


Abstract
Thrombin-binding aptamer (TBA) is a DNA 15-mer of sequence 5′-GGT TGG TGT GGT TGG-3′ that folds into a G-quadruplex structure linked by two T-T loops located on one side and a T-G-T loop on the other. These loops are critical for post-SELEX modification to improve TBA target affinity. With this goal in mind we synthesized a T analog, 5-(indolyl-3-acetyl-3-amino-1-propenyl)-2′-deoxyuridine (W) to substitute one T or a pair of Ts. Subsequently, the affinity for each analog was determined by biolayer interferometry. An aptamer with W at position 4 exhibited about 3-fold increased binding affinity, and replacing both T4 and T12 with W afforded an almost 10-fold enhancement compared to native TBA. To better understand the role of the substituent’s aromatic moiety, an aptamer with 5-(methyl-3-acetyl-3-amino-1-propenyl)-2′-deoxyuridine (K; W without the indole moiety) in place of T4 was also synthesized. This K4 aptamer was found to improve affinity 7-fold relative to native TBA. Crystal structures of aptamers with T4 replaced by either W or K bound to thrombin provide insight into the origins of the increased affinities. Our work demonstrates that facile chemical modification of a simple DNA aptamer can be used to significantly improve its binding affinity for a well-established pharmacological target protein.
INTRODUCTION
Drug discovery for a specific disease often begins with the identification of a target that is involved in steps critical to disease progression, and then proceeds to the design or discovery of molecules that interfere with the function of the protein (1,2). Such molecules are often discovered through screening a large collection of compounds (called a ‘library’) (3–5), and then improved step by step by synthesizing the derivative compounds to optimize their binding affinity for the target and reduce undesirable side effects. Aptamers or modified aptamers identified from a random library of nucleic acids via SELEX (6,7) or other methods (4,5,8–10) seldom have the desired affinity and specificity. Aptamers often need to be optimized by post-SELEX experiments in order to meet the requirements for a desired application (11–14). For example, Pegaptanib sodium (Macugen), a SELEX-derived RNA aptamer approved by the FDA for clinical use to treat age-related macular degeneration (15), was modified post-selection using several different moieties to achieve high affinity binding to human vascular endothelial growth factor-165 (VEGF165) (16).
Thrombin is a serine protease that plays a key role in blood coagulation (17). Mechanistic aspects underlying the multiple functions of thrombin have been revealed by structural features in crystallographic studies of thrombin (18). Because of thrombin’s essential role in the coagulation cascade, achieving the ability to specifically modulate thrombin activity represents a major goal in the development of anticoagulant strategies (19). Thrombin-binding aptamer (TBA), a single-stranded 15-mer DNA with the sequence (5′-GGT TGG TGT GGT TGG-3′), was identified via SELEX (6,7) to bind thrombin with high specificity and affinity from a pool of ∼1013 synthetic oligonucleotides (19). This TBA specifically inhibits clot-bound thrombin and reduces arterial thrombus formation. In addition, it is able to bind both free and clot-bound thrombin (20,21), whereas binding to other serum proteins or proteolytic enzymes is essentially undetectable. Biological analysis shows that TBA binds to exosite-1 but not exosite-2 of thrombin (22). The 3D structure of TBA was analyzed using nuclear magnetic resonance (NMR) solution methods (23,24). As shown in Figure 1A, TBA is characterized by a chair-like structure, consisting of two G-tetrads connected by two T-T loops and a single T-G-T loop. X-ray crystallographic analyses of TBA (25–27) and modified TBAs (28–30) have defined the interactions between amino acid side chains of thrombin and bases in the T-T loops. In addition, site-specific replacement of a thymine with modified or other natural bases confirmed the critical role these residues play in binding (30–38). Native TBA consisting of only natural bases is susceptible to nuclease digestion and has a very short half-life in vivo of 108 s (21). In addition, its binding affinity and selectivity need to be optimized and other off-target interactions limited. These optimization efforts are of critical importance in both diagnostic and therapeutic applications.
Figure 1.

(A) Guanine quadruplex structure adopted by TBA. In this study, T4 was substituted by W or K (X4). (B) Chemical structure of 5-(indolyl-3-acetyl-3-amino-1-propenyl)-2′-deoxyuridine (W). (C) Chemical structure of 5-(methyl-3-acetyl-3-amino-1-propenyl)-2′-deoxyuridine (K).
Hydrophobic and hydrogen bonding interactions have long been recognized as a fundamental driving force behind macromolecular stability and complex formation (39–41). Therefore, such interactions between nucleic acid bases and amino acids play a key role in the sequence-specific recognition of a nucleic acid by a protein in biological systems (42–44). This is also the case for interactions between an in vitro selected nucleic acid aptamer and its protein target (6,7,19,23). Numerous crystal structures show that hydrophobic interactions mainly occur between pyrimidine bases and the side chains of certain hydrophobic amino acids such as the indole ring of tryptophan (45,46). However, relatively little is known about engineering DNA–protein interactions by incorporating amino acid side chains, over-represented in complementary determining regions of antibodies (47,48), into the base portion of an aptamer pyrimidine to improve complex stability.
In an effort to study, the interactions between a protein and an aptamer containing the side chain of tryptophan, we synthesized six modified TBAs in which each individual T was substituted by 5-(indolyl-3-acetyl-3-amino-1-propenyl)-2′-deoxyuridine (W, Figure 1B). The C5-substituted T features an indole moiety that corresponds to the side chain of tryptophan (Supplementary Table S1, ST-1). In addition, we synthesized two modified TBAs containing two W nucleosides (sequences #8 and #9 in ST-1) and determined their binding affinities by biolayer interferometry (BLI). To explain the enhanced binding affinities of T4W (TBA with T4 replaced by W), we determined the crystal structure of thrombin in complex with T4W. In subsequent experiments, TBA with T4 replaced by 5-(methyl-3-acetyl-3-amino-1-propenyl)-2′-deoxyuridine (K, W that lacks the indole moiety; Figure 1C), referred to here as T4K, was also synthesized and its binding affinity investigated. Crystal structures of T4W and T4K in complex with thrombin provide insight into the origins of the increased affinities.
MATERIALS AND METHODS
Materials
Standard solvents and reagents were purchased from either Sigma-Aldrich, Chemgenes or Alfa Aesar. 5′-O-(4,4′-Dimethoxytrityl)-5-(3-trifluoroacetylamino-1-propenyl)-2′-deoxyuridine was purchased from Hongene Biotechnology. 0.5 M (1S)-(+)-(10-Camphorsulfonyl)-oxaziridine (CSO) and biotinTEG phosphoramidite were purchased from Glen Research. Sulfo-NHS-acetate was purchased from ThermoFisher Scientific. Thin layer chromatography was carried out on silica gel 60 F254 from either Selecto Scientific (flexible plates) or Fluka (aluminum plates). Flash chromatography was performed on Fluka silica (230–400 mesh). NMR was performed on either a Varian Inova 500 or Bruker 600 MHz instrument. 1H NMR spectra were referenced to the signal of the solvent, and 31P NMR used 2% phosphoric acid as an external reference. FPLC (fast protein liquid chromatography) and HPLC (high performance liquid chromatography) were performed on an AKTA Basic System from GE Healthcare. The protein used in this investigation was human α-thrombin (Haematologic Technologies). The protein was handled according to the manufacturer's recommendations and aliquots were stored at −80°C. The aptamers were stored at −20°C.
Synthesis of monomer phosphoramidite building block 3 (Figure 2)
Figure 2.

Synthesis of W phosphoramidite building block 3.
Compound 1. 5′-O-(4,4′-Dimethoxytrityl)-5-(3-amino-1-propenyl)-2′-deoxyuridine was synthesized from 5′-O-(4,4′-dimethoxytrityl)-5-(3-trifluoroacetamido-1-propenyl)-2′-deoxyuridine (purchased from Hongene Biotechnology) according to Santoro et al. (49). The product was purified by flash chromatography (1:25:494 to 1:70:429 Et3N/MeOH/CH2Cl2). Rf = 0.21 (3:17 MeOH/CHCl3). 1H NMR (500 MHz, CD3OH, 25°C): δ = 7.91 (s, 1H), 7.43 (d, J = 7.8 Hz, 2H), 7.35–7.23 (m, 7H), 6.90–6.86 (m, 4H), 6.44 (dt, J = 15.8 Hz, 6.5 Hz, 1H), 6.32 (t, J = 6.8 Hz, 1H), 5.54 (d, J = 15.8 Hz, 1H), 4.54–4.51 (m, 1H), 4.05–4.02 (m, 1H), 3.78 (s, 6H), 3.48–3.42 (m, 1H), 3.35–3.30 (m, 1H), 3.10–2.98 (m, 2H), 2.40–2.36 (m, 2H).
