

Abstract
Diffuse axonal injury (DAI) accounts for more than 50% of all traumatic brain injury. In response to the mechanical damage associated with DAI, the abnormal proteins produced in the neurons and axons, namely, β-APP and p-tau, induce endoplasmic reticulum (ER) stress. Curcumin, a major component extracted from the rhizome of Curcuma longa, has shown potent anti-inflammatory, antioxidant, anti-infection, and antitumor activity in previous studies. Moreover, curcumin is an activator of nuclear factor-erythroid 2-related factor 2 (Nrf2) and promotes its nuclear translocation. In this study, we evaluated the therapeutic potential of curcumin for the treatment of DAI and investigated the mechanisms underlying the protective effects of curcumin against neural cell death and axonal injury after DAI. Rats subjected to a model of DAI by head rotational acceleration were treated with vehicle or curcumin to evaluate the effect of curcumin on neuronal and axonal injury. We observed that curcumin (20 mg/kg intraperitoneal) administered 1 h after DAI induction alleviated the aggregation of p-tau and β-APP in neurons, reduced ER-stress-related cell apoptosis, and ameliorated neurological deficits. Further investigation showed that the protective effect of curcumin in DAI was mediated by the PERK/Nrf2 pathway. Curcumin promoted PERK phosphorylation, and then Nrf2 dissociated from Keap1 and was translocated to the nucleus, which activated ATF4, an important bZIP transcription factor that maintains intracellular homeostasis, but inhibited the CHOP, a hallmark of ER stress and ER-associated programmed cell death. In summary, we demonstrate for the first time that curcumin confers protection against abnormal proteins and neuronal apoptosis after DAI, that the process is mediated by strengthening of the unfolded protein response to overcome ER stress, and that the protective effect of curcumin against DAI is dependent on the activation of Nrf2.
Keywords: curcumin, diffuse axonal injury, endoplasmic reticulum stress, nuclear factor-erythroid 2-related factor 2, unfolded protein response
Introduction
Diffuse axonal injury (DAI), a type of traumatic brain injury (TBI), is a significant public health problem that is increasingly being recognized as an important cause of long-term disability and mortality. DAI was clinicopathologically characterized by immediate and prolonged unconsciousness after the mechanical impact to the head 1–3. Axonopathy after DAI has been investigated quite intensively with interesting results, and axonal retraction balls and axonal varicosities have been identified as characteristic changes that mark progressive axonopathy after DAI 4,5. Multiple recent studies have shown that axonal degeneration is connected to increased axolemmal permeability, disruption of axoplasmic transport, mitochondrial swelling, and cytoskeletal (microtubule and neurofilament structures) compaction damage 6,7. Moreover, neuronal cell apoptosis is another important mechanism in the loss of nerve function following DAI and is induced by endoplasmic reticulum (ER) stress, inflammatory mediators, oxygen free radicals, excitatory neurotransmitters, or Ca2+ overload 8,9. Therefore, studies on the effects of drugs on axonal injury or neuronal cell apoptosis after DAI can provide insights into potential treatments for DAI in a clinical setting.
In recent years, ER stress has attracted increasing attention in relation to the pathogenesis of various neurodegenerative disorders, such as TBI, cerebral ischemia, Alzheimer’s disease, and Parkinson’s disease 10,11. The unfolded protein response (UPR) is an evolutionarily conserved process triggered by ER stress, when ER sensors detect excessive accumulation of misfolded or unfolded proteins and/or disruption of Ca2+ homeostasis in the ER lumen 12–14. It is interesting that components of the UPR play an essential role in modulating cellular metabolism and cognitive function. Previous studies indicated that β amyloid precursor protein (β-APP) could be transported in the normal axon by fast axoplasmic transport, but accumulated rapidly in areas of disrupted transport during the formation of axonal bulbs after DAI 15. Moreover, hyperphosphorylated tau (p-tau) complexes could accumulate in the tissue as neurofibrillary tangles and subsequently trigger apoptotic cell death in the aftermath of DAI 16. It is unknown, however, whether the ER stress process participates in axonal injury and/or neuronal apoptosis by β-APP and p-tau accumulation following DAI.
Nuclear factor-erythroid 2-related factor 2 (Nrf2), a nuclear transcription factor, has been shown in recent years to play a critical role in cellular protection after TBI 17,18. In response to oxidative stress, Nrf2 translocates into the nucleus, binds to antioxidant response elements in the nucleus, and regulates the transcription of downstream target genes, including both nonenzymatic and enzymatic antioxidants 19. Curcumin, a major component extracted from the rhizome of Curcuma longa, has shown potent anti-inflammatory, antioxidant, and antitumor activity in previous studies 20,21. It has been shown that curcumin can pass through the blood–brain barrier and maintain high biological activity 22. Recently, curcumin has been shown to upregulate the nuclear translocation of Nrf2 in a cerebral I/R model and a Parkinson’s disease model by counteracting oxidative stress and brain edema 23,24. However, it remains unknown whether exogenous curcumin could modulate the ER stress process by the Nrf2 signaling pathway.
In this study, we investigated the influence of curcumin on axonal injury and neuronal cell apoptosis after DAI and the possible involvement of the PERK/Nrf2-mediated mechanism.
Materials and methods
Animal care and grouping
All experiments were conducted on healthy male Sprague-Dawley rats (weighing 250–300 g, 8–10 weeks of age), which were obtained from the Experimental Animal Center of Xi’an Jiaotong University [license number SCXK (Shaanxi) 2007-001]. The animals were housed and fed in groups of five per cage in a room kept at ∼22°C with a 12/12-h-light/dark cycle. Food and water were available ad libitum. All experiments were conducted in accord with the Guidelines for the Care and Use of Laboratory Animals from the National Institutes of Health (NIH publication #80-23) and were approved by the Biomedical Ethics Committee of the Medical College of Xi’an Jiaotong University of China. All injuries were inflicted under sodium pentobarbital anesthesia and all efforts were made to minimize animal suffering.
The rats were subdivided randomly into four groups (20 animals/group): (i) sham-operated rats+vehicle (control) group; (ii) sham-operated rats+curcumin (control+CUR) group; (iii) DAI rats+vehicle (DAI) group; and (iv) DAI rats+curcumin (DAI+CUR) group (Table 1). Curcumin (purity 94%) was purchased from Sigma-Aldrich (C7727; St Louis, Missouri, USA). One hour after DAI induction, 0.25–0.3 ml curcumin (20 mg/kg)/dimethyl sulfoxide solution was administered intraperitoneally to the curcumin group, as described in a previous report 25. Equal volumes of 10% (w/v) dimethyl sulfoxide solution were administered to the other vehicle groups.
Table 1.
Groups and purposes in our study
Model of DAI in vivo
A DAI model was established using a lateral head rotation device as described previously 5,26,27, which was modified from the method of Xiao-Sheng and colleagues 28,29. After weighing, all rats in the DAI groups were anesthetized by an intraperitoneal injection of 1% (w/v) pentobarbital sodium (35 mg/kg) and placed in a prone position. Under anesthesia, the head of each rat was fixed in the rat instant head rotating injury device; the head was secured horizontally to the lateral device by two lateral ear bars, a head clip and an anterior teeth hole, with the body of the rat 30° oblique to the top of the laboratory table. For the injury group, after the trigger was pushed, the device rapidly rotated the rat head through a 90° angle laterally in the coronal plane. The rats were placed in separate cages at a room temperature of ∼22°C and an indoor relative humidity of 40–70%. Primary coma was observed in all injured rats. Rats that died of their injuries were excluded and later replaced with new rats, and about 5–10% death rate among rats in the model was observed in our study, and the main cause of death was the massive hemorrhage in cerebral parenchyma, subarachnoid space, or ventricle observed by dissecting the brain. The control rats were administered anesthesia and were fixed to the device, but were not subjected to injury.
Neurological assessment
A modified Neurologic Severity Score (mNSS) 30 was used to evaluate the neurological deficits of rats before they were killed in each group, including the time of preinjury in each group and day 2 postinjury in the DAI 3-day subgroups. The mNSS consists of motor, sensory, reflex, and balance tests, and is used to grade the neurological function on a scale of 0–18. A higher score indicates a more severe injury 31. Two observers, blinded to the treatment and grouping, were assigned to assess the mNSS of each rat.
Perfusion fixation, tissue embedding, and sectioning
Euthanasia was performed at 6 h, day 1, and day 3 after DAI in the DAI groups, and rats in the sham-operated group were euthanized at the same time points. The anesthetized rats for western blotting in each group were killed and perfused with 250 ml of normal saline only. The cerebral cortices were collected in sterile tubes and stored in liquid nitrogen. Furthermore, anesthetized rats used for immunohistochemical staining were killed and perfused with 250 ml of normal saline, followed by 400 ml of 4% (w/v) paraformaldehyde in 0.01 M PBS. The whole brains were removed and postfixed in 4% (w/v) paraformaldehyde solution, dehydrated by a graded ethanol series, vitrified with dimethyl benzene, embedded with paraffin, and sectioned into 10-μm thick sections using a microtome and mounted on poly-l-lysine-coated slides (P4981; Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA).
Hematoxylin and eosin staining
Brain sections were stained with hematoxylin and eosin (H&E), followed by dehydration, hyalinization, and fixation, and observed under a high-magnification optical microscope (×400; Olympus, Tokyo, Japan). Five random regions of interest (ROIs) were selected for quantification.
Nissl staining
Brain sections were stained with Nissl solution (Boster Biotech, Wuhan, China) for 10 min. Compared with the cell bodies of normal neurons, those of injured neurons were shrunken and/or contained vacuoles, and the nuclei were stained darker. Sections were observed under a high-magnification optical microscope (×400; Olympus). Five ROIs were selected for quantification.
Congo red staining
Brain sections were rehydrated in a graded ethanol series, immersed in H&E for 2 min, and then dipped in hydrochloric alcohol for 10 s. First, sections were rinsed in running tap water until it turned blue, followed by double-distilled water two times. Second, sections were amyloid stained with Congo red solution (HT60-1KT; Sigma-Aldrich) for 40 min at room temperature before being rinsed in running tap water. Then, sections were dipped in lithium carbonate for 5 s and washed in running tap water. Alcohol (80%, v/v) was used to differentiate the nonspecific background staining, and sections were then rinsed in running tap water for 10 min, dipped in xylene for 20 min, and covered with neutral balsam. After drying, sections were observed under a high-magnification optical microscope (×400; Olympus). Five ROIs were selected for quantification.
