontophoresis (specific for dermal delivery); RT, respiratory tract.
In the present review, we present information regarding recent developments in pulmonary drug administration of peptides and proteins, with emphasis on pulmonary delivery of insulin. The biophysical basis of pulmonary administration, as well as the barrier properties of the lungs, are reviewed in detail. The devices that are available for general drug administration to the lungs are discussed, and a comparative treatise of the pulmonary route and other routes for administration of biopharmaceutical agents is provided. Finally, both recent clinical and toxological findings are discussed.
Biophysical basis for pulmonary drug administration
The anatomical organization of the respiratory tract (characterized by extensive bifurcation) and aerosol characteristics of drug molecules (especially particle size) generally determine the reproducibility of pulmonary drug administration.
The respiratory tract comprises the conducting and respiratory regions. The conducting region essentially consists of nasal cavity, nasopharynx, bronchi and bronchioles. Airways distal to the bronchioles and the alveoli constitute the respiratory region, where rapid solute exchange takes place. According to Wiebel's tracheobronchial classification [4], the conducting airways comprise the first 16 generations, and generations 17–23 include the respiratory bronchioles, the alveolar ducts and the alveolar sacs.
The most important parameter that defines the site of deposition of aerosol drugs, including proteins and peptides, within the respiratory tract is the particle characteristics of the aerosol. The nature of the aerosol droplets is dependent on its MMAD, which is a function of particle size, shape and density. Particle charge and air velocities within the airways are also important attributes. Strict control of MMAD of the particles ensures reproducibility of aerosol deposition and retention within desired regions of the respiratory tract. Good distribution throughout the lung requires particles with an aerodynamic diameter between 1 and 5 μm, and thus most inhaled products are formulated with a high proportion of drug in this size range [5]. In order to target the alveolar region specifically, the aerosol droplet diameter should not be more than 3 μm. Particles with diameters that are greater than 6 μm are deposited in the oropharynx, whereas smaller particles (<1 μm) are exhaled during normal tidal breathing.
Dosage forms and delivery devices
Optimal management of most diseases, including diabetes, requires accurate dosing of the therapeutic compound. Pulmonary drug administration imposes stringent requirements on the delivery device; this is because the particle size of the powder or droplet greatly influences the delivery site, and thus the degree of drug absorption from the lungs.
The devices that are currently available for pulmonary drug administration were mostly developed to achieve local effects of the drug in the conducting airways, such as in asthma. These devices include nebulizers, metered-dose inhalers (MDIs) and dry-powder inhalers (DPIs). With some modification, most of these devices can be used for pulmonary peptide and protein administration.
Use of nebulizers to administer biopharmaceutical agents has many important limitations. Such drugs are often very unstable in aqueous solutions, and are easily hydrolyzed. In addition, the process of nebulization exerts high shear stress on the compounds, which can lead to protein denaturation. This is a particular problem because 99% of the droplets generated are recycled back into the reservoir to be nebulized during the next dosing [6]. Furthermore, the droplets produced by nebulizers are rather heterogeneous, which results in very poor drug delivery to the lower respiratory tract.
MDIs utilize propellants (chlorofluorocarbons and, increasingly, hydrofluoroalkanes) to atomize the drug solution; this results in a more uniform spray than that achieved with nebulizers. However, proteins and peptides are susceptible to denaturation when they come into contact with these propellants or with the large air–liquid interfaces that are constantly being generated during aerosolization [3].
A promising alternative to MDIs and nebulizers is the DPI, in which the biopharmaceutical formulation can be delivered in dry form. Like MDIs, most DPIs that are currently approved are made for pulmonary drug administration of locally acting drugs for the management of asthma and chronic obstructive pulmonary diseases, such as anti-asthmatic agents. Examples of such devices include the Turbohaler (AstraZeneca, Wilmington, DE, USA), Diskhaler (GlaxoSmithKline, Research Triangle Park, NC, USA), Diskus (known as the Accuhaler in some countries, for example the UK; GlaxoSmithKline), Rotahaler (Glaxo-SmithKline) and Aerolizer (Novartis Pharma, Basel, Switzerland), among others. These devices differ not only in their forms of particle generation and delivery, but also with regard to design differences such as discrete or reservoir drug containment, the number of doses and the presence of a dose counter. Compared with discrete types, the performance of reservoir devices is susceptible to environmental humidity and moisture. Additionally, dose-to-dose variations are greater. Furthermore, dose emission for some can be dependent on inhalation flow rates [7,8].
Drug delivery to the lower respiratory tract from these DPIs depends strongly on inspiratory air velocity. Consequently, delivery efficiency in children, elderly persons or adults with certain disease conditions will not always result in reproducible pharmacokinetics and pharmcodynamic responses with some devices. An innovative solution to this problem is offered by the AKITA system (InAmed GmbH, Gauting, Germany). This is a fully electronically controlled device that makes use of vital patient parameters, such as inhalation flow rate, inhaled volume and inhalation duration, among others, to control the exact dose of the drug administered to the patient. This device is particularly suited for drugs that are very expensive and drugs for which accurate dosing is critical, such as insulin, as well as for research use.
In order to deliver amounts of biopharmaceutical agents that are greater than those of steroids and other bronchodilators that are used in asthma therapy, newer devices have been developed. Inhale Therapeutics (San Carlos, CA, USA) and Aradigm Corporation (Hayward, CA, USA) have developed devices that are currently undergoing clinical trials, whereas a delivery device developed by Dura Pharmaceuticals (San Diego, CA, USA) is still at a preclinical trial stage. The Inhale Therapeutics device (Inhance) mechanically compresses a fixed volume of air in order to aerosolize a premetered and sealed dose of the drug into a chamber. The patient inhales the drug within 10 s, during a slow and deep breath. This simple inhalation technique eliminates the complex motor co-ordination that is often required with MDIs, DPIs and nebulizers. The major limitation of this device is its large size. The device from Dura Pharmaceuticals (Spiros motorized blisterdisk) relies on a battery-powered motor/impeller, which is actuated by the patient's breath to aerosolize a preme-tered dose of drug in the chamber. The patient inhales deeply through a mouthpiece that turns on the motor. As with the Inhance device, patient motor co-ordination is not required. The AERx delivery system (Aradigm Corporation) converts large molecules (eg proteins and peptides) into fine-particle aerosols at the time of use. The device has unique features, such as computer-controlled processes and an electronic compliance monitoring system.
Because all of the devices that are currently available have some shortcomings, it is pertinent to present some of the features of an ideal pulmonary delivery device. This device should be portable, discreet and easy to use with minimal patient education. It should be rechargeable, hygienic, incorporate a dose counter, be moisture proof and environmentally friendly. It should also emit a consistent dose to the lungs, and be unaffected by the inhalation rate of the patient.
Barriers to pulmonary absorption of peptides and proteins
Despite the efficiency of modern pulmonary delivery devices and advanced dosage form designs, certain barriers still compromise the absorption of peptides and proteins by the lung. Niven [9] identified respiratory mucus, mucociliary clearance, alveolar lining layer, alveolar epithelium, basement membrane, pulmonary enzymes, macrophages and other cells as barriers to pulmonary absorption of biotherapeutic agents. Although the alveolar epithelium and capillary endothelium have high permeability to water, many gases and lipophilic substances, the permeation of many hydrophilic substances of large molecular size and of ionic species is limited [10]. The molecular weight cutoff of tight junctions for alveolar type I cells is 0.6 nm. Endothelial junctions allow passage of larger molecules (4–6 nm).