Compound 2. (5′-O-(4,4′-Dimethoxytrityl)-5-(indolyl-3-acetyl-3-amino-1-propenyl)-2′-deoxyuridine). Compound 1 (7.01 g, 12.0 mmol) was dissolved in CH2Cl2 (46 ml) and Et3N (5.0 ml, 36 mmol). Succinimido indole-3-acetate (3.9 g, 14.4 mmol) (50) was suspended in CH2Cl2 (25 ml). This suspension was poured into the other solution, and the reaction was stirred at room temperature for 2 h. The reaction was then diluted with CH2Cl2 and washed with saturated NaHCO3 solution. The organic layer was dried over anhydrous Na2SO4, and the liquid was concentrated to a foam. The crude product was purified by flash chromatography (1:20:979 to 1:40:959 Et3N/MeOH/CH2Cl2) to give 6.81 g (76%) of an off-white solid. Rf = 0.31 (1:9 MeOH/CHCl3). 1H NMR (500 MHz, CD3OH, 25°C): δ = 7.75 (s, 1H), 7.52–7.48 (m, 1H), 7.42–7.38 (m, 2H), 7.36–7.32 (m, 1H), 7.31–7.26 (m, 4H), 7.25–7.20 (m, 2H), 7.16–7.07 (m, 3H), 7.02–6.98 (m, 1H), 6.84–6.79 (m, 4H), 6.32–6.27 (m, 1H), 6.24–6.17 (m, 1H), 5.56 (d, J = 15.8 Hz, 1H), 4.51–4.47 (m, 1H), 4.05–4.01 (m, 1H), 3.71 (s, 6H), 3.58 (s, 2H), 3.53–3.49 (m, 2H), 3.40–3.31 (m, 2H), 2.38–2.32 (m, 2H), 2.16 (s, 1H).
Compound 3. (5′-O-(4,4′-Dimethoxytrityl)-3′-O-(2-cyanoethyl-N,N′-diisopropylphosphoramidyl)-5-(indolyl-3-acetyl-3-amino-1-propenyl)-2′-deoxyuridine). Compound 2 (3.71 g, 5.0 mmol) was dissolved in CH2Cl2 (50 ml). Diisopropylethylamine (1.74 ml, 10.0 mmol) was added to the solution, and the flask was immersed in a cool tap water bath. 2-Cyanoethyl N,N′-diisopropylchlorophosphoramidite (1.74 ml, 7.5 mmol) was added dropwise to the solution. The reaction was stirred at room temperature. After 45 min, additional 2-cyanoethyl N,N′-diisopropylchlorophosphoramidite (55 μl, 0.25 mmol) was added to the reaction. After a total reaction time of 1 h, the reaction was diluted with CH2Cl2 and washed with saturated NaHCO3 solution. The organic layer was dried over anhydrous Na2SO4 and diluted with toluene (∼20 ml). The solution was concentrated to a foam. The product was purified by flash chromatography (1:39:60 to 1:0:99 Et3N/hexanes/EtOAc) to give 4.29 g of an off-white foam. Rf = 0.31 (1:19 MeOH/CHCl3). Purity: 98.0% (HPLC). 31P NMR (500 MHz, CD2Cl2, 25°C): δ = 148.78 ppm, 148.66 ppm. Calculated MS: 943.03, observed MS (ESI–): 977.4 [M+Cl-H]–, 1884.1 [2M-H]−.
Oligonucleotide synthesis
TBA and modified TBAs containing W (Figure 1B and Supplementary Table ST-1) were synthesized using standard phosphoramidites and phosphoramidite 3 (Figure 2) on a controlled pore glass support following the β-cyanoethyl phosphoramidite method (51). All standard phosphoramidites were used as 0.1 M acetonitrile solutions. The phosphoramidite 3 was used at a 0.1 M concentration in dichloromethane:acetonitrile (2:3, v/v). Standard detritylation, coupling and capping were used for all standard phosphoramidites. Phosphoramidite 3 was coupled for 6 min. The oxidizing reagent, 0.5 M solution of CSO in acetonitrile, was required for all coupling cycles of an oligonucleotide that contained W. Deprotection was carried out in two steps. First, oligonucleotides were treated with 10% Et2NH in acetonitrile for 10 min and then rinsed with acetonitrile. Second, oligonucleotides were deprotected with aqueous NH3/CH3NH2 for 1 h at 37°C. Oligonucleotide solutions were then diluted with water and lyophilized.
T4K aptamer (Supplementary Table ST-1) was synthesized with commercially available 5-aminoallyl-dU phosphoramidite at a 1 μmol scale. After deprotection, the oligonucleotide was dissolved in NaHCO3 (aq, 0.4 M, 500 μl). Sulfo-NHS-acetate (Pierce, 10.4 mg) was added to the oligonucleotide, and the reaction shaken at room temperature for 2 h.
TBA and all modified TBAs were purified by FPLC (52), desalted by reverse phase (RP)-HPLC and their identity confirmed by mass spectrometry. The concentrations of the samples were determined by measuring the absorbance at a wavelength of 260 nm at room temperature. Supplementary Figure S1 (SF-1) shows the FPLC traces and mass spectra of T4W and T4K, respectively.
Formation of thrombin:aptamer complexes and crystallization experiments
The human D-Phe-Pro-Arg-chloromethylketone (PPACK)-inhibited α-thrombin was purchased from Haematologic Technologies, Inc. Aptamers T4W and T4K were dissolved in 10 mM potassium phosphate buffer (pH 7.1) to a concentration of 2 mM, heated for 10 min at 87°C and slowly cooled to room temperature to induce folding into the quadruplex structure. The complexes with human α-thrombin were prepared by placing a two-fold molar excess of the aptamer onto a frozen sample of inhibited thrombin with a concentration of 1.0 mg/ml and keeping the sample for 3 h at 4°C. The samples were then diluted, extensively washed with buffer containing 50 mM potassium phosphate and 0.1 M KCl (pH 7.1), and concentrated to ca 8–10 mg/ml via an Amicon Ultra miniconcentrator (Merck Millipore Ltd., Ireland) in a refrigerated centrifuge. Screening of crystallization conditions of the human α-thrombin-FPRCK:aptamer complexes was carried out by mixing of 1.5 μl protein/aptamer complex solution and 1.5 μl reservoir solution in 24-well VDX™ plates (Hampton Research) using the hanging drop vapor diffusion technique. Crystallization screening kits Jena Biosciences JBClassic 3 and JBClassic 5 (Jena Biosciences GmbH, Germany) were used for crystallization trials, and a wide range of conditions with various types and concentrations of high molecular weight PEGs versus a wide range of pH values were tested. All crystallization trials were performed at 8°C. After optimization of the most promising conditions, 18% w/v PEG4000, 20% v/v 2-propanol, 0.2 M sodium citrate for T4W (Supplementary Figure SF-2A); 25% w/v PEG4000, 24% v/v 2-propanol, 0.2 M sodium citrate for T4K (Supplementary Figure SF-2B), we obtained crystals of the α-thrombin:T4W and α-thrombin:T4K complexes suitable for diffraction experiments.
X-ray diffraction data collection, processing, structure solution and refinement
Diffraction data for the α-thrombin:T4W complex crystal were collected at a wavelength of 0.9537 Å at −173°C on beamline P13 at the PETRA III/EMBL Deutsches Elektronen-Synchrotron (DESY), Hamburg. Data for the α-thrombin:T4K complex were collected at a wavelength of 0.8950 Å at −173°C on beamline BL14.1 at the Berliner Elektronenspeicherring-Gesellschaft für Synchrotronstrahlung (BESSY), Berlin (53). Crystals grown in PEG4000 conditions did not need cryoprotectant, but had to be mounted on a very thin film from the crystallization buffer. The excess of the liquid was removed by gently touching the mounting loop to the plate surface, and then the crystal was flash-cooled directly in the N2 stream. For the α-thrombin:T4W complex an almost complete dataset (99.7%) was collected to a maximum resolution 1.69 Å. Data for the α-thrombin:T4K complex were collected to a maximum resolution 2.24 Å, with a completeness of 95.4%. All data were processed, integrated and scaled with XDS (54) and AIMLESS (55) from the CCP4 suite. Data collection and processing statistics are summarized in Table 1.