Bielschowsky silver staining
Brain sections were rehydrated in a graded ethanol series before being rinsed in double-distilled water three times. First, sections were immersed in a 4% (w/v) silver nitrate solution for 30–60 min at 37°C in the dark, followed by rinsing in double-distilled water three times to remove the silver nitrate solution. Second, sections were deoxidized with 10% (v/v) formaldehyde until they changed to a faint yellow color. Sections were rinsed in double-distilled water three times, and excess water was removed. Ammoniacal silver solution was added to each section for 10 min, and then excess solution was removed. Then, sections were dipped in 4% (w/v) formalin for 10 min and rotated several times until the yellow coloration stabilized. Subsequently, the sections were fixed in 5% (w/v) sodium thiosulfate for 5 min. Finally, the sections were dried in the open air, cleared in xylene, and covered with neutral balsam.
Immunohistochemical staining and semiquantitative analysis
Brain sections were deparaffinized in xylene and hydrated in a decreasing gradient of alcohol. Endogenous peroxidase activity was blocked with 3% H2O2 (v/v) for 20 min, followed by a brief rinse in double-distilled water and a 15-min wash in PBS. The sections were placed in 0.01 M citrate buffer and heated in a microwave oven at 95°C for 20 min. Subsequently, sections were naturally cooled at room temperature for 20 min and rinsed in PBS for 15 min. Nonspecific protein binding was blocked by 60–120 min of incubation in normal goat serum at room temperature, followed by incubation with the following primary antibodies: mouse anti-NF-H (2836, diluted 1 : 400; Cell Signaling Technology, Boston, Massachusetts, USA), rabbit anti-p-tau (EPR2605, phospho-S404, diluted 1 : 100; Abcam, Cambridge, UK), and anticleaved caspase-3 (AB2302, diluted 1 : 100; Abcam) for 16–24 h at 4°C and a 15-min wash in PBS. Sections were then incubated with goat anti-rabbit or anti-mouse IgG-biotin (both diluted 1 : 200) for 60–120 min at 37°C, after which they were washed with PBS for 15 min. Diaminobenzidine (DAB) was used as the chromogen and H&E was used as the counterstain. Sections incubated with PBS in the absence of primary antibodies were used as negative controls. Microscopic observations of the immunohistochemically stained sections were performed by an experienced pathologist blinded to the experimental conditions.
Immunofluorescence staining
Rats were perfused transcardially with saline under deep anesthesia (100 mg/kg sodium pentobarbital) and the brains were sectioned at a thickness of 20 μm using a cryostat. Sections were blocked in 5% (w/v) normal bull serum albumin diluted in PBS for 1 h at room temperature and then incubated overnight at 4°C with rabbit anti-p-PERK (SC-32577, diluted 1 : 100; Santa Cruz Biotechnology Inc., Europe), mouse anti-Nrf2 (AB89443, diluted 1 : 100; Abcam), rabbit anti-CHOP (AB179823, diluted 1 : 500; Abcam), goat anti-ATF4 (AB1371, diluted 1 : 100; Abcam), rabbit anti-β-APP (Y188, diluted 1 : 100; Abcam), and mouse anti-NF-H (2836, diluted 1 : 400; Cell Signaling Technology) as the primary antibodies. After being washed, brain sections were incubated with goat anti-rabbit and anti-mouse secondary antibodies or donkey anti-rabbit and anti-goat labeled with Alexa Fluor 488 and Alexa Fluor 647 (diluted 1 : 300; Abcam) for 1–2 h at room temperature, and the nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI, AB104139; Abcam) for 10 min. Sections were viewed by confocal microscopy (LSM780, Zeiss, Jena, Germany) and analyzed as individual images for p-PERK/Nrf2/DAPI coexpression, CHOP/ATF4/DAPI coexpression, and β-APP/DAPI coexpression. Immunostained sections were characterized quantitatively by digital image analysis using Image-Pro Plus 6.0 software (Media Cybernetics, Silver Spring, Maryland, USA). P-PERK, Nrf2, ATF4, CHOP, and β-APP were quantified as the average number of positive cells per field. A negative control (no antibody) was included.
Preparation of cytosolic and nuclear fractions
Brain cell nuclear and cytosolic fractions were prepared using the method described by Giufrida et al. 32. Briefly, whole brains were minced and then homogenized in a brain lysis buffer (pH 6.4) containing 0.32 M sucrose, 3.0 mM MgCl2, 1.0 mM KH2PO4, 8.2 mM Brij-35, 50 mM β-mercaptoethanol, and 0.5 mM PMSF using a Dounce tissue grinder. The homogenate was then centrifuged at 11 000g for 20 min and the supernatant was retained as the brain cell cytosolic fraction. To prepare nuclei, we washed the pellet in brain lysis buffer and resuspended it in extraction buffer containing 20 mM HEPES, 420 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, and 25% (v/v) glycerol (pH 7.9). The pellet suspension was shaken gently for 20 min (4°C) and then centrifuged at 11 000g for 20 min. The supernatant was desalted using Amicon Ultra-4 centrifuge filter units (Millipore, Billerica, Massachusetts, USA). The retained filtrate was resuspended in RIPA buffer and was designated the brain nuclear fraction.
Western blot analysis
The cerebral cortices were homogenized on ice in RIPA buffer supplemented with 2.0 mM sodium orthovanadate, 150 mM NaF, and protease inhibitors. The homogenates were centrifuged at 12 000g, 4°C, for 20 min. The protein concentration in the supernatant was measured using the Bradford method. Denatured protein samples (30 μg) were loaded into an SDS-PAGE gel and transferred to nitrocellulose membranes. The membranes were blocked with 5% (w/v) skim milk for 1 h at room temperature, and then incubated with the following primary antibodies: rabbit anti-p-PERK (SC-32577, diluted 1 : 1000; Santa Cruz Biotechnology Inc.), mouse anti-Nrf2 (AB89443, diluted 1 : 1000; Abcam), rabbit anti-p-eIF2α (SC-11386, diluted 1 : 1000; Cell Signaling Technology), rabbit anti-CHOP (AB179823, diluted 1 : 1000; Abcam), goat anti-ATF4 (AB1371, diluted 1 : 1000; Abcam), rabbit anti-GSK-3β (12456, diluted 1 : 400; Cell Signaling Technology), rabbit anti-p-GSK-3β (5558, Ser9; diluted 1 : 400; Cell Signaling Technology), mouse anti-Lamin-B1 (AB8982, diluted 1 : 1000; Abcam), and mouse anti-β-actin (3700, diluted 1 : 1000; Cell Signaling Technology, Danvers, Massachusetts, USA) overnight at 4°C, followed by horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology, Boston, Massachusetts, USA) for 1–2 h at room temperature. The membranes were visualized using a ChemiDoc MP System (Bio-Rad protein assay; Bio-Rad, Segrate, Italy) with ECL substrate (Millipore, Billerica, Massachusetts, USA). The optical density of the bands was quantified using Image-Pro Plus 6.0 software (Media Cybernetics) and was normalized to Lamin-B1 or β-actin.
TUNEL staining
Brain sections were also examined using the DeadEnd colorimetric TUNEL system detection kit (Promega, Madison, Wisconsin, USA). First, sections were deparaffinized with xylene, rehydrated in a graded ethanol series, and rinsed for 15 min in 0.1 M PBS. Second, sections were treated with 20 μg/ml of proteinase K for 20 min at room temperature. Then, sections were treated with 3% (v/v) H2O2 in methanol for 20 min to inactivate endogenous peroxidase. After washing with PBS for 15 min, sections were incubated in the labeling reaction mixture containing terminal deoxynucleotidyl transferase and the deoxynucleotides at 4°C overnight. After incubation, all the sections were rinsed in PBS for 15 min and incubated with horseradish peroxidase (1 : 500) for 30 min at room temperature. Then, sections were washed extensively with PBS for 15 min and treated with DAB solution (30 mg of DAB and 200 μl of H2O2/100 ml of PBS) for 10 min at room temperature in the dark. After washing under running water, all the sections were counterstained with H&E for 30 s. Finally, sections were dehydrated in an increasing graded ethanol series, cleared in xylene, and mounted with a cover slip. By TUNEL staining, the apoptotic nuclei were identified by the presence of dark brown staining.
Transmission electron microscopy
Anesthetized rats were perfused with 0.9% (w/v) saline, followed by fixative solution [4% (w/v) paraformaldehyde and 1% (v/v) glutaraldehyde]. The rat brainstem tissue was removed and cut into 1 mm3 pieces in volume, which were immediately fixed in 2.5% (v/v) glutaraldehyde for 2 h at 4°C, postfixed in 2% (w/v) osmic acid for 2 h, dehydrated in a graded series of alcohol and propylene oxide, and embedded in araldite for morphometric analyses of the semithin sections. Ultrathin sections mounted on copper grids were stained with uranyl acetate and lead citrate, and then examined and photographed under a transmission electron microscope (H-7650; Hitachi Ltd, Tokyo, Japan) at an accelerating voltage of 80 kV. The magnifications of photographs ranged from ×30 000 to ×50 000.
Statistical analysis
All data are presented as the mean±SD. SPSS 18.0 (SPSS Inc., Chicago, Illinois, USA) was used for statistical analysis of the data. One-way analysis of variance was used in combination with least significant difference post-hoc tests in order to determine statistical significance for comparisons of more than two groups. For all analyses, a P value less than 0.05 was considered significant.
Results
Curcumin ameliorated axonal injury and neuronal degeneration after DAI
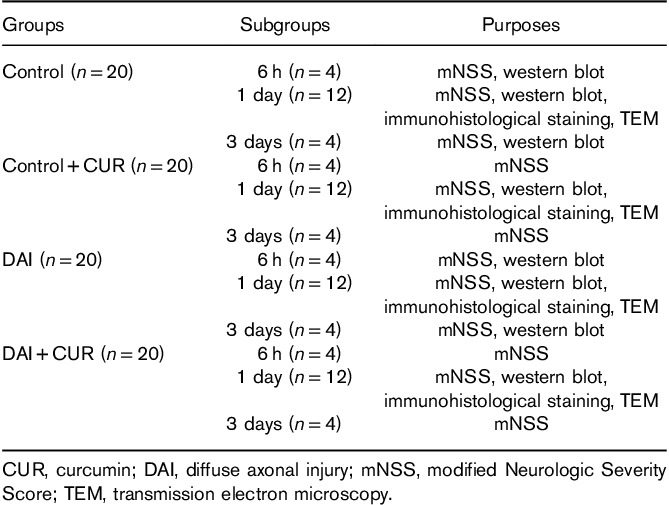
Previous studies indicated that neuronal degeneration and axonal injury after DAI highly affected the degree of consciousness, which is consistent with the severity of the disorder in clinic 33. In our study, the DAI model was produced successfully by the rat instant head rotational injury device. A series of neurological symptoms were discovered in the rats following DAI, including disturbance of consciousness, reduction of physical activity, instability in walking, weakened balance, loss of weight, and even paralysis of limbs. To investigate whether treatment with curcumin could improve neural functional recovery after DAI, we examined the changes in mNSS. No obvious neurological deficits were observed at preinjury in each group. The rats of the control and the control+CUR groups experienced a transient neurological deficit as indicated by the mNSS at 6-h postsham treatment, but returned to a normal score after day 1. This temporal and slight increase in mNSS was solely because of anesthetic. Conversely, the scores in the injury groups peaked at 6 h after DAI and then decreased gradually over time (P<0.05, Fig. 1a). Moreover, a significant improvement from day 1 to day 3 after DAI was observed in the curcumin group compared with the vehicle group (P<0.05, Fig. 1a). These results indicated that curcumin improved neurological recovery after DAI.