On reaching the alveoli most peptides and proteins are either degraded by proteases or removed by alveolar macrophages. The pulmonary macrophages have also been shown to secrete or release short-lived peroxidases, inflammatory and immunomodulatory mediators (including granulocyte colony-stimulating factor, interleukins, leukotrienes and proteases), and other molecules as part of a host defence mechanism. These molecules are able to degrade peptides and proteins [9].
The mucus (1–10 μm thick) that lines the pulmonary epithelium and the surfactant that lines the alveoli (0.1–0.2 μm thick) constitute physical barriers to pulmonary absorption of peptides and proteins. They have high concentrations of protease inhibitors, and presumably protect peptides and proteins from degradation. Nonetheless, this protection appears to be an exception rather than the rule, because membrane-associated (epithelial and endothelial) and intracellular (macrophages, lymphocytes, neutrophils and mast cells) proteases and peptidases readily degrade administered peptides and proteins [11,12,13,14].
Delivery of metabolic hormones
Pulmonary delivery of metabolic hormones, including insulin, calcitonin, growth hormones, somatostatin, TSH and FSH, to humans and experimental animals has been reported, with insulin being the most widely investigated [15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30][31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52].
Insulin
Animal studies
In 1971, Wigley et al [15] used animals to investigate pulmonary insulin absorption, which paved the way for subsequent animal studies. Later, studies focused on improving the reproducibility of the pharmacokinetic and pharmacodynamic profiles of insulin administered via the lung. Colthorpe et al [16] showed that the penetration index (peripheral/central deposition) for aerosolized insulin formulation (1.52) was much greater than that for instilled insulin (0.32) in rabbits. The bioavailable fraction for aerosolized insulin was more than 20-fold greater than that for instilled insulin (57.2% versus 2.6%), although the absorption rate constants were statistically equivalent. Mucociliary clearance of instilled insulin was probably responsible for the lower bioavailability with this method of administration, thus making aerosolization the preferred mode of delivery of insulin.
In a related study in rats, Okumura et al [17] showed that the relative bioavailability of insulin solutions was pH dependent and not higher than 42% (relative to subcutaneous administration), whereas the relative bioavailability of aerosolized insulin was similar to that of subcutaneous administration. In contrast, Sakr [18] reported the relative bioavailability of aerosolized insulin in rabbits to be 50% that of subcutaneous injection. The lower bioavailability after insulin delivery as aerosol reported by Sakr was related to insulin retention in the mini-mist nebulizer.
In another study, Jendle et al [19] studied the effect of pulmonary-delivered insulin in anaesthetized and mechanically ventilated pigs. The nebulized insulin effectively reduced the mean blood glucose level by 39%. The data from this study imply that intrapulmonary administration of insulin in anesthetized and mechanically ventilated animals results in clinically relevant serum insulin levels.
Independent pilot-scale human studies
Published independent human studies of the efficacy of pulmonary-delivered insulin involved either nondiabetic volunteers, or patients with type 1 or type 2 diabetes mellitus. In 1925 Gansslen [20] conducted the first study of the efficacy of insulin after pulmonary administration in humans. According to that study, inhalation of 30–50 einheiten insulin (crude animal pancreas extract) reduced blood glucose level by 26% within 2.5 h. Following this success, many other small-scale studies were conducted later in the century. Wigley et al [15] provided direct evidence of absorption of insulin following aerosol inhalation, and of its efficacy in inducing hypoglycaemia in one nondiabetic and three diabetic persons. A correlation was identified between hypoglycaemia and plasma immunoreactive insulin. Based on comparisons of plasma immunoreactive insulin, only 10% of the aerosolized insulin was recovered. Jendle and Karlberg [21] later showed that the administration of nebulized insulin can induce a significant hypoglycaemia and cause a clinically relevant increase in insulin serum concentration, thus making this route feasible as an alternative to parenteral injections.
In another study, Laube et al [22] demonstrated the efficiency of optimized deposition of aerosolized insulin in normalizing plasma glucose levels in fasting individuals. That study indicated that insulin delivered by inhalation and deposited predominantly within the lung is well tolerated, and can effectively normalize glucose levels in patients with type 2 diabetes mellitus. The feasibility of the lung as an alternative route for insulin administration was further highlighted by Laube et al [23] in patients with type 2 diabetes mellitus. The data from this study showed that, once plasma glucose levels are normalized, postprandial glucose levels may be maintained below diabetic level by delivering insulin into the lung 5 min before the ingestion of a meal.
Although the variability in the metabolic effect of inhaled insulin is of major importance to diabetic patients, only few studies have addressed this issue. In a study in seven patients with type 2 diabetes mellitus, Laube et al [24] showed that the efficacy of inhaled insulin, as observed in animal studies [16], is dependent on the region of deposition in the respiratory tract when compared with subcutaneous injection. Thus, deposition outside the alveolar region results in less reproducible bioavailable fraction in comparison to subcutaneous injections. Those investigators found that the ratios of insulin deposition in the larger central airways versus that in the peripheral airways (expressed as the inner:outer ratio and lung apex:basal ratio) were related to glucose responses after inhalation of insulin. Linear regression analysis identified that the maximum percentage decrease in glucose after insulin administration was correlated with lung apex:basal ratio, whereas no such correlation was found with inner:outer ratio. This means that increasing the distribution of insulin aerosol to the alveolar region of the lung enhances the glucose response in patients with type 2 diabetes mellitus in the fasting state.
In an attempt to reduce the variability of inhaled insulin, Pfützner et al [25] formulated insulin using technospheres. The variability in metabolic effect of technosphere–insulin (TI) formulation in 12 type 2 diabetic patients, in a randomized, four-way, crossover, glucose clamp study, suggested that TI may be superior to recombinant insulin for prandial insulin supplementation in type 2 diabetic patients; TI showed a more rapid onset and shorter duration of action. Also lower intrasubject variability was seen with TI. Similar results were reported Rave et al [26] using this dosage formulation.
In order to identify possible pharmacokinetic and pharmacodynamic differences between children and adults, Elliott et al [27] investigated the pulmonary absorption of insulin in children. To improve the amount of insulin delivered and its reproducibility, those investigators used a Hudson patient-activated nebulizer. This device did not eliminate the variability associated with pulmonary insulin delivery. Within patients, the rise in free plasma insulin concentrations (μU/ml) for a given nebulized dose (0.37–0.49 U dose/day per kg body weight) was variable, indicating either differences in the efficiency of deposition and absorption from time to time, or that some other biological variables (eg variable antibody binding or insulin removal route) were active.
Despite the fact that only a fraction of inhaled insulin is actually absorbed, independent pilot-scale studies suggest that the degree of absorption is reproducible. This is an indication that inhaled insulin is a potential substitute for subcutaneous injections, especially for patients with erratic subcutaneous insulin absorption.
Clinical trials
Innovations in powder processing, protein and small-molecule formulations, and aerosol delivery systems have made clinical trials of aerosol dosage forms of insulin possible. The major products that are in clinical trial stage are inhaled insulin (Inhale Therapeutics) and the AERx insulin delivery system (Aradigm Corporation). The results of phase 2 clinical studies with these products have been reported (Table 1).
Table 1.