Table 1. Crystallographic parameters and data collection statistics for the thrombin:T4W and thrombin:T4K aptamer complexes.
| Complex | α-thrombin:T4W | α-thrombin:T4K |
|---|---|---|
| X-ray source | Beamline P13@PETRA III DESY | Beamline BL14.1@BESSY |
| Wavelength [Å] | 0.9537 | 0.9184 |
| Detector | Dectris Pilatus 6M | Dectris Pilatus 6M |
| Detector dist. [mm] | 350.565 | 573.342 |
| Oscillation width [°] | 0.1 | 0.1 |
| Temperature [K] | 100 | 100 |
| No. of frames | 1010 | 1400 |
| Space group | P3221 | P3221 |
| Unit cell parameters | ||
| a [Å] | 94.1 | 94.58 |
| b [Å] | 94.1 | 94.58 |
| c [Å] | 124.71 | 125.57 |
| α [°] | 90 | 90 |
| β [°] | 90 | 90 |
| γ [°][ | 120 | 120 |
| Total number of reflections | 393 839 (12 279) | 211 313 (8834) |
| Unique reflections | 71 677 (3528) | 30 342 (2117) |
| Completeness [%] | 99.7 (96.6) | 95.4 (73.4) |
| Resolution [Å] | 81.49–1.69 (1.72–1.69) | 47.29–2.24 (2.31–2.24) |
| R merge a | 0.051 (0.563) | 0.091 (0.702) |
| Multiplicity | 5.5 (3.5) | 7.0 (4.2) |
| Mosaicity | 0.04 | 0.05 |
| Wilson B factor | 18.6 | 31.5 |
| Mean I/σ(I) | 16.5 (2.0) | 15.3 (1.7) |
| CC(1/2) | 0.999 (0.822) | 0.998 (0.702) |
a
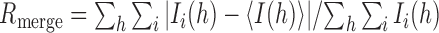 , where Ii(h) is the intensity of an individual measurement of the reflection and 〈I(h)〉 is the mean intensity of the reflection.
, where Ii(h) is the intensity of an individual measurement of the reflection and 〈I(h)〉 is the mean intensity of the reflection.
The structures were solved using the molecular replacement method with the MOLREP software (56), utilizing the structure of human α-thrombin:TBA aptamer complex (PDB ID: 1hao) (26). After calculating an initial electron density map, the structure was completed using alternate cycles of manual building, including rebuilding of main and side chains, construction of alternative residue conformations, adjustment of loop fragments and addition of solvent molecules in the COOT program (57). Because coordinates of modified nucleotides incorporated into the two aptamers were unavailable in structural databases, templates of models were generated using the JLIGAND program (58). Refinement was carried out with the REFMAC5 program (59). All refinement steps were monitored using Rcryst and Rfree values. The stereochemical quality of the resulting models was judged using the MOLPROBITY program (60) and validation tools implemented in COOT. The refinement statistics for both structures are given in Supplementary Table ST-2. All figures were generated using PyMOL v.0.99 (61) or UCSF Chimera v. 1.11.2 (62).
BLI binding analyses
The binding affinities of TBA and modified TBAs were determined by BLI on a fortéBIO Octet Red96 instrument (Pall fortéBIO) at 30°C. All aptamers were chemically labeled with biotin at the 5′-end, allowing immobilization onto a streptavidin (SA)-coated sensor surface and enabling a kinetic analysis of their binding events in a suitable running buffer. Each biotinylated aptamer was diluted to 100 nM in a suitable buffer, heat-denatured at 95°C and slowly cooled to room temperature just prior to the experiment. Samples were stirred at 1000 rpm. Tips were saturated with 100 nM biotinylated aptamers for 1 min, which typically resulted in capture levels of 0.4 ± 0.05 nm within a row of eight tips. Analyte thrombin was prepared in the appropriate buffer as a dilution series (typically in the range from 0.5 to 20 nM in duplicate) along with the buffer blanks. Association was monitored for 300 s and dissociation was followed for 400 s into buffer alone. The data were fit to a 1:1 binding model using fortéBIO Octet data analysis software (Supplementary Figure SF-3). Kinetic constants were determined by integration of the experimental data using the differential rate equation dR/dt = ka·C·(Rmax-R)-kd·R to obtain ka and kd values simultaneously (R = observed response, Rmax = maximum response upon saturation, C = analyte concentration, ka = association rate constant, kd = dissociation rate constant). Then, the ratio between kd and ka gives the reported dissociation constants (kd/ka= KD). The goodness of the fit was judged by the reduced chi-square (χ2) values and by the R2 values approaching 1. The relative KD values (Table 2) of the modified TBA aptamers are also expressed as the ratio of KD values (KDTBA/KDModified TBA).
Table 2. TBA variant sequences, affinities, relative binding affinities and relative free energy changes (−ΔΔGs).
| ID | Sequence | K D(M) | Relative KD* | −ΔΔG[kcal/mol]** |
|---|---|---|---|---|
| TBA | 5′-BioTEG-GGTTGGTGTGGTTGG-3′ | 2.8 × 10−9 | 1.0 | 0.0 |
| T3W | 5′-BioTEG-GGWTGGTGTGGTTGG-3′ | 3.9 × 10−9 | 0.7 | −0.2 |
| T4W | 5′-BioTEG-GGTWGGTGTGGTTGG-3′ | 1.0 × 10−9 | 2.8 | 0.6 |
| T7W | 5′-BioTEG-GGTTGGWGTGGTTGG-3′ | 1.7 × 10−9 | 1.7 | 0.3 |
| T9W | 5′-BioTEG-GGTTGGTGWGGTTGG-3′ | 5.1 × 10−9 | 0.5 | −0.4 |
| T12W | 5′-BioTEG-GGTTGGTGTGGWTGG-3′ | 1.3 × 10−9 | 2.2 | 0.4 |
| T13W | 5′-BioTEG-GGTTGGTGTGGTWGG-3′ | 5.4 × 10−9 | 0.5 | −0.4 |
| T4WT7W | 5′-BioTEG-GGTWGGWGTGGTTGG-3′ | 3.2 × 10−10 | 8.7 | 1.2 |
| T4WT12W | 5′-BioTEG-GGTWGGTGTGGWTGG-3′ | 3.1 × 10−10 | 9.3 | 1.3 |
| T4K | 5′-BioTEG-GGTKGGTGTGGTTGG-3′ | 3.9 × 10−10 | 7.0 | 1.1 |
*Relative KD = KDTBA/KDModified TBA; ** −ΔΔG = ΔGTBA-ΔGModified TBA = RTln[Relative KD].
Circular dichroism experiments
Circular dichroism measurements were performed using a J-815 CD spectrometer (Jasco, Japan). The aptamer samples were prepared at ca. 4 μM concentration in a 10 mM potassium phosphate buffer (KH2PO4/K2HPO4), pH 7.0, with 70 mM KCl. For quadruplex formation, ca. 4 μmol of each selected oligonucleotide was diluted in 100 μl of buffer, heated to 95°C and slowly cooled to room temperature. After annealing the volume of each sample was adjusted to 1000 μl. CD spectra were obtained using a 0.5 cm path length quartz cell. The recording parameters were as follows: scan speed 50 nm/min, response time 0.5 s, bandwidth 1.0 nm and step resolution 0.2 nm. The spectra were recorded at 24°C, in the wavelength range of 220 to 360 nm. The spectrum recorded for the buffer was numerically subtracted from the spectrum for each sample (recorded in five accumulations), and the resultant averaged spectra were smoothed with an Adaptive–Smoothing algorithm (convolution width 10).
Melting temperature (Tm) measurements
All absorption measurements were accomplished in a 1.0 cm path length cell with a Cintra 4040 spectrophotometer equipped with a Peltier Thermocell (GBC, Dandenong, Australia), with the detector set at 260 nm. Tm measurements were performed in a 10 mM potassium phosphate buffer (KH2PO4/K2HPO4, pH 7.0) with 70 mM KCl. The first step of the analysis was annealing from 90 to 15°C, with a temperature gradient of 1°C/min. Melting profiles were measured from 15 to 90°C, with a temperature gradient of 0.5°C/min. Each reported Tm is an average of values from at least three independent experiments.
RESULTS
Synthesis of native and chemically modified TBAs
To the best of our knowledge, the synthesis of phosphoramidite 3 (Figure 2) has not been previously reported. Nucleoside 5′-O-(4,4′-dimethoxytrityl)-5-(3-amino-1-propenyl)-2′-deoxyuridine (1) (49) was used to synthesize the intermediate 5′-O-(4,4′-dimethoxytrityl)-5-(indolyl-3-acetyl-3-amino-1-propenyl)-2′-deoxy-uridine (2) by reacting with the succinimido indole-3-acetate synthesized in the presence of triethylamine (50). Intermediate 2 was converted to phosphoramidite 3 via reaction with β-cyanoethyl-N,N’-diisopropyl-chloro-phosphoramidite according to the published procedures (51,63,64). This monomer was used to automatically synthesize the modified TBA containing W (Figure 1B and Supplementary Table ST-1) using the standard phosphoramidite method (51,65). The length of the coupling time for the phosphoramidite 3 was extended to 6 min. The yields of coupling were similar to those obtained using standard phosphoramidite building blocks. T4K was synthesized using standard phosphoramidites and the commercially available 5-aminoallyl-dU phosphoramidite. After standard deprotection, the oligonucleotide containing 5-aminoallyl-dU was reacted with sulfo-NHS-acetate at room temperature for 2 h in the presence aqueous sodium bicarbonate. All modified TBAs were purified by FPLC, desalted by RP-HPLC and their identities were confirmed by mass spectrometry. Supplementary Figure SF-1 shows the FPLC traces and mass spectra of T4W and T4K, respectively.