Fig. 1.
Neurological dysfunction and pathological changes after DAI. (a) Neurological functions of rats in each group were evaluated by mNSS at preinjury and 6 h, day 1, day 2, and day 3 after DAI. Values are presented as the mean±SD (*P<0.05, compared with the control group; #P<0.05, compared with the DAI+vehicle group). (b) Pathological changes after DAI were confirmed by H&E, Nissl, Congo red, and Bielschowsky silver staining. Scale bar=100 μm. (c) The bar graphs show the results for degenerated, apoptotic, and amyloidotic neurons and axonal injury at day 1 after DAI by H&E, Nissl, Congo red, and Bielschowsky silver staining. Values are presented as the mean±SD (n=4; *P<0.05, compared with the control group; #P<0.05, compared with the DAI+vehicle group). CUR, curcumin; DAI, diffuse axonal injury; H&E, hematoxylin and eosin; mNSS, modified Neurologic Severity Score.
To quantify neurological deficits after DAI, we used H&E, Nissl, Congo red, and Bielschowsky silver staining to visualize neuronal degeneration and axonal injury. H&E-stained sections were used to evaluate neuronal degeneration after DAI. Many rounded eosinophilic corpuscles were observed around injured regions in the cortex after DAI, featuring shriveled cell bodies, hyperchromatic nuclei, absence of nucleoli, and aggregation of chromatin in the injured neurons. The results showed that the DAI group had significantly higher numbers of degenerated neurons in the cortex at day 1 after DAI than the control group (P<0.05, Fig. 1b and c). However, compared with the DAI group, the DAI+CUR group had significantly lower numbers of degenerated neurons (P<0.05, Fig. 1b and c). Nissl staining was used to evaluate neuronal apoptosis in lesioned cortices after DAI. The results showed that the DAI group had significantly higher numbers of apoptotic cells in the cortex than the control group at day 1 after DAI (P<0.05, Fig. 1b and c). However, compared with the DAI group, the DAI+CUR group had significantly lower percentages of apoptotic cells (P<0.05, Fig. 1b and c). Congo red was used to evaluate the amyloid plaques in the brains of rats and the positively stained areas were measured using an image analysis system to determine the quantity of amyloid plaques. The results showed that the DAI group had significantly higher numbers of neurons with amyloidosis in the cortex at day 1 after DAI compared with the control group (P<0.05, Fig. 1b and c). However, compared with the DAI group, the DAI+CUR group had significantly lower percentages of neurons with amyloidosis (P<0.05, Fig. 1b and c). We also used Bielschowsky silver staining to show the pathological changes in axons after DAI. We found that the corpus callosum was thinning and loose at day 1 after DAI. The nerve fibers showed a disorderly arrangement in the cerebral cortex of rats after DAI, but they were arranged neatly in the control group. Diffuse axonal swellings, characteristic of axon retraction balls, were also noted in Bielschowsky silver-stained sections after DAI. Moreover, compared with the vehicle group, the DAI+CUR group showed significantly reduced neuronal degeneration, amyloidosis, and axonal injury at day 1 after DAI (Fig. 1b). These results indicated that curcumin ameliorated axonal injury and neuronal degeneration after DAI.
Time-dependent protein expression of the Nrf2 and UPR pathways after DAI
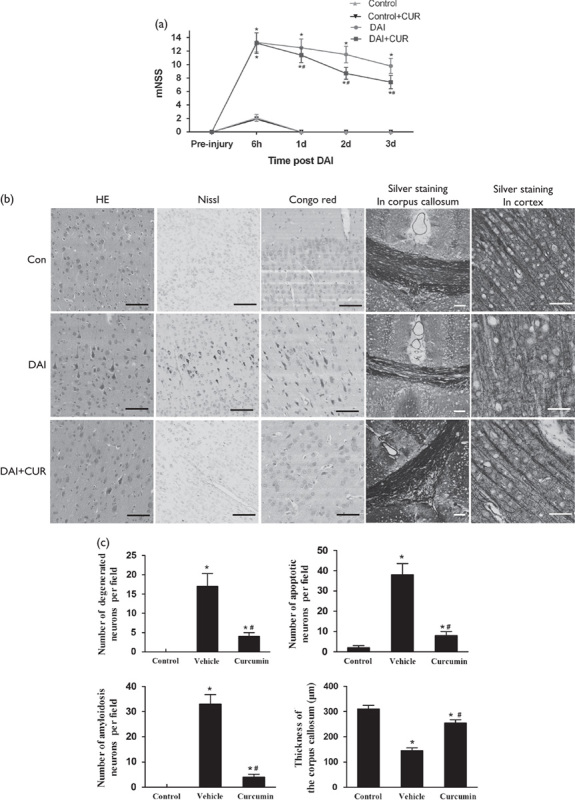
Western blotting was performed to examine the expression of proteins related to the Nrf2 and UPR signaling pathways, including p-PERK, p-eIF2α, ATF4, and CHOP, at different time points (Fig. 2a). The results showed that the expression of Nrf2 was significantly increased in the first 72 h after DAI compared with the control group and reached a peak level at day 1 after DAI. We also found that the proteins related to the UPR pathway were significantly increased in the first 72 h after DAI compared with the control group, except for the expression of CHOP at 6 h after DAI. The expressions of p-PERK, p-eIF2α, and CHOP all reached the peak level at day 3 after DAI, but the expression of ATF4 reached a peak at day 1 and then after day 3 declined toward levels obtained at 6 h after DAI (Fig. 2b). The above results indicated that Nrf2 was upregulated in the initial phase of DAI and may be involved in the pathological changes following DAI by regulating the UPR process.
Fig. 2.
Dynamic expression of proteins related to the Nrf2 and UPR signaling pathway after DAI. (a) Western blotting analysis was carried out to measure the dynamic expression of Nrf2, p-PERK, p-eIF2α, ATF4, and CHOP at 6 h, day 1, and day 3 after DAI. The expression of β-actin was used as an internal control. (b) The bar graphs show the cortical expression results for proteins related to the Nrf2 and UPR signaling pathway at 6 h, day 1, and day 3 after DAI. Values are presented as the mean±SD (n=4; *P<0.05, compared with the control group). DAI, diffuse axonal injury.
Effects of curcumin on nuclear translocation of Nrf2
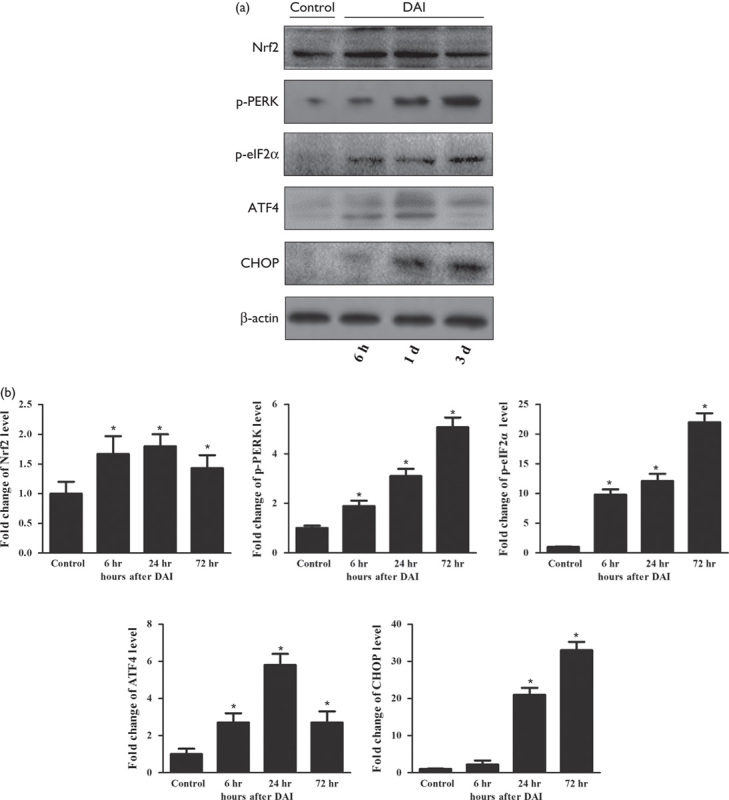
To investigate the effect of curcumin on the activation of Nrf2, we measured Nrf2 protein levels in the cytosol and nucleus by western blotting (Fig. 3a). The expression levels of Nrf2 in the cytosol and nucleus were increased in the DAI groups compared with the control group (P<0.05, Fig. 3b). Moreover, the expression of Nrf2 in the cytosol was decreased in the DAI+CUR group compared with the DAI+vehicle group at day 1 after DAI (P<0.05, Fig. 3b). Meanwhile, the expression of Nrf2 in the nucleus was significantly increased in the DAI+CUR group compared with the DAI+vehicle group at day 1 after DAI (P<0.05, Fig. 3b). These results indicated that curcumin activated Nrf2 and promoted its nuclear translocation.
Fig. 3.
Curcumin promoted the nuclear translocation of Nrf2. (a) The expression levels of Nrf2 in the nucleus and cytosol were measured by western blotting in each group. The expression of Lamin-B1 (nucleus) or β-actin (cytosol) was used as an internal control. (b) The bar graphs show the results for Nrf2 expression as assessed by western blotting in each group. Values are presented as the mean±SD (n=4; *P<0.05, compared with the control group; #P<0.05, compared with the DAI+vehicle group). CUR, curcumin; DAI, diffuse axonal injury.
Curcumin strengthened the ability of the UPR process to overcome ER stress following DAI
To investigate the effect of curcumin on the UPR process, we used western blotting to detect the expression levels of p-PERK, ATF4, and CHOP in the cerebral cortex in each group (Fig. 4a). The p-PERK protein level was significantly increased at day 1 after DAI (P<0.05, Fig. 4b). Moreover, compared with the DAI+vehicle group, the expression of p-PERK was increased in the DAI+CUR group at day 1 after DAI (P<0.05, Fig. 4b). The ATF4 protein level was significantly increased at day 1 after DAI (P<0.05, Fig. 4b). Moreover, compared with the DAI+vehicle group, the expression of ATF4 was increased in the DAI+CUR group at day 1 after DAI (P<0.05, Fig. 4b). The CHOP protein level was significantly increased at day 1 after DAI (P<0.05, Fig. 4b). Moreover, compared with the DAI+vehicle group, the DAI+CUR group had a decreased level of CHOP expression at day 1 after DAI (P<0.05, Fig. 4b). The expression of p-PERK, ATF4, and CHOP showed no difference between the control group and the control+CUR group (P>0.05, Fig. 4b). These data suggested that curcumin strengthened the UPR process by upregulating the coexpression of p-PERK and ATF4, and alleviated ERS-related apoptosis by suppressing the CHOP pathway.