Summary of clinical and related trials of inhaled insulin
| Subjects | ||||
| Dosage forms/ | (diabetics or | |||
| Insulin doses | delivery devices | volunteers) | Pharmacokinetic profiles and therapeutic outcome | Reference |
| 1–2 inhalations per dose | Inhaled insulin | 70 (type 1) | HbA1c (%): 8.51 (INH), 8.53 (SC) | [29] |
| Pulmonary functions: no changes | ||||
| Acceptance/preference of INH: ≥ 80%. | ||||
| 1–2 inhalations per dose | Inhaled insulin | 51 (type 1) | HbA1c (%): 8.7 (INH), 7.8 (SC) | [30] |
| Pulmonary functions: no changes | ||||
| Acceptance/preference of INH: 92% | ||||
| 250 U and 500 U | AERx DMS | 11 (volunteers) | Tmax (min): 7 and 16 for INH 250 and 500 U, respectively | [34] |
| Cmax (μU/ml): 29.7 and 23.8 for INH 250 and 500 U, respectively | ||||
| tGmax (min): 66 and 76 for INH 250 and 500 U, respectively | ||||
| 4–6 inhalations per dose | Inhaled insulin | 16 (type 2) | Baseline glucose change: 100 to 53 mg/dl (INH); | [28] |
| 100 to 57 mg/dl (SC) | ||||
| Pulmonary functions: no change | ||||
| Reproducibility: INH similar to SC | ||||
| 1–2 inhalations per dose | AERx DMS | 20 (type 1) | Glucose change from baseline (mg/dl): 82 (60 min), 79 (120 min) and –11 (300 min) for AERx DMS; | [36] |
| 89 (60 min), 82 (120 min) and –25 (300 min) for SC | ||||
| Deleterious effect: none | ||||
| 1–2 inhalations per dose | Inhaled insulin | 69 (type 2) | Baseline HbA1c (%) before therapy: 9.92 (oral agent alone); 9.78 (oral agent + INH) | [38] |
| Change in HbA1c (%) after 2 weeks: –0.13 (oral agent alone); 2.28 (oral agent + INH) | ||||
| 100 U TI | MedTone inhaler (TI) | 12 (type 2) | GIRmax (mg/kg per min): 5.8 (INH), 2.2 (SC) | [25] |
| (PDC) | GIRtmax (min) = 55 (INH), 276 (SC) | |||
| USA | Early tGIR50% (min): 17 (INH), 122 (SC) | |||
| Late tGIR50% (min): 128 (INH), 335 (SC) | ||||
| Not mentioned | Inhaled insulin | 70 (type 1) | Preference of INH over SC: 81% | [32] |
| Switch from SC to INH: 79% | ||||
| Continuance of SC: 21% | ||||
| Satisfaction: 38% (INH), 14% (SC) | ||||
| Convenience/ease of use: 46% (INH), 12% (SC) | ||||
| Not mentioned | Inhaled insulin | Number not stated | HbA1c (%): 8.9 (baseline), 8.0 (after 3 months), 8.1 (after 12 months), 8.0 (after 18 months), 8.0 (after 24 months) | [33] |
| (type 1 and type 2) | ||||
| FEV1 (l): 3.2 (baseline), 3.1 (after 12 months), 3.1 (after 18 months), 3.2 (after 24 months) | ||||
| DLCO (ml/min per mmHg): 25.6 (baseline), 24.7 (after 12 months), 24.7 (after 18 months), 24.4 (after 24 months) | ||||
| Not mentioned | Inhaled insulin | 56 (type 2) | Mean improvement in patient satisfaction (%): 38 (INH), | [31] |
| 14 (SC) | ||||
| INH preference to SC based on: ease of use, comfort and convenience | ||||
| 0.3–1.8 U/kg | AERx DMS | 18 (type 1) | Tmax (min): for INH 49, 48, 62 and 65 at doses 0.3, 0.6, 1.2 and 1.8 U/kg, respectively; for SC 119 at dose 0.12 U/kg | [37] |
| GIRmax (mg/kg per min): for INH 1.6, 2.5, 4.7 and 6.5 at doses 0.3, 0.6, 1.2 and 1.8 U/kg, respectively; for SC 3.2 at dose 0.12 U/kg | ||||
| tGIRmax (min): for INH 94, 136, 157 and 218 at doses 0.3, 0.6, 1.2 and 1.8 U/kg, respectively; for SC 189 at dose 0.12 U/kg | ||||
| 25–100 U | MedTone inhaler (TI) | 12 (volunteers) | GIRmax (mg/kg per min): concentration dependent | [26] |
| (PDC) | GIRtmax (min): 47, 52, 56 for TI 25, 50 and 100 U, respectively; 192 for SC | |||
| Tmax (min): 12, 18, and 21 for TI 25, 50 and 100 U, respectively; for SC 153 | ||||
| Bioavailability (relative to SC for 3 h): 46, 42 and 28% for TI 25, 50 and 100 U, respectively | ||||
Cmax, maximum insulin concentration; DLCO, diffusion capacity; DMS, diabetic management system; FEV1, forced expiratory volume in 1 s; GIRmax, maximum glucose infusion rate; GIRtmax, time to maximum glucose infusion rate; HbA1c, glycated haemoglobin (glycaemic control index); INH, inhaled insulin; PDC, Pharmaceutical Discovery Corporation (Elmsford, NY, USA); SC, subcutaneous; tGIR50%, time to late half maximum glucose infusion rate; tGmax, time to maximum glycemic effect; Tmax, time to maximum concentration of insulin.
In 16 patients with type 2 diabetes, Gelfand et al [28] demonstrated the reproducibility of rapid-acting insulin in therapeutic amounts of 1–2 inhalations per dose, which resulted in similar efficacy and safety to that of subcutaneous insulin. The insulin was delivered from blister packs containing either 3 or 9 U insulin per dose using an Inhale Therapeutics proprietary delivery system. Using the same delivery device, 3-month, multicenter trials in 121 diabetic patients (70 type 1 and 51 type 2) [29,30] indicated comparable glycaemic control in both groups, and the number of hypoglycaemic events was similar. These studies also indicated that patients were satisfied and preferred inhaled insulin over subcutaneous injections. Capparelli et al [31] also reported improved patient satisfaction with inhaled insulin. Preference of inhaled insulin over subcutaneous injections was based on ease of use, comfort and convenience. This observation is important because improved satisfaction may, in clinical practice, increase willingness of patients to initiate and comply with insulin therapy, and hence achieve better glycaemic control. In another study, Gerber et al [32] demonstrated that patient preference of inhaled insulin over subcutaneous administration did not change with time (1 year), and that the glycaemic control was also stable over that period. In another extended clinical trial (2 years), Cefalu et al [33] emphasized the fact that the efficacy of inhaled insulin identified in short-term clinical trials can be sustained in the long term.
Farr et al [34] used the AERx delivery system for delivery of insulin in 11 healthy volunteers. These investigators showed that inhaled insulin, administered as solution (U250 insulin or U500 insulin), had a faster onset of metabolic effect than did subcutaneous injection. It was concluded that the delivery of inhaled insulin to the vast surface area of the lung could counteract the concentration-dependent absorption that has been reported after subcutaneous injection. Similar results were obtained in a related study using the AERx delivery device [35]. In addition, a clear dose-response was observed, and the system efficiency of AERx diabetic management system was approximately 13% that of subcutaneous treatment. The AERx delivery system reduced the dose-to-dose variability in the pharmacokinetic and pharmacodynamic response to inhaled insulin. Another study [36] emphasized the reproducibility and safety of inhaled insulin using the device. Administration of regular insulin with this device resulted in a reproducible pharmacodynamic effect, similar to subcutaneous injections, and intrasubject variability did not differ significantly from that seen with subcutaneous administration. Reproducibility using this system was also reported by Brunner et al [37].