Affinity of modified TBAs to thrombin and relative binding free energy
BLI was used to analyze the affinity of the modified TBAs for thrombin (Table 2). For these assays, all TBA variants contained a biotin group at the 5′-end to facilitate attachment to the surface of a SA-coated sensor. Kinetic constants were obtained at thrombin concentrations ranging from 0.5 to 20 nM. The kinetic profile of the unmodified TBA revealed a dissociation constant (KD) of 2.8 nM. Each T nucleotide was substituted individually with the corresponding W nucleotide, and the affinities of six synthetic variants of TBA were measured. Results are illustrated in Table 2. TBA variants with W at positions 4, 7 or 12 all exhibited increased binding, while W at all other positions 3, 9 or 13 exhibited decreased binding to thrombin compared to native TBA. TBA variants containing two W nucleotides, T4WT7W (both T4 and T7 were substituted by W) and T4WT12W (both T4 and T12 were substituted by W), exhibited 8.7- and 9.3-fold tighter binding, respectively, relative to that of the native TBA. T4K afforded 7.0-fold enhanced binding affinity, relative to that of the native TBA. Table 2 shows all the affinity ratios (KDTBA/KDmodified TBA] and relative binding free energy changes.
CD spectra and thermal stability
The effect of T4W and T4K on the G-quadruplex structure was tested by means of CD. As shown in Figure 3, the CD spectrum of the native TBA exhibits two positive bands at 295 and 247 nm, and a negative band at 266 nm. Similar features in the spectra of the T4W and T4K aptamers support the notion that they also adopt the antiparallel G-quadruplex structure in which anti and syn guanosines alternate along the strand (66–68). However, slight differences in the intensity and position of bands are consistent with subtle effects of the base-modified nucleotide on the conformation of the G-quadruplex (31,33,34,36,69,70).
Figure 3.

CD spectra of TBA (solid line), T4K (dashed line) and T4W (dotted line) at ca. 4 μM concentration in a 10 mM potassium phosphate buffer (KH2PO4/K2HPO4, pH 7.0) with 70 mM KCl.
To investigate the thermal stability of the folded G-quadruplexes, we performed UV melting experiments in the 15–90°C range at 260 nm with a temperature gradient of 0.5°C/min. The melting temperatures of the T4K and T4W aptamers are very close to that of TBA under the conditions used (Supplementary Table ST-3). Thus, these modifications do not affect the thermal stability of the TBA G-quadruplex in a significant fashion.
Overall structures of the modified aptamer:thrombin complexes
We obtained crystals of the α-thrombin:T4W and α-thrombin:T4K aptamer complexes and determined their structures by molecular replacement using the complex of the native TBA aptamer (10) as the search model. The two crystals are isomorphous and belong to the trigonal space group P3221 (Table 1). For crystallization, thrombin was covalently modified with the protease active site inhibitor D-Phe-Pro-Arg chloromethylketone (PPACK) in order to prevent proteolysis. The structure of the complex containing the T4W aptamer has a resolution of 1.69 Å and was refined to values for Rwork and Rfree of 0.161 and 0.176, respectively. The structure of the complex with the T4K aptamer has a resolution of 2.24 Å and was refined to values for Rwork and Rfree of 0.160 and 0.208, respectively. Detailed refinement statistics are reported in Supplementary Table ST-2. The final electron density maps for the T4W and T4K aptamer complexes are of excellent quality; examples for both are depicted in the supporting information (Supplementary Figure SF-4). One Na+ ion is octahedrally coordinated by carbonyl oxygen atoms of Arg-596 and Lys-599 of the α-thrombin heavy chain as well as four well-ordered water molecules. In both complexes, the crystallographic asymmetric unit contains one thrombin and one aptamer molecule (Figure 4). Both T4W and T4K as well as TBA (10) bind the thrombin jointly via the T12-T13 and T3-X4 loops (X = W, T4W; X = K, T4K; X = T, TBA). The heavy chain (H; residues 364–622) and the light chain (L; 332–361) of the thrombin molecule are well defined, with the exception of the residue 511–517 loop in the heavy chain. Residues from the N-(328–332) and C-(362–363) termini of the L chain were not included in the model. The thrombin molecules of the T4W and T4K complexes can be superimposed with a root mean square (r.m.s.) deviation of 0.31 Å (260 Cα pairs) and the r.m.s. deviations for overlays of the thrombin molecules from T4W and TBA (PDB ID: 1hao (10)) and T4K and TBA are 0.58 Å and 0.62 Å, respectively (253 Cα pairs). These numbers were obtained using the program UCSF Chimera with the match option (62). Overlays of the thrombin molecules from the T4W and T4K complexes with the protein portion from another TBA complex (PDB ID: 4dii (27)) also result in relatively minor r.m.s. deviations of 0.60 Å and 0.55 Å, respectively (250 Cα pairs). These comparisons demonstrate that neither the T4W nor the T4K aptamer bound to thrombin causes any significant structural changes in the protein backbone.
Figure 4.

Overall structures of the α-thrombin complexes with (A) T4W (PDB ID: 6EO6; this work), (B) T4K (PDB ID: 6EO7; this work) and (C) TBA (PDB ID: 1hao). Only a portion of the thrombin molecule (gray ribbon) is shown in the panels, and aptamer molecules are colored by atom, with carbon atoms of T4W, T4K and TBA colored in tan, light-blue and pink, respectively. The modified nucleotide W is highlighted in yellow, the modified nucleotide K is highlighted in magenta and selected residues are labeled.
Comparison between the conformations of the TBA, T4W and T4K aptamers
As shown in Figure 4, the T4W and T4K aptamers adopt similar conformations for the most part. Except for the region in the immediate vicinity of modified Ts, the structures also show close resemblance to published TBA crystal structures (24,27). Thus, they fold into a chair-like G-quadruplex with two G-quartets surrounded by a T-G-T loop on one side and the T-T and T-W/K loops on the other, whereby the two latter interact with thrombin exosite I. Inspection of electron density maps shows the presence of a strong peak, located between the two G-quartets and consistent with coordination of a potassium cation (Figure 5). Two well-ordered water molecules are located above and below the potassium cation in both structures. One of these water molecules (below K+ in both Figure 5 panels) exhibits contacts to O6 atoms of G2, G5, G11 and G14 as well as the N3-H groups of T13 and W/K4. The second water molecule (above K+ in both Figure 5 panels) bridges G6 (O6), G8 (N7) and G10 (O6) as well as T7 (N3-H). In the T4K complex, the second water displays only partial occupancy, as apparent from relatively weak electron density (Figure 5B).
Figure 5.
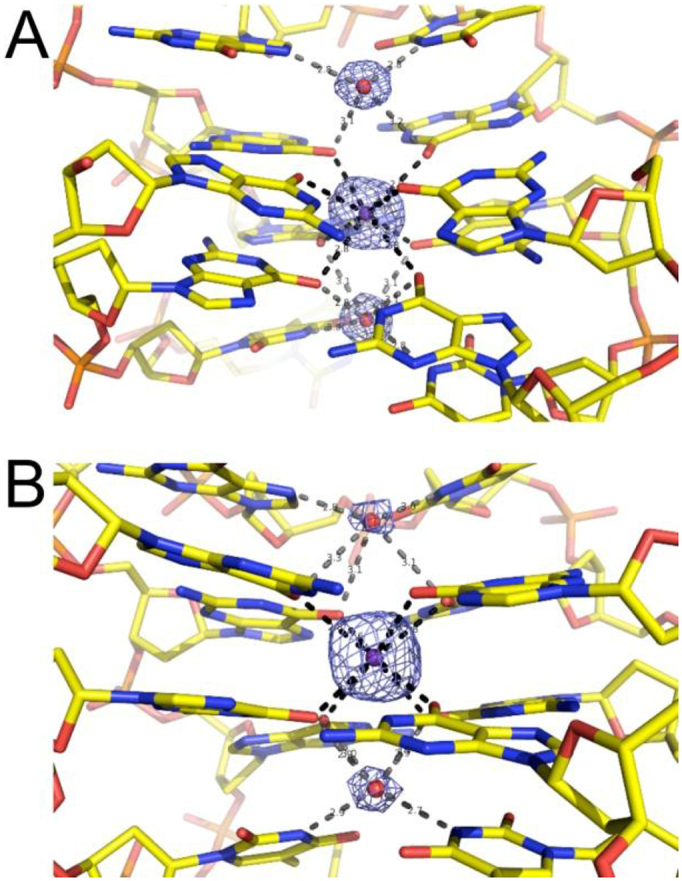
Fourier 2Fo–Fc sum electron density contoured at the 1.0σ level around the potassium ion (indicated as a purple sphere) coordinated between aptamer G-quartets, and around the closest water molecules (indicated as red spheres) in (A) the α-thrombin:T4W and (B) the α-thrombin:T4K complex structures. Potassium coordination and water H bonds are depicted as dashed lines in black and gray, respectively.