Fig. 4.
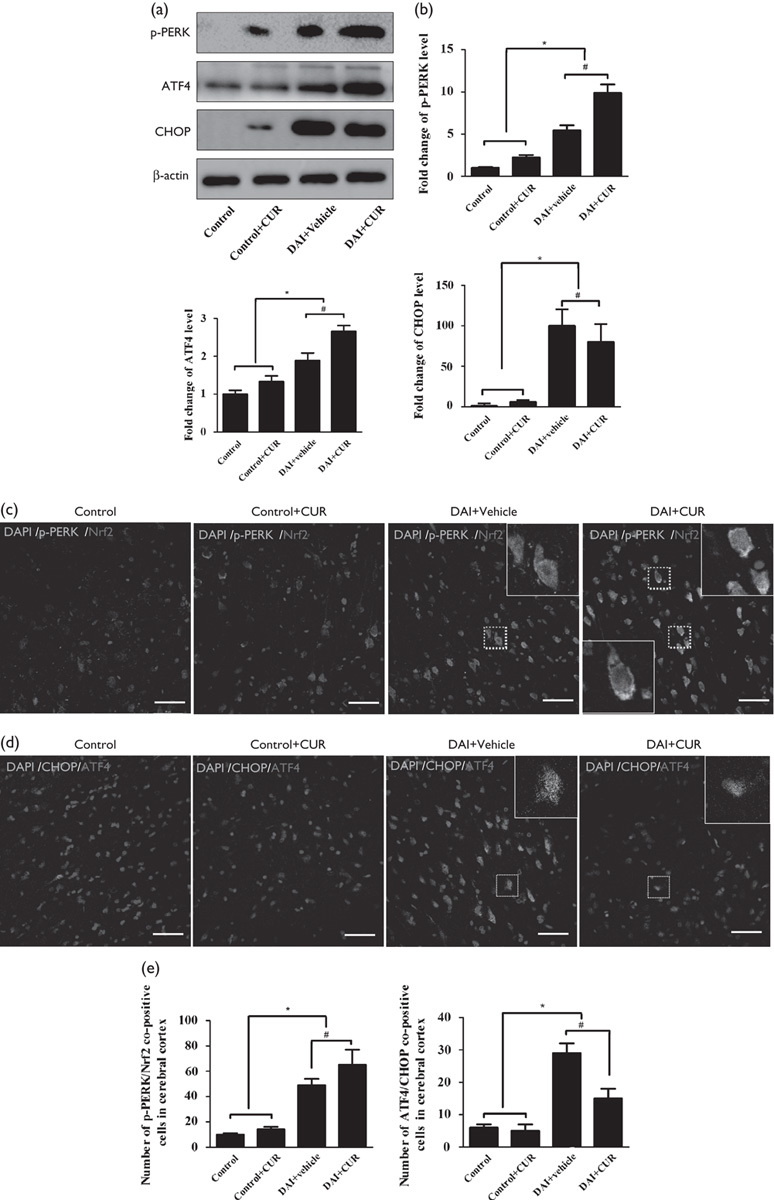
Curcumin strengthened the UPR process to overcome ER stress after DAI by the nuclear translocation of Nrf2. (a) The expression levels of p-PERK, ATF4, and CHOP were measured by western blotting in each group. The expression of β-actin was used as an internal control. (b) The bar graphs show the results for p-PERK, ATF4, and CHOP expression, as assessed by western blotting in each group. Values are presented as the mean±SD (n=4; *P<0.05, compared with the control group; #P<0.05, compared with the DAI+vehicle group). (c) Double immunofluorescence staining was performed with an antibody against Nrf2 and another against p-PERK in each group. Scale bar=100 μm. (d) Double immunofluorescence staining was performed with an antibody against CHOP and another against ATF4 in each group. Scale bar=100 μm. (e) The bar graphs show the results for the numbers of p-PERK/Nrf2 copositive cells in each group. Values are presented as the mean±SD (n=4; *P<0.05, compared with the control group; #P<0.05, compared with the DAI+vehicle group). The bar graphs show the results for the numbers of CHOP/ATF4 copositive cells in each group. Values are presented as the mean±SD (n=4; *P<0.05, compared with the control group; #P<0.05, compared with the DAI+vehicle group). CUR, curcumin; DAI, diffuse axonal injury.
To investigate the role of curcumin in the UPR-Nrf2 pathway, we also used immunofluorescence analysis to detect cells copositive for PERK phosphorylation and Nrf2 in the cerebral cortex in each group. The abundance of cells copositive for p-PERK and Nrf2 was increased at day 1 after DAI (P<0.05, Fig. 4c and e). Moreover, the number of p-PERK and Nrf2 copositive cells was significantly increased in the DAI+CUR group compared with the DAI+vehicle group at day 1 after DAI (P<0.05, Fig. 4c and e). Interestingly, the nuclear translocation of Nrf2 by curcumin was also observed in the neurons surrounding the injured areas (in the white dotted frame, Fig. 4c). Further, to investigate the role of curcumin in ATF4/CHOP, the downstream targets of ERS-related apoptosis, we used immunofluorescence analysis to detect cells copositive for ATF4 and CHOP in the cerebral cortex in each group (Fig. 4d). The number of cells copositive for ATF4 and CHOP was also increased at day 1 after DAI (P<0.05, Fig. 4d and e), but the number of ATF4 and CHOP copositive cells was significantly decreased in the DAI+CUR group compared with the DAI+vehicle group at day 1 after DAI (P<0.05, Fig. 4d and e). These results indicated that curcumin promoted PERK phosphorylation following DAI and then promoted the nuclear translocation of Nrf2 by combination with Nrf2. The translocation of Nrf2 into the nucleus inhibited the increased expression of CHOP, which was induced by ATF4, and thereby alleviated the apoptosis related to ER stress.
Effects of curcumin on apoptosis after DAI
Few TUNEL-positive cells were observed in the control group, whereas DAI induced a marked increase in neuronal apoptosis relative to the control group (P<0.05, Fig. 5a and b). However, the administration of curcumin significantly reduced the number of TUNEL-positive cells compared with the DAI+vehicle group (P<0.05, Fig. 5a and b). Consistent with the results of TUNEL staining, the expression of cleaved caspase-3 was significantly higher in neurons after DAI than in neurons from the control group (P<0.05, Fig. 5a and b). Curcumin treatment significantly decreased the expression of cleaved caspase-3 compared with the DAI+vehicle group (P<0.05, Fig. 5a and b).
Fig. 5.
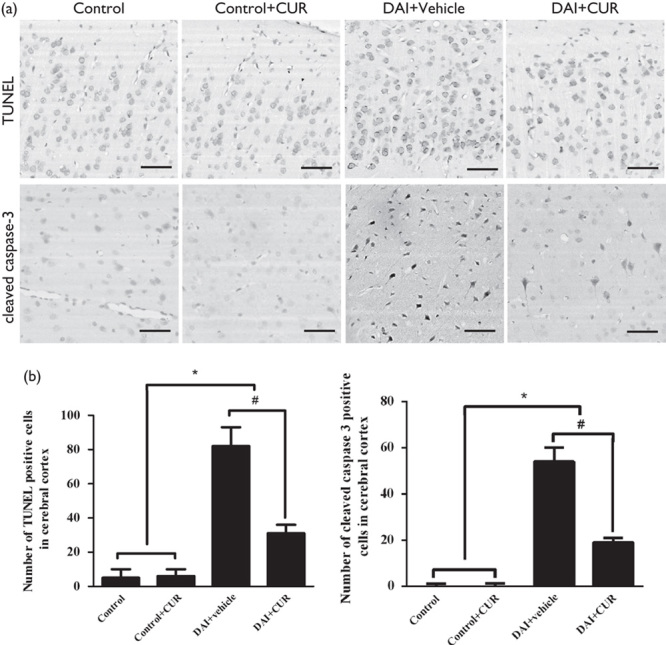
Curcumin significantly attenuated apoptosis after DAI. (a) TUNEL assay was used to detect apoptotic cells in the cortex of rats in each group. Immunohistochemistry of cleaved caspase-3 was performed to assess apoptosis in each group. Scale bar=100 μm. (b) The bar graphs show the results for the numbers of TUNEL and cleaved caspase-3-positive cells in each group. Values are presented as the mean±SD (n=4; *P<0.05, compared with the control group; #P<0.05, compared with the DAI+vehicle group). CUR, curcumin; DAI, diffuse axonal injury.
Curcumin reduced the accumulation of p-tau, β-APP, and NF-H after DAI
Glycogen synthase kinase-3β (GSK-3β) is the principal kinase that mediates tau phosphorylation and promotes NFT formation. GSK-3β activity is inhibited by phosphorylation at Ser9. Therefore, the expression levels of GSK-3β and p-GSK-3β were measured by immunoblotting. Compared with the control group, the p-GSK-3β/GSK-3β ratio was significantly decreased after DAI (P<0.05, Fig. 6a), but increased after the intervention with curcumin (P<0.05, Fig. 6a).
Fig. 6.