It has been proposed that inhaled insulin may serve as adjunctive therapy to oral therapy in type 2 diabetic patients in whom oral agents are not effective. In a 3-month, multicenter, phase 2 trial that involved 69 patients in nine sites, Weiss et al [38] demonstrated this possibility. That study showed that, in patients with type 2 diabetes who were not benefiting from oral agents, a no-injection regimen with adjunctive inhaled insulin therapy markedly improved glycaemic control, with low risk of hypoglycaemia.
Similar pharmacokinetic and pharmacodynamic profiles have been reported for subcutaneous and inhaled insulin, but the latter required as high as 15 times the subcutaneous dose (1.8 U/kg body weight for inhaled administration versus 0.12 U/kg body weight for subcutaneous administration) [37]. Therefore, much formulation and delivery refinement is necessary before inhalational delivery of insulin can replace subcutaneous administration.
Calcitonin
Very few attempts have been made to deliver calcitonin through the lung. Patton et al [39] reported an absolute bioavailability of approximately 17% for both human and salmon calcitonin after intratracheal instillation using catethers in rats. In another study, Komada et al [40] reported an absolute bioavailability of approximately 12% when administered as powder to rats and humans. The difference in bioavailability reported by Patton et al [39] and Komada et al [40] could be due to the different formulations used (solution versus powder).
Growth hormones
In rats, Folkesson et al [41] demonstrated the passage of human growth factor (hGH) across the lung to the blood following administration by instillation. The absorption was sex-specific, with female rats showing higher bioavailability than male rats. Colthorpe et al [42], in another study, compared the pharmacokinetics of pulmonary administered hGH in the form of aerosol (MMAD <5.5 μm) and instillate in rats. The bioavailable fraction for aerosolized hGH (45%) was greater than that for instilled hGH (16%). As suggested for insulin solutions [16], lower bioavailability for instillate was due to mucociliary clearance.
Using a different approach, Smith et al [53] showed that pulmonary administration via endotracheal tube of a hexapeptide (His-D-Trp-Ala-Trp-D-Phe-Lys-NH2, SK&F 110679), which elicits growth hormone release in animals and humans, caused dose-related increase in plasma growth hormone concentrations. In dogs, the bioavailability was approximately 45% that of intravenous administration.
Thyroid-stimulating hormone, follicle-stimulating hormone, parathyroid hormone and somatostatin
Pulmonary delivery of parathyroid hormone (PTH) 1–84 and 1–34, TSH, FSH and somatostatin have been investigated [37,38]. Bioavailabilities following pulmonary administration by instillation using silicone tubing or catheters were as follows: PTH 84, >23%; PTH 34, approximately 40%; and somatostatin, <1%. The bioavailabilities of TSH and FSH delivered in solution of neutral pH were similar (2.5 and 2.3%, respectively). The bioavailabilities of TSH and FSH in alkaline conditions were two to 30 times greater than those in neutral pH conditions. On the other hand, the bioavailabilities of TSH and FSH when given intratracheally as dry powder were 1.6 and 0.6%, respectively.
Delivery of luteinizing hormone-releasing hormone agonist/antagonists
The pulmonary delivery of leuprolide (luteinizing hormone-releasing hormone agonist), detirelix and cetrorelix (luteinizing hormone-releasing hormone antagonists) by intratracheal intubation or aerosol administration (MMAD 2.6 μm) has been reported.
Adjei and coworkers [54,55,56] clearly showed that leuprolide acetate attains high plasma concentrations after pulmonary aerosol delivery or instillation. A corresponding decrease in plasma gonadotrophin, with sequential increases in plasma leuprolide concentrations, was observed in these studies. In one of the studies [55], the investigators reported decreases in bioavailability when solution aerosols were administered. Subsequently they showed [56] that using a suspension aerosol instead of a cosolvent solution aerosol formulation of leuprolide resulted in a dose-related increase in plasma concentration. According to the authors, decreases in lung bioavailability were related to mild microscopic and inflammatory reactions of the lung tissue caused by the cosolvent (alcohol), which affected absorption when the cosolvent was used.
Pulmonary administration of detirelix has been investigated in anaesthetized dogs [57] and awake sheep [58]. In dogs the relative bioavailability was 29% following aerosol administration, and a similar profile was seen with instillation. In unanaesthethized sheep the average bioavailability following pulmonary administration as instillate or aerosol was approximately 10%. No significant changes in pharmacokinetic or systemic uptake of detirelix were observed during the 5-month period of repeated pulmonary administration.
Lizio et al [59] investigated the bioavailability of cetrorelix after pulmonary instillation in rats. When compared with intravenous administration, the absolute bioavailabilty of intratracheal cetrorelix was more than 70%. According to that study, pulmonary administration of 0.5–2.5 mg/kg body weight cetrorelix decreased plasma concentrations of testosterone to subnormal levels (≤ 1 ng/ml) within 72 h.
Delivery of cardiovascular peptides: vasopressin analogue
Folksson and coworkers [60,61] showed that high plasma concentrations of an analogue of vasopressin (1-deamino-8-D-arginine vasopressin [dDAVP]) could be attained following administration via the lung by instillation. These investigators found that the passage of dDAVP aerosol and instillate via the rat lung was age dependent, and was significantly increased in inflammatory conditions [60]. They also demonstrated pulmonary absorption of dDAVP in pigs [61]. As in previous studies, a significant decrease in total passage of dDAVP was observed with age: 74.6 ± 9.4% in the newborn, 44.1 ± 13.3% in 2 day old pigs and 23.6 ± 7.1% in 70 day old pigs. These data indicate that proteins and peptides may traverse the lung epithelium via different routes that are differently affected during postnatal development.
Delivery to induce systemic response
Recent studies have shown that a systemic response may be achieved following pulmonary administration of certain macromolecules. This has been demonstrated for immunoglobulins, CsA, r-huG-CSF, pancreatic islet autoantigen insulin and interferons.
Immunoglobulins
Delivery of specific antibodies or immunoglobulin constructs to the respiratory tract may be useful for prophylaxis or active treatment of local or systemic disorders. Folkesson et al [60] showed the possibility of systemic delivery of immunoglobulins via the lung. In their studies, the passage of bovine immunoglobulin was extremely slow, except in inflammation, with a transferred amount of 1.5 ± 0.3% after 16 h in young rats. For older rats, similar passage time curves were obtained, but the amount of bovine immunoglobulin transferred was lower. Using an entirely different approach, Bot et al [62] investigated pulmonary delivery of human immunoglobulin (MMAD 4.6 μm) using microparticles (Pulmospheres; Alliance Pharmaceutical Corporation, San Diego, CA, USA) as a platform for delivery. Instillation of nonaqueous human immunoglobulin formulated in Pulmospheres to the respiratory tract of BALB/c mice resulted in systemic biodistribution. The formulation triggered enhanced local and systemic immune responses against xenotypic epitopes, and was associated with receptor-mediated loading of alveolar macrophages. Thus, local and systemic delivery of immunoglobulins via the respiratory mucosa may be used to trigger or modulate immune responses.