Crystal structures of the native TBA aptamer in complex with thrombin revealed two distinct T7-G8-T9 loop arrangements. In one, the base portions of G8 and T9 are stacked against the G-quartet that is farther removed from the thrombin surface (Figure 4C, PDB ID: 1hao (10)), with the third loop residue T7 projecting away from the quadruplex. In the other structure, T7 and G8 are stacked against the outer G-quartet and T9 is detached from the quadruplex (PDB ID: 4dii (12)). This second loop conformation is also seen in the structures of the T4W and T4K aptamer complexes (Figure 4A and B). To calculate r.m.s. deviations between the TBA and the T4W and T4K aptamers, we therefore relied on the second native TBA complex with residue T9 extruded and the program UCSF Chimera (62). Accordingly, the r.m.s. deviation between TBA and T4W is 0.97 Å for 275 atom pairs and excluding atoms of residues T3 and T/W4 as the former undergoes a significant reorientation and the latter entails the modified base (vide infra). Similarly, the r.m.s. deviation between TBA and T4K is 1.25 Å for 295 atom pairs, whereby only the modified residue T/K4 was omitted. Both outcomes support the notion that the conformational changes as a consequence of modification are chiefly local and leave unaffected the core quadruplex structure adopted by the DNA 15mer and the T12-T13 loop.
Interaction between the T4W and T4K aptamers and thrombin
The DNA aptamer-thrombin interface is comprised of the T3-T4 and T12-T13 loops and residues from a loop region of the protein that inserts itself between these two thymidine loops (Figure 6). By comparison, the T7-G8-T9 loop on the opposite side of the G-stack is not involved in any significant interactions with thrombin. A tandem of arginines (Arg-433 and Arg-436) plays a key role among the various interactions. The two residues stack atop the inner G-quartet, alongside T4 and T13, thereby also establishing multiple H-bonds (with T4, T12 and T13 (Arg-433), and with T13 and G14 (Arg-436)). Additional H-bonds are formed between Glu-435 and T12, Asn-437 and G14 as well as Tyr-477 and T12. Both T3 and T12 that jut outward from the G-quadruplex are involved in further contacts to thrombin. T3 stacks onto Tyr-434 but also engages in a hydrophobic interaction with Ile-441. The latter contact is replicated by T12 and Ile-372 on the other side, but instead of a stacking interaction like T3, T12 forms an additional hydrophobic interaction with His-429. The aforementioned interactions are depicted in Figure 6 and are seen in the native TBA and the T4W and T4K thrombin complexes.
Figure 6.

Comparison between the T4W- and T4K-thrombin interfaces. Close-up view of the overlaid T4W:thrombin (W4 carbon atoms are colored in yellow, remaining residues and thrombin backbone ribbon and side chain carbons are tan) and T4K:thrombin complex binding regions (carbon atoms are colored in magenta, remaining residues and thrombin backbone ribbon and side chain carbons are light-blue). Important side chains that interact with the modified residue in one (Arg-436…K4 H-bond) or both structures (Tyr-434 and Ile-441 engage in stacking with T3 and W4) are highlighted in green. Arrows indicate key interactions between W4 or K4 and thrombin side chains, and H-bonds that occur in both the T4W and T4K complex structures are depicted as thin black lines.
However, the introduction of the W4 and K4 residues in place of T4 results in locally altered interactions. The most significant among these in terms of the conformational adjustments is the stacking interaction between T3 and Tyr-434 that is replaced by a partial stack between the W4 indole moiety and the tyrosine in the T4W aptamer:thrombin complex. As a result, T3 undergoes a considerable shift and slight rotation, allowing it to stack onto the W4 indole moiety from the opposite side of Tyr-434 (Figure 6). Overall, the pyrimidine ring and 2′-deoxyribose of T3 in T4W have undergone shifts of ca. 6.5 Å and 3.9 Å, respectively, and the 5′-phosphates of T3 and W4 have moved by ca. 2.2 Å, relative to the corresponding moieties in the structure of the native complex (25). A movie showing the morphing of the native into the T4W aptamer complex structure is provided in the supporting information (Supplementary Movie, SM). These conformational changes are accompanied by formation of multiple new interactions between the T4W aptamer and thrombin that are mainly of a hydrophobic nature. The stack formed by the pyrimidine ring of the T3 and the indole moiety of W4 sits in a hydrophobic cleft formed by Phe-382, Leu-423, Ile-441 and Tyr-434 (Supplementary Figure SF-4). The observed differences in the interactions at the interface obviously account for the enhanced affinity of the T4W aptamer to thrombin relative to the native TBA (Table 2).
In the T4K complex, the presence of the modified nucleotide does not significantly affect T3 that remains stacked on top of Tyr-434 (Figure 6). The substituent at the thymine C5 position of K4 snakes along the upper G-quartet and the head group is directed toward Arg-436 that sits between K4 and T13 and H-bonds to O2 of the latter as well as to the phosphate group and the deoxyribose O4′ of G14. This allows the keto oxygen of the K4 substituent to establish an H-bond to the guanidino moiety of Arg-436 (Figure 6). The methyl group of the K4 substituent forms hydrophobic contacts with Tyr-434 and T3. Although these interactions may at first appear to be less significant than those observed for W4 at the T4W-thrombin binding interface, replacement of T4 by K4 more than doubles the thrombin affinity compared to replacement of T4 by W4 (Table 2).
DISCUSSION
The development of new classes of inhibitors aimed at validated targets for pharmacological intervention is a central tenet of drug discovery. Aptamers selected from combinatorial nucleic acid libraries represent a novel class of binding reagents (4,8,9,71). They possess several properties such as high affinity and specificity toward the target protein and non-immunogenicity among other features that make them very promising molecules (5). They are also readily produced and easily modified by chemical synthesis (5,13,72,73). Post-SELEX modifications are generally aimed at increasing binding affinity (4,32,73–78), thermal and nuclease stabilities (4,13,64,72), and cellular delivery to the target (79–82).
TBA is the first example of a potential aptamer therapeutic agent targeted to a key enzyme of the coagulation cascade (19). Indeed a few years ago the Phase I clinical trial of TBA was started (83). In recent years, many attempts to study the pharmacological properties of TBA have also been described, including introducing locked nucleic acid residues (84), unlocked nucleic acid residues (38) and many other types of acyclic thymidines and L-residues (31–36,69,70,85–87) to substitute individual nucleotides. In addition, since the loops of the TBA are important in the folding of the aptamer and in the interactions with thrombin, modification of the TBA loop composition by natural and/or non-natural nucleosides were also evaluated (37,84,85,87,88). Despite these efforts, a clear understanding of the changes in the interactions between thrombin and TBA as a result of the introduction of chemically modified nucleosides is currently missing. Our research has focused on the development of modified nucleic acids containing amino acid side chains such as the indole moiety of tryptophan to mimic natural protein-protein interactions (72,89–91) by bead-based selection method (8,9) or post-SELEX optimization (13,32,76,92).
In the present study, we incorporated the W nucleotide with an extended substituent containing the indole moiety (mimics the side chain of tryptophan) at the C5 position to replace the native methyl group (Figure 1B). Deoxyguanosine residues fold into a G quadruplex and form the core of the TBA, whereas thymidines make up the loop regions and occupy positions at the binding interface with thrombin. The increased flexibility of loop residues, their key role in recognizing and binding the target protein and facile modification of thymine at the C5 position justify a focus of the modification strategy on thymidine in the case of TBA. When a single T in the native TBA was replaced by the W nucleotide and the affinities of modified aptamers to thrombin measured using BLI, the aptamer exhibiting the highest increase in affinity for the target protein was the TBA with W in place of T4 (T4W) (Table 2). The limited binding interface area of the TBA with thrombin compared with more conventional nucleic acid-protein or aptamer–protein interactions (two T-T loops versus ca. 12 bp) may account for the moderate binding affinity improvement. Another factor that is worth noting concerns the affinity of the originally selected TBA that was already quite high (KD = 2.8 nM). Subsequently, we tested combinations and found that the T4W/T7W and T4W/T12W dual modifications yielded 8.7- and 9.3-fold binding affinity enhancements, respectively. The observed increases in binding affinity are consistent with similar C5-substituted T derivatives recently described in the literature (93–95).