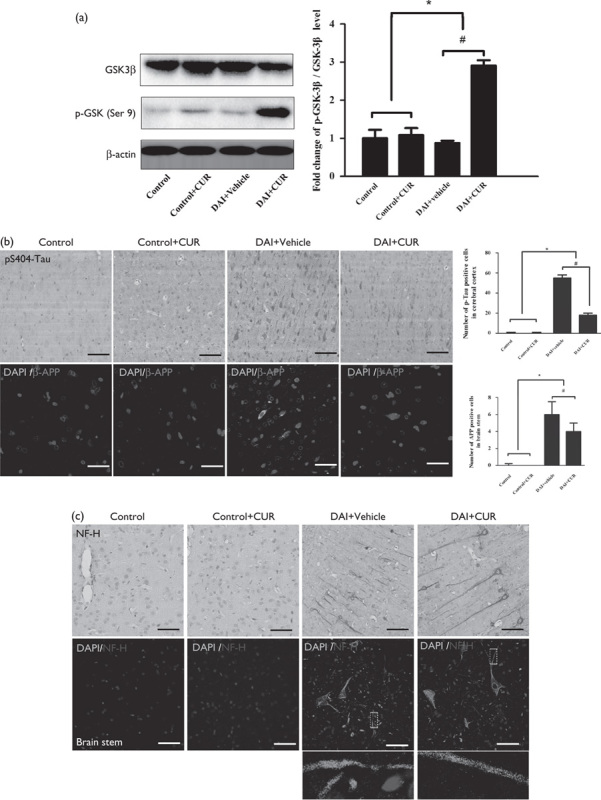
Curcumin reduced the accumulation of p-tau, β-APP, and NF-H after DAI. (a) The expression levels of GSK-3β and p-GSK-3β (Ser9) in cortical samples were detected by western blotting in each group. The expression of β-actin was used as an internal control. The bar graphs show the results for the ratio of p-GSK-3β (Ser9) to GSK-3β in each group. Values are presented as the mean±SD (n=4; *P<0.05, compared with the control group; #P<0.05, compared with the DAI+vehicle group). (b) Immunohistochemistry of p-tau (S404) and β-APP was performed to assess the aggregation of abnormal proteins in axons in each group. Scale bar=100 μm. The bar graphs show the numbers of p-tau-positive and β-APP-positive cells in each group. Values are presented as the mean±SD (n=4; *P<0.05 compared with the control group; #P<0.05 compared with the DAI+vehicle group). (c) Immunohistochemistry of NF-H was performed to assess axonal degeneration in the cerebral cortex and the brainstem in the each group. Scale bar=100 μm. CUR, curcumin; DAI, diffuse axonal injury.
Few p-tau-positive cells were observed in the control group, whereas the number of p-tau-positive neurons was increased at day 1 after DAI (P<0.05, Fig. 6b). However, after intervention with curcumin, the number of p-tau-positive neurons was significantly decreased compared with the DAI+vehicle group (P<0.05, Fig. 6b). Few β-APP-positive cells were observed in the control group, whereas the number of β-APP-positive neurons was increased at day 1 after DAI (P<0.05, Fig. 6b). However, after intervention with curcumin, the number of β-APP-positive neurons was significantly decreased compared with the DAI+vehicle group (P<0.05, Fig. 6b). We further investigated the axonal damage in the cerebral cortex and brainstem at day 1 after DAI by immunocytochemical staining and immunofluorescence analysis for NF-H. The axons in the control group showed no NF-H immunolabeling, but abundant expression of NF-H was detected in the cerebral cortex and the brainstem at day 1 after DAI. More importantly, the injured axons with NF-H accumulation showed multiple prominent varicosities and disconnections, which are characteristics of diffuse axonal injury 34,35. In contrast, in the DAI+CUR group, axons in the cerebral cortex and brainstem showed significantly less axonal swelling and disconnection on morphological examination compared with the DAI+vehicle group (Fig. 6c).
Curcumin reduced axonal injury after DAI in terms of ultrastructural changes
The ultrastructural features of myelinated nerve fibers in the brainstem were detected by transmission electron microscopy at day 1 after DAI. In the control group, the myelinated fibers showed a normal axoplasm with a well-preserved cellular structure. The multiple layers of the myelin sheath also showed a normal appearance: regularly and tightly organized. At day 1 after DAI, we found that the myelin sheath became loose and swollen, and the layers of the myelin sheath shrank, showing an onion-like appearance (Fig. 7). In the control group and the control+vehicle group, the MT structure aligned with the longitudinal axis of the axon (black arrowheads, Fig. 7), but in the DAI group, we observed that the MT structure was depolymerized and dissolved into puncta (white arrows, Fig. 7), the mitochondria were swollen (asterisks, Fig. 7) in the injured axons, but the pathological changes were alleviated after curcumin intervention after DAI.
Fig. 7.
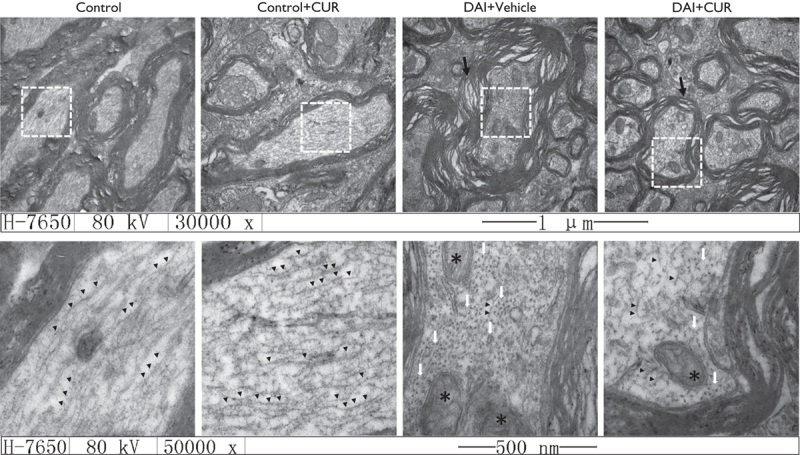
The protective effect of curcumin against nerve injury after DAI in terms of ultrastructural changes. The ultrastructural changes in the sheath of nerves were observed in the brainstem after DAI: Fibers in the brainstem were investigated using a transmission electron microscope (n=4 per group; magnifications ×30 000× and ×50 000; black arrowheads: the MT structure aligned with the longitudinal axis of the axon; white arrows: MT structure depolymerized and dissolved into puncta; black arrows: swollen and damaged myelin; asterisks: swollen mitochondria). CUR, curcumin; DAI, diffuse axonal injury.
Discussion
The DAI model for this study was based on the DAI device described in a past publication by Xiao-Sheng and colleagues 28,29, operating by instant rotational acceleration of the head 26. This model produced a wide range of neuronal degeneration and axonal injuries in the rats’ brain at day 1 after injury, which can effectively simulate the acute pathophysiological changes of DAI in the clinic. Moreover, these injuries affect the degree of consciousness and the motor or sensory functions, consistent with the severity of the disorder in the clinic. Therefore, studies of the effects of drugs on neuronal and axonal injury following DAI can provide insights into potential treatments for DAI in clinic. In an encephalomyelitis model and a sciatic nerve repair model, it has been confirmed that curcumin enhances neuroprotection and myelin repair 36,37. In this study, it was primarily found that the administration of curcumin could alleviate neuronal degeneration and axonal injury, and facilitate the recovery of neurological function after DAI.
The ER, an important cellular organelle, plays a role in the post-translational processing of newly synthesized proteins and in proper protein folding and assembly 38. ER stress triggered by types of damage, such as oxidative stress, disturbed calcium homeostasis, and deprivation of glucose and oxygen induces the expression of chaperone proteins and triggers many rescue responses, including the UPR 39. The UPR strengthens the mechanisms of misfolded protein degradation to reduce cell damage, restore cellular environmental homeostasis, and promote cell survival 40. TBI constitutes one of the epigenetic risk factors for the development of neurodegenerative disorders, such as Alzheimer’s disease and Parkinson’s disease 41,42. Both of these neurodegenerative diseases show a characteristic regulatory imbalance, leading to the abnormal accumulation of proteins as production outpaces degradation. In addition, accumulation of several key proteins, including β-APP, p-tau, NF proteins, or synuclein, reflects disruption of axonal transport as a result of extensive axonal injury after TBI 43. To date, most studies on TBI-induced ER stress or UPR have focused on its association with acute neuronal cell death 44. Therapeutic approaches that attenuate ER stress by reducing aberrant protein accumulation may be a potential therapeutic option to promote neuronal recovery after DAI. However, the impact of sustained ER stress on DAI remains unknown. In our study, we focused on the role of the UPR pathway in neuronal apoptosis and axonal injury after DAI, which were caused by the formation and aggregation of misfolded proteins (mainly β-APP and p-tau).
PERK is an ER transmembrane protein that is activated during ER stress. In an adaptive response stage of ER stress, activated PERK indirectly reduces the quantity of unfolded polypeptides within the ER to enable more efficient chaperone-mediated protein folding in a well-saturated ER lumen 39. PERK first phosphorylates the eukaryotic translation initiation factor 2α (eIF2α) at serine 51 45,46. The increase in p-eIF2α subsequently suppresses the translation of 90% of nuclear-encoded mRNAs by compromising the formation of the GTP·eIF2α·Met–tRNAi ternary complex. This, in turn, prevents the assembly of the preinitiation complex at the 5′-end of mRNA 47. Delaying translation reinitiation in this manner results in the increased translation of some specific mRNAs, which mostly encodes ATF4 48. ATF4, an important bZIP transcription factor, maintains intracellular homeostasis through the upregulation of UPR-target genes involved in more efficient protein folding, amino acid biosynthesis and transport, and antioxidant response 49. In addition to promoting an adaptive response, ATF4 also binds to promoter/enhancer regions and transcriptionally augments the expression of C/EBP homologous protein (CHOP or GADD153), which impacts the control of cell death/survival outputs on ER stress 39,49. Moreover, CHOP also binds to promoter elements associated with growth arrest and DNA damage-inducible protein 34 to recruit protein phosphatase 1 to dephosphorylate eIF2α, which is essential for cells to restore global mRNA translation after an acute insult 50. However, in the later stage of prolonged ER stress, when the UPR cannot overcome the ER stress from a persistent or an intense stressor, the overexpression of CHOP and growth arrest and DNA damage-inducible protein 34 elevates the protein load and restores global translation of proteins involved in reactive oxygen species production and apoptosis 51. Thus, the expression of CHOP is a hallmark of prolonged ER stress and ER stress-induced apoptosis 52,53. Previous research has shown that, under stress, CHOP promotes apoptosis by elevating the expression of caspase-12 and reducing the expression of Bcl-2 52,54–57. In this study, we first detected the persistent activation of the UPR process at 6–72 h after DAI. The expression of p-PERK was increased at 6 h after DAI and remained elevated at day 3 after DAI, as with the downstream factor p-eIF2α. These results indicated that the UPR pathway was persistently activated from the acute stage after DAI. However, the expression of ATF4 began to decrease at day 3 after injury, and the expression of CHOP was distinctly increased at day 1 after DAI, which was slightly later than the expression of PERK pathway components, and progressively elevated at day 3 after DAI. These results suggest that ER stress persisted continuously through at least day 3 after DAI and that the UPR process could not overcome the ER stress beginning day 1 after DAI because of the increased expression of CHOP.