Pancreatic islet autoantigen insulin
Delivery of aerosols that contain soluble immunologically active self-antigens such as collagen and myelin-basic proteins to the respiratory tract has been suggested as therapy for autoimmune diseases such as rheumatoid arthritis and multiple sclerosis, as a result of induction of systemic tolerance. Such an approach may be extended to diabetes mellitus. In a study of mucosal tolerance in autoimmune diabetes using the nonobese diabetic mouse model, Hänninen and Harrison [63] reported that treatment of prediabetic mice with the pancreatic islet autoantigen insulin by aerosol (MMAD <5.8 μm) inhalation reduced the incidence of diabetes. The reduction was associated with induction of CD8+ (αα) γδ T cells, small numbers of which prevent adoptive transfer of diabetes. Regulatory γδ T cells secrete interleukin-10 in pancreatic lymph nodes, which could account for the antidiabetic and bystander suppressor effect of nasorespiratory insulin [64].
Interferons
High plasma concentrations of recombinant-methionyl interferon consensus (rCon-IFN) and interferon-α have been attained following pulmonary administration to animals.
Patton et al [39] found the absolute bioavailability of interferon-α in rats to be greater than 56% following intratracheal instillation. Altrock et al [65] reported that, in hamsters infected with encephalomyelocarditis virus, significant protection was conferred following intratracheal instillation of rCon-IFN (5 μg/kg body weight). On the other hand, Niven et al [66] compared the pharmacokinetics and bioavailability of rCon-IFN and a modified lactose-conjugated consensus interferon in rat and hamster. After aerosol administration to rat, the estimated bioavailability of both compounds approached 70%, and rCon-IFN (5 μg/kg body weight) was effective in reducing the incidence of paralysis in the hamsters. These results demonstrate the feasibility of treating systemic viral infections with interferon administered directly to the lung.
Recombinant-methionyl human granulocyte colony-stimulating factor
Systemic delivery of r-huG-CSF can readily induce an increase in circulating levels of natural granulocyte colony-stimulating factor to approximately three to five times greater than baseline. Niven et al [67] showed that r-huG-CSF induced systemic response after delivery by aerosol in hamsters. The absorption from the lung was rapid, with a concomitant increase in white blood cells to four times baseline. The bioavailability was 45.9% of the administered dose, and 62.0% of the dose reached the lung lobes. In a study that compared pulmonary administration of r-huG-CSF powder with solution [68] a normal systemic response was obtained, indicating that r-huG-CSF retains its activity in the solid state after formulation. Dissolution and absorption of r-huG-CSF from powders were not rate limiting, because the plasma concentration versus time profiles peaked at similar times in both powder and solution administration.
Cyclosporin A
CsA is widely used in organ transplantation and abnormal immune reactions. After instillation of CsA with micelles-forming surfactant (Cremophor EL) in adult and young rats, Taljanski et al [69] showed that the plasma levels peaked at 5 min, with bioavailability of 77.5 ± 7.2% and 66.3 ± 4.5%, respectively. The bioavailability of aerosolized CsA was 80.1 ± 4.1% in adults. The investigators concluded that CsA was absorbed by the lungs into the systemic circulation in rats in high concentrations, independent of age and type of delivery system. This may be of clinical relevance to treatment of chronic rejection.
Delivery with absorption enhancers and enzyme inhibitors
Physical barriers and enzymatic degradation, among other factors, limit pulmonary absorption of peptides and proteins. Part of the strategy to improve pulmonary absorption of macromolecules via the lung includes co-administration with absorption enhancers and enzyme inhibitors. This approach has been shown to improve the bioavailabilities and pharmacodynamic response of biotherapeutic agents, including insulin, calcitonin and others.
Shao et al [43] reported the effectiveness of cyclodextrins as pulmonary absorption promoters. The relative effectiveness of cyclodextrins in enhancing pulmonary insulin absorption, as measured by pharmacodynamics, and relative efficiency is according to the following rank order: dimethyl-β-cyclodextrin > α-cyclodextrin > β-cyclodextrin > γ-cyclodextrin > hydroxypropyl-β-cyclodextrin. In another study, Shen et al [44] showed that lanthanide ions are effective in promoting pulmonary insulin absorption in rats. The effectiveness of absorption enhancers for pulmonary insulin delivery was confirmed by Heinemann et al [45], in a single-centre, open euglycaemic glucose clamp study in 13 healthy male volunteers. Insulin inhaled with bile salt as an absorption enhancer led to a considerably greater metabolic effect than was seen with inhalation of insulin alone, although the intra-individual variabilities were comparable.
Furthermore, the potential usefulness of enzyme inhibition to improve absorption of biotherapeutic agents via the lung has been demonstrated [12,13,14]. In addition to insulin, the pulmonary absorption of salmon calcitonin [46] and (ASU1,7)-Eel calcitonin [47,48] have been shown to be more efficient with absorption enhancers (oleic acid, polyoxyethylene oleyl ether, sodium glycocholate) and enzyme inhibitors (bacitracin, bestatin, nafanostat mesilate, soybean trypsin inhibitor, chymostatin, potato carboxypeptidase inhibitor, phosphoramidon).
Delivery with microparticles
The human lung has efficient mechanisms to remove deposited particles by mucociliary clearance and phagocytosis. When peptide and protein drugs are formulated using microparticles as vehicles, the influence of these clearance mechanisms may be attenuated, and more efficient absorption and a sustained therapeutic effect may be achieved. Edwards et al [49] showed that inhalation of large porous insulin particles (formulated with poly[lactic acid-co-glycolic acid]) resulted in elevated systemic levels of insulin and suppressed systemic glucose levels for 96 h, whereas small, nonporous insulin particles had this effect for only 4 h. Similarly, Kawashima et al [50] reported that the pulmonary delivery of insulin with nebulized DL-lactide/glycocholide copolymer nanospheres resulted in prolonged hypoglycaemia (48 h) as compared with the nebulized aqueous solution (6 h). The observed prolonged insulin concentrations and hypoglycaemic effect in these studies was attributed to the sustained release of insulin from the polymers.
Other studies have shown enhancement of pulmonary insulin absorption using liposomes as carriers. Liu et al [51] reported that intratracheal administration of insulin liposomes (dipalmitoylphosphatidyl choline:cholesterol, 7:2) led to facilitated pulmonary uptake of insulin and enhanced hypoglycaemic effect. The ability of liposomes to promote pulmonary insulin absorption depends on the concentration, charge and acyl chain length of the phospholipid [52]. In another study, Niven et al [70] emphasized the use of polyethyleneglycol for systemic delivery of r-huG-CSF. According to these authors, the pulmonary absorption of polyethylene glycolated r-huG-CSF in rat generated a more intense response and extended white blood cell response, as compared with r-huG-CSF alone.
Safety aspects of pulmonary peptide and protein delivery
The major concern regarding pulmonary administration of therapeutic peptides and proteins via the lung is the possibility of immunological reactions, because the body may recognize these molecules as antigens. A recent review by Wolff [71] suggested that pulmonary delivery of most therapeutic peptides and proteins is safe, at least after short-term use. In type 1 and type 2 diabetic patients, Cefalu et al [33] recently showed that pulmonary insulin administration over a 2 year period is safe.
Safety in use of absorption enhancers and enzyme inhibitors is of major concern. Yamamoto et al [48] showed a correlation between pulmonary absorption of calcitonin and local toxicity in the presence of absorption enhancers. The improved pharmacodynamic response seen with insulin in the presence of bile salt [45] may not be clinically advantageous, especially for chronic use, as bile salts erode epithelial surfaces.
Microparticles used to improve systemic delivery of peptides and proteins via the lung may have a detrimental effect, although the safety of some of them (eg liposomes) has been demonstrated [72]. Nevertheless, Dokka et al [73] recently reported that pulmonary administration of some liposomes may be detrimental, depending on the charge of the liposome. Reactive oxygen species were implicated in cationic lipid-mediated toxicity. Polyvalent cationic liposomes cause a release of reactive oxygen species, which are responsible for pulmonary toxicity.