To investigate whether the enhanced binding affinity was caused by the indole moiety of the W nucleotide, we designed the K nucleotide in which the indole moiety was absent (Figure 1C). Surprisingly, the T4K was found to more than double the affinity for thrombin exhibited by T4W (7- versus 2.8-fold, respectively). Interestingly, neither modification triggered a significant change in the thermodynamic stability of TBA (Supplementary Table ST-3) or its CD spectrum (Figure 3). Clearly, the W or K nucleotides do not assist in stabilizing the internal structure of the TBA aptamer. Conversely, crystallographic analyses of the T4W and T4K aptamers bound to thrombin revealed considerable local conformational adjustments at the DNA:protein interface in the case of the former. Thus, the indole moiety of W4 was found to be inserted between the thymine moiety of T3 and Tyr-434 that engage in direct stacking interactions in the native complex. The rearrangement of T3 results in an extended stack now involving Tyr-434, W4 indole and the T3 base (Supplementary Figure SF-5). The two latter moieties are surrounded by several hydrophobic thrombin side chains (Supplementary Figure SF-6), but these interactions do not boost affinity beyond a factor of about three relative to native TBA. By comparison, the presence of the K4 nucleotide causes virtually no changes in the conformation of the aptamer but creates a hydrogen-bonding interaction. Most likely H-bonding contributes to the enhanced binding affinity, since the corresponding 1.1 kcal/mol of relative binding free energy caused by the T4K modification is in the H-bond energy range (1–3 kcal/mol). The absence of structural disruptions paired with additional stabilizing contacts at the aptamer:protein interface thus result in higher affinity of the T4K aptamer for thrombin compared to T4W.
Examining the effects of W modification at other loop positions reveals that the T3W aptamer exhibits slightly diminished binding affinity relative to TBA (Table 2). The methyl group of T3 is directed away from the thrombin binding interface, and the extended C5 substituent of W at that site apparently is unable to loop back and occupy a position that would lead to an improved affinity. Conversely, T12W has a binding affinity that is just slightly lower than that of T4W and thus binds thrombin about twice as tightly as native TBA. Inspection of the structures shows that several thrombin residues that could interact productively with the W substituent lie in vicinity of the T12 nucleobase, among them Ser-72, Ser-521 and Val-522. These same residues are also potentially within reach of the indole-containing substituent of T7W, as the C5-methyl carbons of T7 and T12 only lie some 10 Å apart (KD values of 1.7 nM and 1.3 nM for T7W and T12W, respectively). When we combined the T4W and T12W substitutions into the same sequence (entry # 10 in Table 2), a significant binding affinity gain (9.3-fold) was observed. Thus, T4W and T12W act as a pincer-like system to catch the protruding region of thrombin at exosite I. Similarly, the significantly enhanced binding affinity (8.7-fold) can be accounted for by the synergistic interactions of T4W and T7W. Unfortunately, we were unable to grow crystals of the T7W or T12W aptamer:thrombin complexes after many trials. Nevertheless, the structures help rationalize the enhanced binding affinities resulting from combined T4W and T12W modification.
The work described in this paper demonstrates that a relatively simple post-SELEX modification strategy that introduces an amino acid side chain-like substituent at the C5 position of thymine can be used to improve the binding affinity of the native DNA aptamer toward its target. The co-crystal structures of modified TBAs bound to thrombin illustrate structural details of the interaction between these molecules and their target that can account for the improved affinity of the aptamers. Considering that the TBA is just 15 nt long and lacks the structural and dynamic complexity encoded by much longer DNA and RNA aptamers, the observed affinity gains are certainly noteworthy. Overall, this work shows that a relatively simple post-SELEX modification strategy that introduces amino acid side chain-like substituents at the C5 position of thymine can be used for fine-tuning the aptamer binding affinity to a well established protein target, even with the arguably simplest DNA aptamer identified to date.
DATA AVAILABILITY
Coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers PDB IDs: 6EO6 (α-thrombin:T4W complex) and 6EO7 (α-thrombin:T4K complex).
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr David Gorenstein for his useful comments on the manuscript. Diffraction data have been collected on BL14.1 beamline at the BESSY II electron storage ring operated by the Helmholtz-Zentrum Berlin (HZB), Germany (96), and on P13 beamline at the PETRA III storage ring operated by EMBL Hamburg (DESY, Hamburg, Germany) (97). We would particularly like to acknowledge the help and support of Manfred S. Weiss (from HZB) and Guillaume Pompidor (from EMBL) during the experiments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [GM108110]; National Natural Science Foundation of China [21372207]; CMMS PAS, Lodz, Poland, statutory funds. The open access publication charge for this paper has been waived by Oxford University Press—NAR Editorial Board members are entitled to one free paper per year in recognition of their work on behalf of the journal.
Conflict of interest statement. X.Y. and C.H.L. are employees of AM Biotechnologies LLC. The remaining authors declare no competing financial interests.
REFERENCES
- 1. Drews J. Drug discovery: a historical perspective. Science. 2000; 287:1960–1964. [DOI] [PubMed] [Google Scholar]
- 2. Evans W.J. Drug discovery and development for ageing: opportunities and challenges. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011; 366:113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu R., Li X., Lam K.S.. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol. 2017; 38:117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang X., Gorenstein D.G.. Progress in thioaptamer development. Curr. Drug Targets. 2004; 5:705–715. [DOI] [PubMed] [Google Scholar]
- 5. Yang X., Li N., Gorenstein D.G.. Strategies for the discovery of therapeutic Aptamers. Expert. Opin. Drug Discov. 2011; 6:75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ellington A.D., Szostak J.W.. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990; 346:818–822. [DOI] [PubMed] [Google Scholar]
- 7. Tuerk C., Gold L.. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990; 249:505–510. [DOI] [PubMed] [Google Scholar]
- 8. Yang X., Li X., Prow T.W., Reece L.M., Bassett S.E., Luxon B.A., Herzog N.K., Aronson J., Shope R.E., Leary J.F. et al. . Immunofluorescence assay and flow-cytometry selection of bead-bound aptamers. Nucleic Acids Res. 2003; 31:e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yang X., Bassett S.E., Li X., Luxon B.A., Herzog N.K., Shope R.E., Aronson J., Prow T.W., Leary J.F., Kirby R. et al. . Construction and selection of bead-bound combinatorial oligonucleoside phosphorothioate and phosphorodithioate aptamer libraries designed for rapid PCR-based sequencing. Nucleic Acids Res. 2002; 30:e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang H., Lam C.H., Li X., West D.L., Yang X.. Selection of PD1/PD-L1 X-Aptamers. Biochimie. 2018; 145:125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kimoto M., Nakamura M., Hirao I.. Post-ExSELEX stabilization of an unnatural-base DNA aptamer targeting VEGF165 toward pharmaceutical applications. Nucleic Acids Res. 2016; 44:7487–7494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Eaton B.E., Gold L., Hicke B.J., Janjic N., Jucker F.M., Sebesta D.P., Tarasow T.M., Willis M.C., Zichi D.A.. Post-SELEX combinatorial optimization of aptamers. Bioorg. Med. Chem. 1997; 5:1087–1096. [DOI] [PubMed] [Google Scholar]
- 13. Yang X., Fennewald S., Luxon B.A., Aronson J., Herzog N.K., Gorenstein D.G.. Aptamers containing thymidine 3′-O-phosphorodithioates: synthesis and binding to nuclear factor-kappaB. Bioorg. Med. Chem. Lett. 1999; 9:3357–3362. [DOI] [PubMed] [Google Scholar]