Previous research has indicated that Nrf2, a bZIP transcription factor, is a relevant target of pharmacological therapy to activate endogenous cytoprotection in the central nervous system and is required for free radical scavenging, detoxification of xenobiotics, and maintenance of redox potential 58–60. In our study, the expression of Nrf2 also increased at 6 h after DAI, and the increase was sustained until day 3 after DAI. This result suggests that Nrf2 is activated after DAI and may also play a role in cytoprotection after DAI. Because ER stress is also triggered by excitotoxicity and oxidative stress in neurotrauma conditions 61–63, Nrf2 may be involved in the ER stress process following DAI. Upon ER stress, in addition to transautophosphorylating and phosphorylating, the translation initiation factor eIF2α PERK also phosphorylates Nrf2 and promotes its nuclear translocation 64,65. Activated Nrf2 controls the antioxidant response through the upregulation of antioxidant genes, such as heme oxygenase 1 66,67. To investigate the role of Nrf2 in ER stress after DAI, we focused on curcumin, which is used as an activator of Nrf2 and significantly upregulates nuclear translocation of Nrf2 68. Curcumin is used as a pigment or a spice because of its diverse pharmacological activity, very low toxicity, and widespread availability. The bioavailability of curcumin has been reported in many articles to be much better by an intraperitoneal injection than by the oral route 21,69. Thus, we chose an intraperitoneal injection at 1-h post-DAI in our study because of the comparatively high bioavailability and the limited effective window of curcumin. In experimental TBI, curcumin has been found to cross the blood–brain barrier and maintain high biological activity, and it also improves patient outcome by reducing acute activation of microglia/macrophages and neuronal apoptosis 21. In our study, curcumin upregulated the expression of Nrf2 and promoted the nuclear translocation of the protein. It is interesting that the expression of p-PERK, used here as an indicator of the early UPR process, was also increased after the intervention with curcumin. The coexpression of p-PERK and Nrf2 in neurons was also increased after the intervention with curcumin. Therefore, we believed that the activation of Nrf2 was closely related to the strengthening of the UPR process. Previous studies have established the role of Nrf2 activation during the UPR following the identification of Nrf2 as a PERK substrate. Under basal conditions, Nrf2 is normally sequestered within the cytosol by kelch-like ECH-associated protein 1 (Keap1). Under oxidative stress conditions, PERK promotes the phosphorylation of Nrf2 and its dissociation from Keap1, and then Nrf2 translocates into the nucleus to upregulate the expression of genes involved in redox homeostasis to promote cell survival 70. However, without the presence of reactive oxygen species or reactive nitrogen species, PERK-dependent Nrf2 activation may contribute toward protein degradation during ER stress conditions. PERK phosphorylation also acts on the Nrf2–Keap1 complex at the initiation of UPR 67,71, and the dissociated Nrf2 directly promotes the transcription of several genes encoding proteasome subunits and promoting proteasome assembly 72. Moreover, the study found that apoptosis increased in a PERK−/− mouse model, probably mediated by a lack of dissociated Nrf2 67. In our study, we suggest that curcumin strengthens the UPR process by promoting the phosphorylation of PERK and the nuclear translocation of dissociated Nrf2 after DAI.
ATF4 is a major downstream target of eIF2α phosphorylation, but Nrf2-mediated ATF4 induction is present during oxidative stress 73,74, indicating that Nrf2 is a potent activator of the ATF4 promoter. Antioxidant response element-dependent gene transcription can be induced by Nrf2 and ATF4 jointly, leading to the possibility that both arms of the PERK signaling pathway converge 75. The complement of target genes for both Nrf2 and ATF4 overlaps somewhat 76. One potential point for convergence or coregulation of target genes is CHOP 72,73,77. CHOP transcription is inhibited by Nrf2 and its target genes such as heme oxygenase 1 78. In this study, we found that the activation of Nrf2 by curcumin promoted ATF4 expression to maintain intracellular homeostasis and inhibited the downstream CHOP expression to inhibit neuronal apoptosis.
Previous studies have confirmed that neuronal cell apoptosis is an important mechanism during DAI 6,79. Caspase signaling exerts an important effect on cell apoptotic protease cascade, especially caspase-3, which plays an important role in the ER pathway, mitochondrial pathway, and the death receptor pathway 80. ER stress is an adaptive cellular response to internal and external cell stress and features the upregulation of the UPR pathway, which is involved in protein quantity control and calcium homeostasis. However, above a certain threshold, ER stress results in cell apoptosis 81. The mechanism pathways involved in apoptosis under irreversible ER damage are now partially understood, and one of them is the PERK-mediated apoptosis signaling pathway 82. In addition to the expression of CHOP, we also assessed the apoptosis by TUNEL staining and measured the expression of cleaved caspase-3. On the basis of our results, we suggest that curcumin alleviates ER-stress-related apoptosis after DAI by downregulating the expression of CHOP and cleaved caspase-3.
The feature of widespread and disseminated damage of the axons in the brain occurs after DAI and leads to severe brain failure, vegetative state, or even death 83. However, axonal injury following DAI was not associated with direct mechanical tearing of axons, but rather a delayed impairment of axoplasmic transport. According to this theory, we focused on the axons when their function was disrupted, but not permanently damaged, searching for a potential time window for therapeutic intervention following DAI 84,85. It is known that β-APP is a transmembrane glycoprotein synthesized in neurons and plays a role in cell adhesion, growth, and response to injury. β-APP is synthesized in the perikaryon, and then moves through the neuron by fast anterograde transport 86. Therefore, it can accumulate rapidly in areas of disrupted transport after injury. β-APP has been shown to be highly sensitive and specific as a selective marker of damaged axons. In the control group in our study, β-APP was not accumulated and detected in the brain tissue, but the accumulation of β-APP was detected in the proximal and distal axonal segments following DAI. After intervention with curcumin, the number of β-APP-positive neurons was decreased. Microtubule-associated protein tau, a cytoskeletal protein in axons, plays an important role in maintaining the stability of the microtubule assembly and function. Under normal conditions, protein tau is only transported inside the axons, but in the pathological cases, the abnormal phosphorylation of tau can inhibit microtubule assembly and promote microtubule depolymerization, followed by axon fracture 87. In our study, we found that the accumulated amount of p-tau in the brain following DAI was decreased after the intervention with curcumin. Neurofilament markers, which are composed of light-chain (NF-L), medium-chain (NF-M), and heavy-chain (NF-H) subunits, are useful cytoskeletal injury markers after TBI 88. The subunits are transported within the axons, and in axonal injury, they accumulate in areas of disrupted transport. In our study, we also found the accumulation of NF-H following DAI, likely reflecting the effect of injury on the cytoskeleton, and this decreased after the intervention with curcumin. Moreover, we observed the protection of axons after curcumin intervention in the ultrastructural changes following DAI. We suggest that curcumin mitigates the pathological changes of axons after DAI by alleviating abnormal protein accumulation, and the process may be associated with the intensive UPR process by Nrf2 nuclear translocation and the downregulation of abnormal protein expression of upstream genes, such as GSK-3β 89.
Conclusion
We found that Nrf2 was activated and underwent nuclear translocation following DAI. In addition to counteracting oxidative stress, Nrf2 also acted as a protective factor by regulating the UPR pathway after DAI. We also indicated that curcumin successfully inhibited excessive ER stress and effectively alleviated pathological changes, including neuronal apoptosis and axonal injury, in a rat model of DAI (Fig. 8).
Fig. 8.
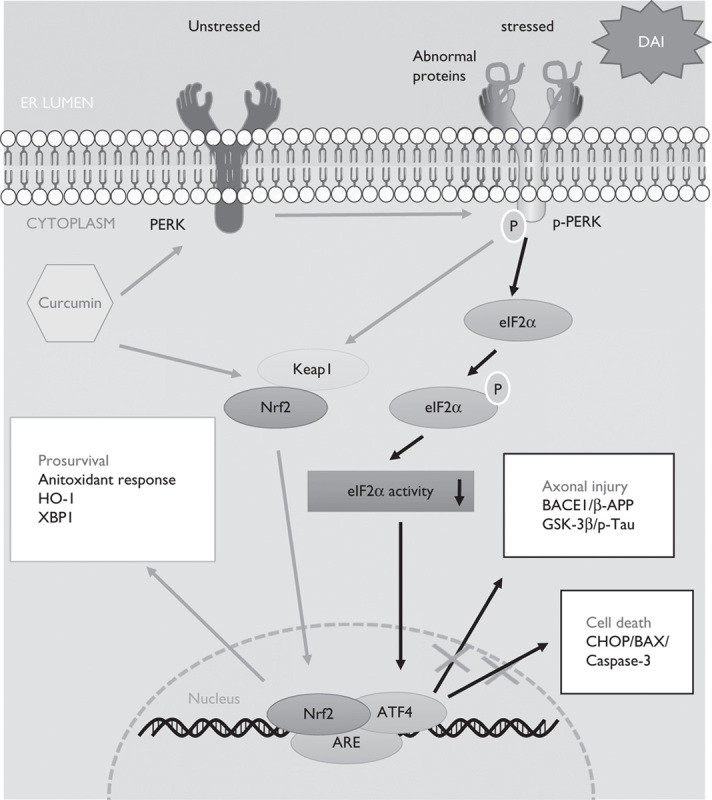
The protective mechanism of curcumin by the UPR-Nrf2 pathway after DAI. When ER stress appeared as a result of abnormal protein aggregation after DAI, curcumin increased the expression of Nrf2 and promoted its nuclear translocation. Moreover, curcumin also promoted PERK phosphorylation to dissociate Nrf2 from Keap1 in the cytoplasm and enable its translocation to the nucleus. Nrf2 in the nucleus combined with ARE and upregulated the expression of ATF4 to maintain intracellular homeostasis and promote cell survival by the following mechanisms: (i) The upregulation of the antioxidant response, such as HO-1, and other unfolded protein response-target genes such as XBP1; (ii) inhibition of the expression of BACE1 and GSK-3β to reduce β-APP and p-tau aggregation in neurons; and (iii) inhibition of the expression of ER-stress-associated apoptosis genes, such as CHOP, Bax, and caspase-3. On the basis of our study, we suggest that curcumin strengthens the unfolded protein response process by promoting the phosphorylation of PERK and the nuclear translocation of dissociated Nrf2. DAI, diffuse axonal injury; ER, endoplasmic reticulum.
Acknowledgements
Conflicts of interest
There are no conflicts of interest.