When peptides and proteins are coadministered with absorption enhancers or enzyme inhibitors, or are delivered using microparticles, the safety of the adjuvant must be ascertained, both in short-term and long-term use.
Conclusion
Pulmonary drug delivery offers the opportunity for systemic administration of peptides and proteins that are at present usually administered parenterally. It is expected that the continued research interest in this route of administration will lead to more breakthroughs in several areas of both formulation and device design, and as such the market and benefits to patients will improve. Pulmonary drug administration research should be integrated. Those who are developing formulations with very high drug absorption should keep in mind the importance of safety and convenience. This is the only way to ensure, on a long-term basis, the success of a particular formulation in a given disease state. Although the current status of pulmonary administration of insulin is promising, the possible side effects following chronic use (10–20 years) are yet to be ascertained.
Abbreviations
CsA = cyclosporin A; dDAVP = 1-deamino-8-D-arginine vasopressin; DPI = dry-powder inhaler; FSH = follicle-stimulating hormone; hGH = human growth hormone; MDI = metered-dose inhaler; MMAD = mass median aerodynamic diameter; PTH = parathyroid hormone; r-Con-IFN = recombinant-methionyl interferon consensus; r-huG-CSF = recombinant-methionyl human granulocyte colony-stimulating factor; TI = technosphere–insulin formulation; TSH = thyroid-stimulating hormone.
Acknowledgments
Acknowledgement
We are grateful to Patrick Rombaut for his assistance in constructing the scheme for routes of delivery of proteins and peptides.
References
- Wearley LL. Recent progress in protein and peptide delivery by non-invasive routes. Crit Rev Ther Drug Carrier Syst. 1991;8:1331–1394. [PubMed] [Google Scholar]
- Wilson CG, Washington N. Physiological Pharmaceutics England: John Wiley; 1989.
- Banga AK. Therapeutic Peptides and Proteins: Formulation, Processing and Delivery Systems Lancaster: Technomic; 1995.
- Weibel ER. Morphometry of the Human Lung Berlin: Springer Verlag; 1963.
- Chrystyn H. Is total particle dose more important than particle distribution. Respir Med. 1997;91(suppl):17–19. doi: 10.1016/s0954-6111(97)90100-1. [DOI] [PubMed] [Google Scholar]
- Gupta S, Moussy F, Dalby RN, Meikka SI, Bruley DF. Pulmonary delivery of human protein C and factor IX. Textbook of Advances in Experimental Medicine and Biology or Oxygen Transport to Tissue, vol 411. Edited by Nemoto EM, LaManna J. New York: Plenum Press; 1997. pp. 429–435. [DOI] [PubMed]
- Engel T, Heinig JH, Malling HJ, Scharling B, Nikander K, Madsen F. Clinical comparison of inhaled busesonide delivered either via pressurized metered dose inhaler or Turbuhaler®. Allergy. 1989;44:220–225. doi: 10.1111/j.1398-9995.1989.tb02266.x. [DOI] [PubMed] [Google Scholar]
- Nielsen KG, Skov M, Klug B, Ifversen M, Bisgaard H. Flow-dependent effect of formoterol dry powder inhaled from the Aerolizer®. Eur Respir J. 1997;10:2105–2109. doi: 10.1183/09031936.97.10092105. [DOI] [PubMed] [Google Scholar]
- Niven RW. Delivery of biotherapeutics by inhalation aerosol. Crit Rev Ther Drug Carrier Syst. 1995;12:151–231. doi: 10.1615/critrevtherdrugcarriersyst.v12.i2-3.20. [DOI] [PubMed] [Google Scholar]
- Sayani AP, Chien YW. Systemic delivery of peptides and proteins across absorptive mucosae. Crit Rev Ther Drug Carrier Syst. 1996;13:85–184. [PubMed] [Google Scholar]
- Zhou XH. Overcoming enzymatic and absorption barriers to non-parenterally administered protein and peptide drugs. J Contr Rel. 1994;29:239–252. [Google Scholar]
- Liu FY, Kildsig DO, Mitra AK. Pulmonary biotransformation of insulin in rat and rabbit. Life Sci. 1992;51:1683–1689. doi: 10.1016/0024-3205(92)90313-e. [DOI] [PubMed] [Google Scholar]
- Shen Z, Zhang Q, Wei S, Nagai T. Proteolytic enzymes as a limitation for pulmonary absorption of insulin: in vitro and in vivo investigations. Int J Pharm. 1999;192:115–121. doi: 10.1016/s0378-5173(99)00295-1. [DOI] [PubMed] [Google Scholar]
- Fukuda F, Tsuji T, Fujita T, Yamamoto A, Muranishi S. Susceptibility of insulin to proteolysis in rat lung homogenates and its protection from proteolysis by various protease inhibitors. Biol Pharm Bull. 1995;18:891–894. doi: 10.1248/bpb.18.891. [DOI] [PubMed] [Google Scholar]
- Wigley FM, Londono JH, Wood SH, Shipp JC, Waldman RH. Insulin across respiratory mucosa by aerosol delivery. Diabetes. 1971;20:552–556. doi: 10.2337/diab.20.8.552. [DOI] [PubMed] [Google Scholar]
- Colthorpe P, Farr SJ, Taylor G, Smith IJ, Wyatt D. The pharmacokinetics of pulmonary-delivered insulin: a comparison of intratracheal and aerosol administration to the rabbit. Pharm Res. 1992;9:764–768. doi: 10.1023/a:1015851521551. [DOI] [PubMed] [Google Scholar]
- Okumura k, Iwaka S, Tsuguchika Y, Toshimitsu S, Komada F. Intratracheal delivery of insulin absorption from solution and aerosol by rat lung. Int J Pharm. 1992;88:63–73. [Google Scholar]
- Sakr FM. A new approach for insulin delivery via the pulmonary route: design and pharmacokinetics in non-diabetics rabbits. Int J Pharm. 1992;86:1–7. [Google Scholar]
- Jendle JH, Karlberg BE, Arborelius M. An exploration of intrapulmonary insulin administration in anaesthetized and mechanically ventilated pigs. Scand J Clin Lab Invest. 1996;56:251–258. doi: 10.3109/00365519609088614. [DOI] [PubMed] [Google Scholar]
- Gansslen M. About inhalation of insulin [abstract; in German]. Klin Wochenschr. 1925;4:71. [Google Scholar]
- Jendle JH, Karlberg BE. Intrapulmonary administration of insulin to healthy volunteers. J Intern Med. 1996;240:93–98. doi: 10.1046/j.1365-2796.1996.502836000.x. [DOI] [PubMed] [Google Scholar]
- Laube BL, Georgopoulos A, Adams GK. Preliminary study of the efficiency of insulin aerosol delivered by oral inhalation in diabetic patients. JAMA. 1993;269:2106–2109. [PubMed] [Google Scholar]
- Laube BL, Benedict GW, Dobs AS. The lung as an alternative route of delivery for insulin in controlling postprandial glucose levels in patients with diabetes. Chest. 1998;114:1734–1739. doi: 10.1378/chest.114.6.1734. [DOI] [PubMed] [Google Scholar]