- 14. Gao S., Zheng X., Jiao B., Wang L.. Post-SELEX optimization of aptamers. Anal. Bioanal. Chem. 2016; 408:4567–4573. [DOI] [PubMed] [Google Scholar]
- 15. Ng E.W., Shima D.T., Calias P., Cunningham E.T. Jr, Guyer D.R., Adamis A.P.. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discov. 2006; 5:123–132. [DOI] [PubMed] [Google Scholar]
- 16. Ruckman J., Green L.S., Beeson J., Waugh S., Gillette W.L., Henninger D.D., Claesson-Welsh L., Janjic N.. 2′-Fluoropyrimidine RNA-based aptamers to the 165-amino acid form of vascular endothelial growth factor (VEGF165). Inhibition of receptor binding and VEGF-induced vascular permeability through interactions requiring the exon 7-encoded domain. J. Biol. Chem. 1998; 273:20556–20567. [DOI] [PubMed] [Google Scholar]
- 17. Huntington J.A. Molecular recognition mechanisms of thrombin. J. Thromb. Haemost. 2005; 3:1861–1872. [DOI] [PubMed] [Google Scholar]
- 18. Stubbs M.T., Bode W.. A player of many parts: the spotlight falls on thrombin's structure. Thromb. Res. 1993; 69:1–58. [DOI] [PubMed] [Google Scholar]
- 19. Bock L.C., Griffin L.C., Latham J.A., Vermaas E.H., Toole J.J.. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature. 1992; 355:564–566. [DOI] [PubMed] [Google Scholar]
- 20. Li W.X., Kaplan A.V., Grant G.W., Toole J.J., Leung L.L.. A novel nucleotide-based thrombin inhibitor inhibits clot-bound thrombin and reduces arterial platelet thrombus formation. Blood. 1994; 83:677–682. [PubMed] [Google Scholar]
- 21. Griffin L.C., Tidmarsh G.F., Bock L.C., Toole J.J., Leung L.L.. In vivo anticoagulant properties of a novel nucleotide-based thrombin inhibitor and demonstration of regional anticoagulation in extracorporeal circuits. Blood. 1993; 81:3271–3276. [PubMed] [Google Scholar]
- 22. Wu Q., Tsiang M., Sadler J.E.. Localization of the single-stranded DNA binding site in the thrombin anion-binding exosite. J. Biol. Chem. 1992; 267:24408–24412. [PubMed] [Google Scholar]
- 23. Wang K.Y., McCurdy S., Shea R.G., Swaminathan S., Bolton P.H.. A DNA aptamer which binds to and inhibits thrombin exhibits a new structural motif for DNA. Biochemistry. 1993; 32:1899–1904. [DOI] [PubMed] [Google Scholar]
- 24. Macaya R.F., Schultze P., Smith F.W., Roe J.A., Feigon J.. Thrombin-binding DNA aptamer forms a unimolecular quadruplex structure in solution. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:3745–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Padmanabhan K., Tulinsky A.. An ambiguous structure of a DNA 15-mer thrombin complex. Acta Crystallogr. D Biol. Crystallogr. 1996; 52:272–282. [DOI] [PubMed] [Google Scholar]
- 26. Padmanabhan K., Padmanabhan K.P., Ferrara J.D., Sadler J.E., Tulinsky A.. The structure of alpha-thrombin inhibited by a 15-mer single-stranded DNA aptamer. J. Biol. Chem. 1993; 268:17651–17654. [DOI] [PubMed] [Google Scholar]
- 27. Russo Krauss I., Merlino A., Randazzo A., Novellino E., Mazzarella L., Sica F.. High-resolution structures of two complexes between thrombin and thrombin-binding aptamer shed light on the role of cations in the aptamer inhibitory activity. Nucleic Acids Res. 2012; 40:8119–8128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Russo Krauss I., Spiridonova V., Pica A., Napolitano V., Sica F.. Different duplex/quadruplex junctions determine the properties of anti-thrombin aptamers with mixed folding. Nucleic Acids Res. 2016; 44:983–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Russo Krauss I., Merlino A., Giancola C., Randazzo A., Mazzarella L., Sica F.. Thrombin-aptamer recognition: a revealed ambiguity. Nucleic Acids Res. 2011; 39:7858–7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pica A., Russo Krauss I., Merlino A., Nagatoishi S., Sugimoto N., Sica F.. Dissecting the contribution of thrombin exosite I in the recognition of thrombin binding aptamer. FEBS J. 2013; 280:6581–6588. [DOI] [PubMed] [Google Scholar]
- 31. Scuotto M., Persico M., Bucci M., Vellecco V., Borbone N., Morelli E., Oliviero G., Novellino E., Piccialli G., Cirino G. et al. . Outstanding effects on antithrombin activity of modified TBA diastereomers containing an optically pure acyclic nucleotide analogue. Org. Biomol. Chem. 2014; 12:5235–5242. [DOI] [PubMed] [Google Scholar]
- 32. Cai B., Yang X., Sun L., Fan X., Li L., Jin H., Wu Y., Guan Z., Zhang L., Yang Z.. Stability and bioactivity of thrombin binding aptamers modified with D-/L-isothymidine in the loop regions. Org. Biomol. Chem. 2014; 12:8866–8876. [DOI] [PubMed] [Google Scholar]
- 33. Esposito V., Scuotto M., Capuozzo A., Santamaria R., Varra M., Mayol L., Virgilio A., Galeone A.. A straightforward modification in the thrombin binding aptamer improving the stability, affinity to thrombin and nuclease resistance. Org. Biomol. Chem. 2014; 12:8840–8843. [DOI] [PubMed] [Google Scholar]
- 34. Virgilio A., Petraccone L., Scuotto M., Vellecco V., Bucci M., Mayol L., Varra M., Esposito V., Galeone A.. 5-Hydroxymethyl-2′-deoxyuridine residues in the thrombin binding aptamer: investigating anticoagulant activity by making a tiny chemical modification. Chembiochem. 2014; 15:2427–2434. [DOI] [PubMed] [Google Scholar]
- 35. Varizhuk A.M., Tsvetkov V.B., Tatarinova O.N., Kaluzhny D.N., Florentiev V.L., Timofeev E.N., Shchyolkina A.K., Borisova O.F., Smirnov I.P., Grokhovsky S.L. et al. . Synthesis, characterization and in vitro activity of thrombin-binding DNA aptamers with triazole internucleotide linkages. Eur. J. Med. Chem. 2013; 67:90–97. [DOI] [PubMed] [Google Scholar]
- 36. Borbone N., Bucci M., Oliviero G., Morelli E., Amato J., D’Atri V., D’Errico S., Vellecco V., Cirino G., Piccialli G. et al. . Investigating the role of T7 and T12 residues on the biological properties of thrombin-binding aptamer: enhancement of anticoagulant activity by a single nucleobase modification. J. Med. Chem. 2012; 55:10716–10728. [DOI] [PubMed] [Google Scholar]
- 37. Nagatoishi S., Isono N., Tsumoto K., Sugimoto N.. Loop residues of thrombin-binding DNA aptamer impact G-quadruplex stability and thrombin binding. Biochimie. 2011; 93:1231–1238. [DOI] [PubMed] [Google Scholar]
- 38. Pasternak A., Hernandez F.J., Rasmussen L.M., Vester B., Wengel J.. Improved thrombin binding aptamer by incorporation of a single unlocked nucleic acid monomer. Nucleic Acids Res. 2011; 39:1155–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kellis J.T. Jr, Nyberg K., Sali D., Fersht A.R.. Contribution of hydrophobic interactions to protein stability. Nature. 1988; 333:784–786. [DOI] [PubMed] [Google Scholar]
- 40. Clackson T., Wells J.A.. A hot spot of binding energy in a hormone-receptor interface. Science. 1995; 267:383–386. [DOI] [PubMed] [Google Scholar]
- 41. Eriksson A.E., Baase W.A., Zhang X.J., Heinz D.W., Blaber M., Baldwin E.P., Matthews B.W.. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science. 1992; 255:178–183. [DOI] [PubMed] [Google Scholar]
- 42. Rohs R., Jin X., West S.M., Joshi R., Honig B., Mann R.S.. Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 2010; 79:233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jankowsky E., Harris M.E.. Specificity and nonspecificity in RNA-protein interactions. Nat. Rev. Mol. Cell Biol. 2015; 16:533–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bewley C.A., Gronenborn A.M., Clore G.M.. Minor groove-binding architectural proteins: structure, function, and DNA recognition. Annu. Rev. Biophys. Biomol. Struct. 1998; 27:105–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Watkins D., Hsiao C., Woods K.K., Koudelka G.B., Williams L.D.. P22 c2 repressor-operator complex: mechanisms of direct and indirect readout. Biochemistry. 2008; 47:2325–2338. [DOI] [PubMed] [Google Scholar]
- 46. Aggarwal A.K., Rodgers D.W., Drottar M., Ptashne M., Harrison S.C.. Recognition of a DNA operator by the repressor of phage 434: a view at high resolution. Science. 1988; 242:899–907. [DOI] [PubMed] [Google Scholar]
- 47. Mian I.S., Bradwell A.R., Olson A.J.. Structure, function and properties of antibody binding sites. J. Mol. Biol. 1991; 217:133–151. [DOI] [PubMed] [Google Scholar]