References
- 1.Zhang P, Zhu S, Li Y, Zhao M, Liu M, Gao J, et al. Quantitative proteomics analysis to identify diffuse axonal injury biomarkers in rats using iTRAQ coupled LC-MS/MS. J Proteomics 2015; 133:93–99. [DOI] [PubMed] [Google Scholar]
- 2.Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil 2005; 20:76–94. [DOI] [PubMed] [Google Scholar]
- 3.Meythaler JM, Peduzzi JD, Eleftheriou E, Novack TA. Current concepts: diffuse axonal injury-associated traumatic brain injury. Arch Phys Med Rehabil 2001; 82:1461–1471. [DOI] [PubMed] [Google Scholar]
- 4.Ryu J, Horkayne-Szakaly I, Xu L, Pletnikova O, Leri F, Eberhart C, et al. The problem of axonal injury in the brains of veterans with histories of blast exposure. Acta Neuropathol Commun 2014; 2:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pang H, Huang T, Song J, Li D, Zhao Y, Ma X. Inhibiting HMGB1 with glycyrrhizic acid protects brain injury after DAI via its anti-inflammatory effect. Mediators Inflamm 2016; 2016:4569521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Su E, Bell M. Laskowitz D, Grant G. Diffuse axonal injury. Translational research in traumatic brain injury. Boca Raton, FL: CRC Press/Taylor and Francis Group; 2016. 41–85. [Google Scholar]
- 7.Zhao Y, Zhao J, Zhang M, Zhao Y, Li J, Ma X, et al. Involvement of toll like receptor 2 signaling in secondary injury during experimental diffuse axonal injury in rats. Mediators Inflamm 2017; 2017:1570917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boone DR, Micci MA, Taglialatela IG, Hellmich JL, Weisz HA, Bi M, et al. Pathway-focused PCR array profiling of enriched populations of laser capture microdissected hippocampal cells after traumatic brain injury. PLoS One 2015; 10:e0127287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuruker V, Naziroglu M, Senol N. Reduction in traumatic brain injury-induced oxidative stress, apoptosis, and calcium entry in rat hippocampus by melatonin: possible involvement of TRPM2 channels. Metab Brain Dis 2015; 30:223–231. [DOI] [PubMed] [Google Scholar]
- 10.Begum G, Yan HQ, Li L, Singh A, Dixon CE, Sun D. Docosahexaenoic acid reduces ER stress and abnormal protein accumulation and improves neuronal function following traumatic brain injury. J Neurosci 2014; 34:3743–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang HY, Wang ZG, Lu XH, Kong XX, Wu FZ, Lin L, et al. Endoplasmic reticulum stress: relevance and therapeutics in central nervous system diseases. Mol Neurobiol 2015; 51:1343–1352. [DOI] [PubMed] [Google Scholar]
- 12.Duan Z, Zhao J, Fan X, Tang C, Liang L, Nie X, et al. The PERK-eIF2alpha signaling pathway is involved in TCDD-induced ER stress in PC12 cells. Neurotoxicology 2014; 44:149–159. [DOI] [PubMed] [Google Scholar]
- 13.Imaizumi K. [Endoplasmic reticulum stress response involved in osteogenesis and chondrogenesis]. Clin Calcium 2013; 23:1759–1766. [PubMed] [Google Scholar]
- 14.Urra H, Dufey E, Lisbona F, Rojas-Rivera D, Hetz C. When ER stress reaches a dead end. Biochim Biophys Acta 2013; 1833:3507–3517. [DOI] [PubMed] [Google Scholar]
- 15.Kaur B, Rutty GN, Timperley WR. The possible role of hypoxia in the formation of axonal bulbs. J Clin Pathol 1999; 52:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y, Zhao Y, Zhang M, Zhao J, Ma X, Huang T, et al. Inhibition of TLR4 signalling-induced inflammation attenuates secondary injury after diffuse axonal injury in rats. Mediators Inflamm 2016; 2016:4706915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu G, Liu Z. Nuclear factor erythroid 2-related factor 2 (Nrf2) mediates neuroprotection in traumatic brain injury at least in part by inactivating microglia. Med Sci Monit 2016; 22:2161–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller DM, Singh IN, Wang JA, Hall ED. Nrf2-ARE activator carnosic acid decreases mitochondrial dysfunction, oxidative damage and neuronal cytoskeletal degradation following traumatic brain injury in mice. Exp Neurol 2015; 264:103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang R, Xu M, Wang Y, Xie F, Zhang G, Qin X. Nrf2 – a promising therapeutic target for defensing against oxidative stress in stroke. Mol Neurobiol 2016; 54:6006–6017. [DOI] [PubMed] [Google Scholar]
- 20.Wu A, Ying Z, Gomez-Pinilla F. Dietary curcumin counteracts the outcome of traumatic brain injury on oxidative stress, synaptic plasticity, and cognition. Exp Neurol 2006; 197:309–317. [DOI] [PubMed] [Google Scholar]
- 21.Zhu HT, Bian C, Yuan JC, Chu WH, Xiang X, Chen F, et al. Curcumin attenuates acute inflammatory injury by inhibiting the TLR4/MyD88/NF-kappaB signaling pathway in experimental traumatic brain injury. J Neuroinflammation 2014; 11:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem 2005; 280:5892–5901. [DOI] [PubMed] [Google Scholar]
- 23.Li W, Suwanwela NC, Patumraj S. Curcumin by down regulating NF-kB and elevating Nrf2, reduces brain edema and neurological dysfunction after cerebral I/R. Microvasc Res 2015; 106:117–127. [DOI] [PubMed] [Google Scholar]
- 24.Cui Q, Li X, Zhu H. Curcumin ameliorates dopaminergic neuronal oxidative damage via activation of the Akt/Nrf2 pathway. Mol Med Rep 2016; 13:1381–1388. [DOI] [PubMed] [Google Scholar]
- 25.Kuo CP, Lu CH, Wen LL, Cherng CH, Wong CS, Borel CO, et al. Neuroprotective effect of curcumin in an experimental rat model of subarachnoid hemorrhage. Anesthesiology 2011; 115:1229–1238. [DOI] [PubMed] [Google Scholar]
- 26.Li Y, Song J, Liu X, Zhang M, An J, Sun P, et al. High expression of STIM1 in the early stages of diffuse axonal injury. Brain Res 2013; 1495:95–102. [DOI] [PubMed] [Google Scholar]
- 27.Huang TQ, Song JN, Zheng FW, Pang HG, Zhao YL, Gu H, et al. Protection of FK506 against neuronal apoptosis and axonal injury following experimental diffuse axonal injury. Mol Med Rep 2017; 15:3001–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiao-Sheng H, Sheng-Yu Y, Xiang Z, Zhou F, Jian-ning Z. Diffuse axonal injury due to lateral head rotation in a rat model. J Neurosurg 2000; 93:626–633. [DOI] [PubMed] [Google Scholar]
- 29.Xiaoshengi H, Guitao Y, Xiang Z, Zhou F. A morphological study of diffuse axonal injury in a rat model by lateral head rotation trauma. Acta Neurol Belg 2010; 110:49–56. [PubMed] [Google Scholar]
- 30.Lu M, Chen J, Lu D, Yi L, Mahmood A, Chopp M. Global test statistics for treatment effect of stroke and traumatic brain injury in rats with administration of bone marrow stromal cells. J Neurosci Methods 2003; 128:183–190. [DOI] [PubMed] [Google Scholar]
- 31.Germano AF, Dixon CE, d’Avella D, Hayes RL, Tomasello F. Behavioral deficits following experimental subarachnoid hemorrhage in the rat. J Neurotrauma 1994; 11:345–353. [DOI] [PubMed] [Google Scholar]
- 32.Giufrida AM, Cox D, Mathias AP. RNA polymerade activity in various classes of nuclei from different regions of rat brain during postnatal development. J Neurochem 1975; 24:749–755. [PubMed] [Google Scholar]
- 33.Ma J, Zhang K, Wang Z, Chen G. Progress of research on diffuse axonal injury after traumatic brain injury. Neural Plast 2016; 2016:9746313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vajtr D, Benada O, Linzer P, Samal F, Springer D, Strejc P, et al. Immunohistochemistry and serum values of S-100B, glial fibrillary acidic protein, and hyperphosphorylated neurofilaments in brain injuries. Soud Lek 2012; 57:7–12. [PubMed] [Google Scholar]
- 35.Chen XH, Meaney DF, Xu BN, Nonaka M, McIntosh TK, Wolf JA, et al. Evolution of neurofilament subtype accumulation in axons following diffuse brain injury in the pig. J Neuropathol Exp Neurol 1999; 58:588–596. [DOI] [PubMed] [Google Scholar]
- 36.Mohajeri M, Sadeghizadeh M, Najafi F, Javan M. Polymerized nano-curcumin attenuates neurological symptoms in EAE model of multiple sclerosis through down regulation of inflammatory and oxidative processes and enhancing neuroprotection and myelin repair. Neuropharmacology 2015; 99:156–167. [DOI] [PubMed] [Google Scholar]
- 37.Zhao Z, Li X, Li Q. Curcumin accelerates the repair of sciatic nerve injury in rats through reducing Schwann cells apoptosis and promoting myelinization. Biomed Pharmacother 2017; 92:1103–1110. [DOI] [PubMed] [Google Scholar]
- 38.Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol 2015; 10:173–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sprenkle NT, Sims SG, Sánchez CL, Meares GP. Endoplasmic reticulum stress and inflammation in the central nervous system. Mol Neurodegener 2017; 12:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pluquet O, Pourtier A, Abbadie C. The unfolded protein response and cellular senescence. A review in the theme: cellular mechanisms of endoplasmic reticulum stress signaling in health and disease. Am J Physiol Cell Physiol 2015; 308:C415–C425. [DOI] [PubMed] [Google Scholar]
- 41.Hoozemans JJ, van Haastert ES, Nijholt DA, Rozemuller AJ, Scheper W. Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neurodegener Dis 2012; 10:212–215. [DOI] [PubMed] [Google Scholar]
- 42.Dash PK, Hylin MJ, Hood KN, Orsi SA, Zhao J, Redell JB, et al. Inhibition of eukaryotic initiation factor 2 alpha phosphatase reduces tissue damage and improves learning and memory after experimental traumatic brain injury. J Neurotrauma 2015; 32:1608–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Smith DH, Uryu K, Saatman KE, Trojanowski JQ, McIntosh TK. Protein accumulation in traumatic brain injury. Neuromolecular Med 2003; 4:59–72. [DOI] [PubMed] [Google Scholar]
- 44.Krajewska M, Xu L, Xu W, Krajewski S, Kress CL, Cui J, et al. Endoplasmic reticulum protein BI-1 modulates unfolded protein response signaling and protects against stroke and traumatic brain injury. Brain Res 2011; 1370:227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peidis P, Papadakis AI, Muaddi H, Richard S, Koromilas AE. Doxorubicin bypasses the cytoprotective effects of eIF2alpha phosphorylation and promotes PKR-mediated cell death. Cell Death Differ 2011; 18:145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su Q, Wang S, Gao HQ, Kazemi S, Harding HP, Ron D, et al. Modulation of the eukaryotic initiation factor 2 alpha-subunit kinase PERK by tyrosine phosphorylation. J Biol Chem 2008; 283:469–475. [DOI] [PubMed] [Google Scholar]