- Laube BL, Benedict GW, Dobs AS. Time to peak insulin level, relative bioavailability, and effect of site of deposition of nebulized insulin in patients with noninsulin-dependent diabetes mellitus. J Aerosol Med. 1998;11:153–173. doi: 10.1089/jam.1998.11.153. [DOI] [PubMed] [Google Scholar]
- Pfützner A, Heise T, Steiner S, Heinemann L, Rave K. Inhaled technosphere: insulin shows a low variability in metabolic action in type 2 diabetic patients [abstract]. Diabetes. 2000;49 (suppl):A121. [Google Scholar]
- Rave KM, Heise T, Pfützner A, Steiner S, Heinemann L. Results of a dose-response study with a new insulin inhaler formulation [abstract]. Diabetes. 2000;49(suppl):A75. [Google Scholar]
- Elliot RB, Edgar BW, Pitcher CC, Quested C, McMaster J. Parenteral absorption of insulin from the lung in diabetic children. Aust Paediatr J. 1987;23:293–297. doi: 10.1111/j.1440-1754.1987.tb00275.x. [DOI] [PubMed] [Google Scholar]
- Gelfand AG, Schwartz SL, Horton M, Law CG, Pun EF. Pharmacological reproducibility of inhaled human insulin pre-meal dosing in patients with type 2 diabetes mellitus (NIDDM) [abstract]. Diabetes. 1998;47(suppl):A99. [Google Scholar]
- Skyler JS, Gelfand RA, Kourides IA. Treatment of type 1 diabetes mellitus with inhaled human insulin: a 3-month, multicenter trial [abstract]. Diabetes. 1998;47(suppl):A61. [Google Scholar]
- Cefalu WT, Gelfand RA, Kourides I. Treatment of type 2 diabetes mellitus with inhaled human insulin: a 3-month, multicenter trial [abstract]. Diabetes. 1998;47(suppl):A61. [Google Scholar]
- Cappelleri JC, Gerber RA, Bell-farrow AD, English JS, Agramonte RF, Kourides IA. Improved patient satisfaction with inhaled insulin in subjects with type 2 diabetes mellitus: results from a multicenter randomized controlled trial [abstract]. Diabetes. 2000;49(suppl):A100. [Google Scholar]
- Gerber RA, Cappelleri JC, Bell-farrow AD, English JS, Agramonte RF, Gelfand RA. Improved patient satisfaction with inhaled insulin in subjects with type 1 diabetes mellitus after one year: results from a multicenter extension trial [abstract]. Diabetes. 2000;49(suppl):A108. [Google Scholar]
- Cefalu WT, Balagtas CC, Landschulz WH, Gelfand RA. Sustained efficacy and pulmonary safety of inhaled insulin during two years of outpatient therapy [abstract]. Diabetes. 2000;49 (suppl):A101. [Google Scholar]
- Farr S, McElduff A, Ward E, Okumu F, Mather L, Gonda I, Rubsamen R. A comparison of the pharmacokinetics and pharmacodynamics of inhaled insulin administered as different strength solutions to healthy volunteers [abstract]. Diabetes. 1998;47(suppl):A61. [Google Scholar]
- Farr S, McElduff A, Ward E, Okikawa J, Lloyd P, Schuster J, Mather L, Gonda I. The mode of inhalation influences the pharmacokinetics and pharmacodynamics of pulmonary delivered insulin in healthy fasted volunteers [abstract]. Pharm Res. 1997;14(suppl):S136. [Google Scholar]
- Kipnes M, Otulana B, Clauson P, Fischer J, Farr S, Hatorp V, Schwartz S. A comparison of the pharmacodynamic effects of inhaled insulin versus subcutaneous insulin in type 1 diabetic patients [abstract]. Diabetes. 1999;48(suppl):A95. [Google Scholar]
- Brunner GA, Balent B, Sendlhofer G, Ellmerer M, Schaupp L, Jendle JH, Kristensen A, Okikawa J, Pieber TR. Pharmacokinetics and pharmcodynamics of inhaled versus subcutaneous insulin in subjects with type 1 diabetes: a glucose clamp study [abstract]. Diabetes. 2000;49(suppl):A76. [Google Scholar]
- Weiss SR, Berger S, Cheng S, Kourides I, Landschulz WH, Gelfand R. Adjunctive therapy with inhaled human insulin in type 2 diabetic patients failing oral agents: a multicenter phase II trial [abstract]. Diabetes. 1999;48(suppl):A12. [Google Scholar]
- Patton JS, Trinchero P, Platz RM. Bioavailability of pulmonary delivered peptides and proteins: α-interferon, calcitonins and parathyroid hormone. J Contr Rel. 1994;28:79–85. [Google Scholar]
- Komada F, Iwakawa S, Yamamoto N, Sakakibara H, Okumura K. Intratracheal delivery of peptide and protein agents: absorption from solution and dry powder by rat lung. J Pharm Sci. 1994;83:863–867. doi: 10.1002/jps.2600830621. [DOI] [PubMed] [Google Scholar]
- Folkesson HG, Hedin L, Westrom BR, Pierzynowski SG, Karlsson BW. Lung to blood passage of human growth hormone after intratracheal instillation: stimulation of growth in hypophysectomized rats. J Endocrinol. 1992;134:197–203. doi: 10.1677/joe.0.1340197. [DOI] [PubMed] [Google Scholar]
- Colthorpe P, Farr SJ, Smith IJ, Wyatt D, Taylor G. The influence of regional deposition on the pharmacokinetics of pulmonary-delivered human growth hormone in rabbits. Pharm Res. 1995;12:356–359. doi: 10.1023/a:1016292232513. [DOI] [PubMed] [Google Scholar]
- Shao Z, Li Y, Mitra AK. Cyclodextrins as mucosal absorption promoters of insulin III: pulmonary route of delivery. Eur J Pharm Biopharm. 1994;40:283–288. [Google Scholar]
- Shen Z, Cheny Y, Zhang Q. Lanthanides enhance pulmonary absorption of insulin. Biol Trace Element Res. 2000;75:215–225. doi: 10.1385/BTER:75:1-3:215. [DOI] [PubMed] [Google Scholar]
- Heinemann L, Klappoth W, Rave K, Hompesch B, Linkeschowa R, Heis T. Intra-individual variability of metabolic effect of inhaled insulin together with an absorption enhancer. Diabetes Care. 2000;23:1343–1347. doi: 10.2337/diacare.23.9.1343. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Kondo S, Juni K. Study of pulmonary delivery of salmon calcitonin in rats: effects of protease inhibitors and absorption enhancers. Pharm Res. 1994;11:1239–1243. doi: 10.1023/a:1018926007902. [DOI] [PubMed] [Google Scholar]
- Morita T, Yamamoto A, Takakura Y, Hashida M, Sekaki H. Improvement of pulmonary absorption of (ASU1,7)-Eel calci-tonin by various protease inhibitors in rats. Pharm Res. 1994;11:909–913. doi: 10.1023/a:1018950429341. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Okumura S, Fukuda Y, Fukui M, Takahashi K, Muran-ishi S. Improvement of the pulmonary absorption of (ASU1,7)-Eel calcitonin by various absorption enhancers and their pulmonary toxicity in rats. J Pharm Sci. 1997;86:1144–1147. doi: 10.1021/js9603764. [DOI] [PubMed] [Google Scholar]