- 47.Tenkerian C, Krishnamoorthy J, Mounir Z, Kazimierczak U, Khoutorsky A, Staschke KA, et al. mTORC2 balances AKT activation and eIF2alpha serine 51 phosphorylation to promote survival under stress. Mol Cancer Res 2015; 13:1377–1388. [DOI] [PubMed] [Google Scholar]
- 48.Blais JD, Filipenko V, Bi M, Harding HP, Ron D, Koumenis C, et al. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol 2004; 24:7469–7482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taniguchi M, Yoshida H. Endoplasmic reticulum stress in kidney function and disease. Curr Opin Nephrol Hypertens 2015; 24:345–350. [DOI] [PubMed] [Google Scholar]
- 50.Liu CL, He YY, Li X, Li RJ, He KL, Wang LL. Inhibition of serine/threonine protein phosphatase PP1 protects cardiomyocytes from tunicamycin-induced apoptosis and I/R through the upregulation of p-eIF2alpha. Int J Mol Med 2014; 33:499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cong XQ, Piao MH, Li Y, Xie L, Liu Y. Bis(maltolato)oxovanadium(IV) (BMOV) attenuates apoptosis in high glucose-treated cardiac cells and diabetic rat hearts by regulating the unfolded protein responses (UPRs). Biol Trace Elem Res 2016; 173:390–398. [DOI] [PubMed] [Google Scholar]
- 52.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 2004; 11:381–389. [DOI] [PubMed] [Google Scholar]
- 53.Fan XF, Wang XR, Yuan GS, Wu DH, Hu LG, Xue F, et al. Effect of safflower injection on endoplasmic reticulum stress-induced apoptosts in rats with hypoxic pulmonary hypertension. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2012; 28:561–567. [PubMed] [Google Scholar]
- 54.Pahl HL. Signal transduction from the endoplasmic reticulum to the cell nucleus. Physiol Rev 1999; 79:683–701. [DOI] [PubMed] [Google Scholar]
- 55.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 2001; 21:1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen F, Wu R, Zhu Z, Yin W, Xiong M, Sun J, et al. Wogonin protects rat dorsal root ganglion neurons against tunicamycin-induced ER stress through the PERK-eIF2alpha-ATF4 signaling pathway. J Mol Neurosci 2015; 55:995–1005. [DOI] [PubMed] [Google Scholar]
- 57.Rao J, Qin J, Qian X, Lu L, Wang P, Wu Z, et al. Lipopolysaccharide preconditioning protects hepatocytes from ischemia/reperfusion injury (IRI) through inhibiting ATF4-CHOP pathway in mice. PLoS One 2013; 8:e65568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang C, Zhang X, Fan H, Liu Y. Curcumin upregulates transcription factor Nrf2, HO-1 expression and protects rat brains against focal ischemia. Brain Res 2009; 1282:133–141. [DOI] [PubMed] [Google Scholar]
- 59.Kim JK, Jang HD. Nrf2-mediated HO-1 induction coupled with the ERK signaling pathway contributes to indirect antioxidant capacity of caffeic acid phenethyl ester in HepG2 cells. Int J Mol Sci 2014; 15:12149–12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee BW, Chun SW, Kim SH, Lee Y, Kang ES, Cha BS, et al. Lithospermic acid B protects beta-cells from cytokine-induced apoptosis by alleviating apoptotic pathways and activating anti-apoptotic pathways of Nrf2-HO-1 and Sirt1. Toxicol Appl Pharmacol 2011; 252:47–54. [DOI] [PubMed] [Google Scholar]
- 61.Zhang C, Wang C, Ren J, Guo X, Yun K. Morphine protects spinal cord astrocytes from glutamate-induced apoptosis via reducing endoplasmic reticulum stress. Int J Mol Sci 2016; 17:10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Logsdon AF, Lucke-Wold BP, Nguyen L, Matsumoto RR, Turner RC, Rosen CL, et al. Salubrinal reduces oxidative stress, neuroinflammation and impulsive-like behavior in a rodent model of traumatic brain injury. Brain Res 2016; 1643:140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patel S, Sharma D, Kalia K, Tiwari V. Crosstalk between endoplasmic reticulum stress and oxidative stress in schizophrenia: the dawn of new therapeutic approaches. Neurosci Biobehav Rev 2017; 83:589–603. [DOI] [PubMed] [Google Scholar]
- 64.Periyasamy P, Shinohara T. Age-related cataracts: role of unfolded protein response, Ca2+ mobilization, epigenetic DNA modifications, and loss of Nrf2/Keap1 dependent cytoprotection. Prog Retin Eye Res 2017; 60:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aydin Y, Chedid M, Chava S, Danielle Williams D, Liu S, Hagedorn CH, et al. Activation of PERK-Nrf2 oncogenic signaling promotes Mdm2-mediated Rb degradation in persistently infected HCV culture. Sci Rep 2017; 7:9223. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 66.Zhu YF, Li XH, Yuan ZP, Li CY, Tian RB, Jia W, et al. Allicin improves endoplasmic reticulum stress-related cognitive deficits via PERK/Nrf2 antioxidative signaling pathway. Eur J Pharmacol 2015; 762:239–246. [DOI] [PubMed] [Google Scholar]
- 67.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 2003; 23:7198–7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jiang H, Tian X, Guo Y, Duan W, Bu H, Li C. Activation of nuclear factor erythroid 2-related factor 2 cytoprotective signaling by curcumin protect primary spinal cord astrocytes against oxidative toxicity. Biol Pharm Bull 2011; 34:1194–1197. [DOI] [PubMed] [Google Scholar]
- 69.Ravindranath V, Chandrasekhara N. Absorption and tissue distribution of curcumin in rats. Toxicology 1980; 16:259–265. [DOI] [PubMed] [Google Scholar]
- 70.Cullinan SB, Diehl JA. Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int J Biochem Cell Biol 2006; 38:317–332. [DOI] [PubMed] [Google Scholar]
- 71.Wang J, Hu X, Jiang H. ERS-PERK signaling pathway-mediated Nrf2/ARE-HO-1 axis: a novel therapeutic target for attenuating myocardial ischemia and reperfusion injury. Int J Cardiol 2016; 203:779–780. [DOI] [PubMed] [Google Scholar]
- 72.Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J Biol Chem 2004; 279:20108–20117. [DOI] [PubMed] [Google Scholar]
- 73.Tao TQ, Wang XR, Liu M, Xu FF, Liu XH. Myofibrillogenesis regulator-1 attenuated hypoxia/reoxygenation-induced apoptosis by inhibiting the PERK/Nrf2 pathway in neonatal rat cardiomyocytes. Apoptosis 2015; 20:285–297. [DOI] [PubMed] [Google Scholar]
- 74.Luis A, Martins JD, Silva A, Ferreira I, Cruz MT, Neves BM. Oxidative stress-dependent activation of the eIF2alpha-ATF4 unfolded protein response branch by skin sensitizer 1-fluoro-2,4-dinitrobenzene modulates dendritic-like cell maturation and inflammatory status in a biphasic manner [corrected]. Free Radic Biol Med 2014; 77:217–229. [DOI] [PubMed] [Google Scholar]
- 75.He CH, Gong P, Hu B, Stewart D, Choi ME, Choi AM, et al. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J Biol Chem 2001; 276:20858–20865. [DOI] [PubMed] [Google Scholar]
- 76.Ehren JL, Maher P. Concurrent regulation of the transcription factors Nrf2 and ATF4 mediates the enhancement of glutathione levels by the flavonoid fisetin. Biochem Pharmacol 2013; 85:1816–1826. [DOI] [PubMed] [Google Scholar]
- 77.Zong ZH, Du ZX, Li N, Li C, Zhang Q, Liu BQ, et al. Implication of Nrf2 and ATF4 in differential induction of CHOP by proteasome inhibition in thyroid cancer cells. Biochim Biophys Acta 2012; 1823:1395–1404. [DOI] [PubMed] [Google Scholar]
- 78.Kim KM, Pae HO, Zheng M, Park R, Kim YM, Chung HT. Carbon monoxide induces heme oxygenase-1 via activation of protein kinase R-like endoplasmic reticulum kinase and inhibits endothelial cell apoptosis triggered by endoplasmic reticulum stress. Circ Res 2007; 101:919–927. [DOI] [PubMed] [Google Scholar]
- 79.Smith DH, Hicks R, Povlishock JT. Therapy development for diffuse axonal injury. J Neurotrauma 2013; 30:307–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sato Y, Ishihara N, Nagayama D, Saiki A, Tatsuno I. 7-ketocholesterol induces apoptosis of MC3T3-E1 cells associated with reactive oxygen species generation, endoplasmic reticulum stress and caspase-3/7 dependent pathway. Mol Genet Metab Rep 2017; 10:56–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen WJ, Xiong ZA, Zhang M, Yao CG, Zhao ZY, Hua YY, et al. Picosecond pulsed electric fields induce apoptosis in HeLa cells via the endoplasmic reticulum stress and caspase-dependent signaling pathways. Int J Oncol 2013; 42:963–970. [DOI] [PubMed] [Google Scholar]
- 82.Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. The role of the PERK/eIF2alpha/ATF4/CHOP signaling pathway in tumor progression during endoplasmic reticulum stress. Curr Mol Med 2016; 16:533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hill CS, Coleman MP, Menon DK. Traumatic axonal injury: mechanisms and translational opportunities. Trends Neurosci 2016; 39:311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Davceva N, Basheska N, Balazic J. Diffuse axonal injury-a distinct clinicopathological entity in closed head injuries. Am J Forensic Med Pathol 2015; 36:127–133. [DOI] [PubMed] [Google Scholar]
- 85.Davceva N, Janevska V, Ilievski B, Spasevska L, Popeska Z. Dilemmas concerning the diffuse axonal injury as a clinicopathological entity in forensic medical practice. J Forensic Leg Med 2012; 19:413–418. [DOI] [PubMed] [Google Scholar]
- 86.Hayashi T, Ago K, Ago M, Ogata M. Two patterns of beta-amyloid precursor protein (APP) immunoreactivity in cases of blunt head injury. Leg Med (Tokyo) 2009; 11 (Suppl 1):S171–S173. [DOI] [PubMed] [Google Scholar]
- 87.Zhao YL, Song JN, Ma XD, Zhang BF, Li DD, Pang HG. Rosiglitazone ameliorates diffuse axonal injury by reducing loss of tau and up-regulating caveolin-1 expression. Neural Regen Res 2016; 11:944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shaw G, Yang C, Ellis R, Anderson K, Parker Mickle J, Scheff S, et al. Hyperphosphorylated neurofilament NF-H is a serum biomarker of axonal injury. Biochem Biophys Res Commun 2005; 336:1268–1277. [DOI] [PubMed] [Google Scholar]
- 89.King MK, Pardo M, Cheng Y, Downey K, Jope RS, Beurel E. Glycogen synthase kinase-3 inhibitors: rescuers of cognitive impairments. Pharmacol Ther 2014; 141:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]