- Edwards DA, Hanes J, Caponetti G, Hrkach J, Ben-Jebria A, Eskew ML, Mintzes J, Deaver D, Lotan N, Langer R. Large porous particles for pulmonary drug delivery. Science. 1997;276:1868–1871. doi: 10.1126/science.276.5320.1868. [DOI] [PubMed] [Google Scholar]
- Kawashima Y, Yamamoto H, Takeuchi H, Fujoka S, Hino T. Pulmonary delivery of insulin with nebulized DL-lactide/glycolide copolymer (PLGA) nanosphere to prolong hypoglycemic effect. J Contr Rel. 1999;62:279–287. doi: 10.1016/s0168-3659(99)00048-6. [DOI] [PubMed] [Google Scholar]
- Liu F, Shao Z, Kildsig DO, Mitra AK. Pulmonary delivery of free and liposomal insulin. Pharm Res. 1993;10:228–232. doi: 10.1023/a:1018934810512. [DOI] [PubMed] [Google Scholar]
- Li Y, Mitra AK. Effect of phospholipid chain length, concentration, charge, and vesicle size on pulmonary insulin absorption. Pharm Res. 1996;13:76–79. doi: 10.1023/a:1016029317299. [DOI] [PubMed] [Google Scholar]
- Smith PL, Yeulet SE, Citerone DR, Drake F, Cook K, Wall DA, Marcello J. SK&F 110679: comparison of absorption following oral or respiratory administration. J Contr Rel. 1994;28:67–77. [Google Scholar]
- Ajei A, Garren J. Pulmonary delivery of peptide drugs: effect of particle size on bioavailability of leuprolide acetate in healthy voluteers. Pharm Res. 1990;7:565–569. doi: 10.1023/a:1015853824722. [DOI] [PubMed] [Google Scholar]
- Adjei A, Johnson E, Doyle R, Sims K. Formulations of a non-apeptide: in vitro and in vivo studies [abstract]. J Pharm Sci. 1987;76(suppl):PS47. [Google Scholar]
- Adjei A, Hui J, Finley R, Lin T, Lancaster J, Fort F. Pulmonary bioavailability of leuprolide acetate following multiple dosing to beagle dogs: some pharmacokinetic and clinical issues. Int J Pharm. 1994;107:57–66. [Google Scholar]
- Bennett DB, Tyson E, Nuremberg CA, Mah S, De Groot JS, Teitelbaum Z. Pulmonary delivery of detirelix by intratracheal instillation and aerosol inhalation in briefly anesthetized dog. Pharm Res. 1994;11:1048–1055. doi: 10.1023/a:1018999707476. [DOI] [PubMed] [Google Scholar]
- Schreier H, McNicol KJ, Bennette DB, Teitelbaum Z, Derendorf H. Pharmacokinetics of detirelix following intratracheal instillation and aerosol inhalation in the unanesthethized awake sheep. Pharm Res. 1994;11:1056–1059. doi: 10.1023/a:1018951824315. [DOI] [PubMed] [Google Scholar]
- Lizio R, Klenner T, Borchard G, Romeis P, Sarlikiotis AW, Reissmann T, Lehr C. Systemic delivery of the GnRH antagonist cetrorelix by intratracheal instillation in anesthetized rat. Eur J Pharm Sci. 2000;9:253–258. doi: 10.1016/s0928-0987(99)00067-6. [DOI] [PubMed] [Google Scholar]
- Folkesson HG, Westrom BR, Karlsson BW. Permeability of the respiratory tract to different sized macromolecules after intratracheal instillation in young and adult rats. Acta Physiol Scand. 1990;139:347–354. doi: 10.1111/j.1748-1716.1990.tb08933.x. [DOI] [PubMed] [Google Scholar]
- Folkesson HG, Westrom BR, Pierzynowski SG, Svendsen J, Karlsson BW. Lung to blood passage of albumin and a nonapeptide after intratracheal instillation in the young developing pig. Acta Physiol Scand. 1993;147:173–178. doi: 10.1111/j.1748-1716.1993.tb09486.x. [DOI] [PubMed] [Google Scholar]
- Bot AI, Tarara TE, Smith DJ, Bot SR, Woods CM, Weers JG. Novel lipid-based hollow-porous microparticles as a platform for immunoglobulin delivery to the respiratory tract. Pharm Res. 2000;17:275–283. doi: 10.1023/a:1007544804864. [DOI] [PubMed] [Google Scholar]
- Hänninen A, Harisson LC. γδ T cells as mediators of mucosal tolerance: the autoimmune diabetes model. Immunol Rev. 2000;173:109–119. doi: 10.1034/j.1600-065x.2000.917303.x. [DOI] [PubMed] [Google Scholar]
- Harrison LC, Dempsey-Collier , Kramer DR, Takahashi K. Aerosol insulin induces regulatory CD8 γδ T cells that prevent murine insulin-dependent diabetes. J Exp Med. 1996;184:2167–2174. doi: 10.1084/jem.184.6.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altrock BW, Fagin KD, Hockman HR, Fish EN, Goldstein L, Chang D, Duker K, Stebbing N. Antiviral and antitumor effects of human interferon analog, IFN-α Con1, assessed in hamsters. J Interferon Res. 1986;6:405–415. doi: 10.1089/jir.1986.6.405. [DOI] [PubMed] [Google Scholar]
- Niven RW, Whitcomb KL, Woodwards M, Liu J, Jornacion C. Systemic absorption and activity of recombinant consensus interferons after intratracheal intillation and aerosol administration. Pharm Res. 1995;12:1889–1895. doi: 10.1023/a:1016279503631. [DOI] [PubMed] [Google Scholar]
- Niven RW, Lott FD, Cribbs JM. Pulmonary absorption of recombinant methionyl human granulocyte colony stimulating factor (r-huG-CSF) after intratracheal instillation to the hamster. Pharm Res. 1993;10:1604–1610. doi: 10.1023/a:1018920619424. [DOI] [PubMed] [Google Scholar]
- Niven RW, Lott FD, Ip AY, Cribbs JM. Pulmonary delivery of powders and solutions containing granulocyte colony-stimulating factor (r-huG-CSF) to the rabbit. Pharm Res. 1994;11:1101–1109. doi: 10.1023/a:1018924512928. [DOI] [PubMed] [Google Scholar]
- Taljanski W, Pierzynowki SG, Lundin PD, Westrom BR, Eirefelt S, Podlesny J, Dahlback M, Siwinska-Golebiowska H, Karlsson BW. Pulmonary delivery of intratracheally instilled and aerosolized cyclosporin A to young and adult rats. Drug Metab Dis. 1997;25:917–920. [PubMed] [Google Scholar]
- Niven RW, Whitcomb KL, Shaner LD, Ralph LD, Habberfield AD, Wilson JV. Pulmonary absorption of polyethylene glycolated recombinant human granulocyte colony stimulating factor (PEGr-huG-CSF). J Contr Rel. 1994;32:177–189. [Google Scholar]
- Wolff RK. Safety of inhaled proteins for therapeutic use. J Aerosol Med. 1998;11:197–219. doi: 10.1089/jam.1998.11.197. [DOI] [PubMed] [Google Scholar]
- Myers MA, Thomas DA, Straub L, Soucy DW, Niven RW, Kaltenback M, Hood CI. Pulmonary effect of chronic exposure to liposome aerosols in mice. Exp Lung Res. 1993;19:1–19. doi: 10.3109/01902149309071077. [DOI] [PubMed] [Google Scholar]
- Dokka S, Toledo D, Shi X, Castranova V, Rojanasakul Y. Oxygen radical-mediated pulmonary toxicity induced by some cationic liposomes. Pharm Res. 2000;17:521–525. doi: 10.1023/a:1007504613351. [DOI] [PubMed] [Google Scholar]