- 48. Ramaraj T., Angel T., Dratz E.A., Jesaitis A.J., Mumey B.. Antigen-antibody interface properties: composition, residue interactions, and features of 53 non-redundant structures. Biochim. Biophys. Acta. 2012; 1824:520–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Santoro S.W., Joyce G.F., Sakthivel K., Gramatikova S., Barbas C.F. 3rd. RNA cleavage by a DNA enzyme with extended chemical functionality. J. Am. Chem. Soc. 2000; 122:2433–2439. [DOI] [PubMed] [Google Scholar]
- 50. Pfeiffer M.J., Hanna S.B.. Aminolysis of activated esters of indole-3-acetic acid in acetonitrile. J. Org. Chem. 1993; 58:735–740. [Google Scholar]
- 51. Caruthers M.H., Barone A.D., Beaucage S.L., Dodds D.R., Fisher E.F., McBride L.J., Matteucci M., Stabinsky Z., Tang J.Y.. Chemical synthesis of deoxyoligonucleotides by the phosphoramidite method. Methods Enzymol. 1987; 154:287–313. [DOI] [PubMed] [Google Scholar]
- 52. Yang X., Hodge R.P., Luxon B.A., Shope R., Gorenstein D.G.. Separation of synthetic oligonucleotide dithioates from monothiophosphate impurities by anion-exchange chromatography on a mono-q column. Anal. Biochem. 2002; 306:92–99. [DOI] [PubMed] [Google Scholar]
- 53. Mueller U., Darowski N., Fuchs M.R., Forster R., Hellmig M., Paithankar K.S., Puhringer S., Steffien M., Zocher G., Weiss M.S.. Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J. Synchrotron Radiat. 2012; 19:442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Evans P.R., Murshudov G.N.. How good are my data and what is the resolution?. Acta Crystallogr. D Biol. Crystallogr. 2013; 69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vagin A., Teplyakov A.. Molecular replacement with MOLREP. Acta Crystallogr D Biol. Crystallogr. 2010; 66:22–25. [DOI] [PubMed] [Google Scholar]
- 57. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lebedev A.A., Young P., Isupov M.N., Moroz O.V., Vagin A.A., Murshudov G.N.. JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr. D Biol. Crystallogr. 2012; 68:431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Murshudov G.N., Skubak P., Lebedev A.A., Pannu N.S., Steiner R.A., Nicholls R.A., Winn M.D., Long F., Vagin A.A.. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011; 67:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen V.B., Arendall W.B. 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C.. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. DeLano W.L. Unraveling hot spots in binding interfaces: progress and challenges. Curr. Opin. Struct. Biol. 2002; 12:14–20. [DOI] [PubMed] [Google Scholar]
- 62. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 63. Beaucage S.L., Iyer R.P.. Advances in the Synthesis of Oligonucleotides by the phosphoramidite Approach. Tetrahedron. 1992; 48:2223–2311. [Google Scholar]
- 64. Yang X., Misiura K., Sochacki M., Stec W.J.. Deoxyxylothymidine 3′-O-phosphorothioates: synthesis, stereochemistry and stereocontrolled incorporation into oligothymidylates. Bioorg. Med. Chem. Lett. 1997; 7:2651–2656. [Google Scholar]
- 65. Caruthers M.H. Chemical synthesis of DNA and DNA analogues. Acc. Chem. Res. 1991; 24:278–284. [Google Scholar]
- 66. Paramasivan S., Rujan I., Bolton P.H.. Circular dichroism of quadruplex DNAs: applications to structure, cation effects and ligand binding. Methods. 2007; 43:324–331. [DOI] [PubMed] [Google Scholar]
- 67. Vorlickova M., Kejnovska I., Bednarova K., Renciuk D., Kypr J.. Circular dichroism spectroscopy of DNA: from duplexes to quadruplexes. Chirality. 2012; 24:691–698. [DOI] [PubMed] [Google Scholar]
- 68. Vorlickova M., Kejnovska I., Sagi J., Renciuk D., Bednarova K., Motlova J., Kypr J.. Circular dichroism and guanine quadruplexes. Methods. 2012; 57:64–75. [DOI] [PubMed] [Google Scholar]
- 69. Virgilio A., Petraccone L., Vellecco V., Bucci M., Varra M., Irace C., Santamaria R., Pepe A., Mayol L., Esposito V. et al. . Site-specific replacement of the thymine methyl group by fluorine in thrombin binding aptamer significantly improves structural stability and anticoagulant activity. Nucleic Acids Res. 2015; 43:10602–10611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Scuotto M., Rivieccio E., Varone A., Corda D., Bucci M., Vellecco V., Cirino G., Virgilio A., Esposito V., Galeone A. et al. . Site specific replacements of a single loop nucleoside with a dibenzyl linker may switch the activity of TBA from anticoagulant to antiproliferative. Nucleic Acids Res. 2015; 43:7702–7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bouchard P.R., Hutabarat R.M., Thompson K.M.. Discovery and development of therapeutic aptamers. Annu. Rev. Pharmacol. Toxicol. 2010; 50:237–257. [DOI] [PubMed] [Google Scholar]
- 72. Wang H., Lam C.H., Li X., West D.L., Yang X.. Selection of PD1/PD-L1 X-Aptamers. Biochimie. 2017; 145:125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yang X. Solid-phase synthesis of oligodeoxynucleotide analogs containing phosphorodithioate linkages. Curr. Protoc. Nucleic Acid Chem. 2016; 70:4.71.1–4.71.14. [DOI] [PubMed] [Google Scholar]
- 74. Yang X. Solid-phase synthesis of RNA analogs containing phosphorodithioate linkages. Curr. Protoc. Nucleic Acid Chem. 2017; 70:4.77.1–4.77.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yang X., Dinuka Abeydeera N., Liu F.W., Egli M.. Origins of the enhanced affinity of RNA-protein interactions triggered by RNA phosphorodithioate backbone modification. Chem. Commun. 2017; 53:10508–10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Abeydeera N.D., Egli M., Cox N., Mercier K., Conde J.N., Pallan P.S., Mizurini D.M., Sierant M., Hibti F.E., Hassell T. et al. . Evoking picomolar binding in RNA by a single phosphorodithioate linkage. Nucleic Acids Res. 2016; 44:8052–8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lou X., Egli M., Yang X.. Determining functional aptamer-protein interaction by biolayer interferometry. Curr. Protoc. Nucleic Acid Chem. 2016; 67:7. 25.1–7.25.15. [DOI] [PubMed] [Google Scholar]
- 78. Yang X., Wang H., Beasley D.W., Volk D.E., Zhao X., Luxon B.A., Lomas L.O., Herzog N.K., Aronson J.F., Barrett A.D. et al. . Selection of thioaptamers for diagnostics and therapeutics. Ann. N. Y. Acad. Sci. 2006; 1082:116–119. [DOI] [PubMed] [Google Scholar]
- 79. Ferguson M.R., Rojo D.R., Somasunderam A., Thiviyanathan V., Ridley B.D., Yang X., Gorenstein D.G.. Delivery of double-stranded DNA thioaptamers into HIV-1 infected cells for antiviral activity. Biochem. Biophys. Res. Commun. 2006; 344:792–797. [DOI] [PubMed] [Google Scholar]
- 80. Pica A., Russo Krauss I., Parente V., Tateishi-Karimata H., Nagatoishi S., Tsumoto K., Sugimoto N., Sica F.. Through-bond effects in the ternary complexes of thrombin sandwiched by two DNA aptamers. Nucleic Acids Res. 2017; 45:461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gilboa E., Berezhnoy A., Schrand B.. Reducing toxicity of immune therapy using aptamer-targeted drug delivery. Cancer. Immunol. Res. 2015; 3:1195–1200. [DOI] [PubMed] [Google Scholar]
- 82. McNamara J.O. 2nd, Andrechek E.R., Wang Y., Viles K.D., Rempel R.E., Gilboa E., Sullenger B.A., Giangrande P.H.. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006; 24:1005–1015. [DOI] [PubMed] [Google Scholar]
- 83. Avino A., Fabrega C., Tintore M., Eritja R.. Thrombin binding aptamer, more than a simple aptamer: chemically modified derivatives and biomedical applications. Curr. Pharm. Des. 2012; 18:2036–2047. [DOI] [PubMed] [Google Scholar]
- 84. Bonifacio L., Church F.C., Jarstfer M.B.. Effect of locked-nucleic acid on a biologically active g-quadruplex. A structure-activity relationship of the thrombin aptamer. Int. J. Mol. Sci. 2008; 9:422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Coppola T., Varra M., Oliviero G., Galeone A., D’Isa G., Mayol L., Morelli E., Bucci M.R., Vellecco V., Cirino G. et al. . Synthesis, structural studies and biological properties of new TBA analogues containing an acyclic nucleotide. Bioorg. Med. Chem. 2008; 16:8244–8253. [DOI] [PubMed] [Google Scholar]
- 86. Varizhuk A.M., Kaluzhny D.N., Novikov R.A., Chizhov A.O., Smirnov I.P., Chuvilin A.N., Tatarinova O.N., Fisunov G.Y., Pozmogova G.E., Florentiev V.L.. Synthesis of triazole-linked oligonucleotides with high affinity to DNA complements and an analysis of their compatibility with biosystems. J. Org. Chem. 2013; 78:5964–5969. [DOI] [PubMed] [Google Scholar]
- 87. Zaitseva M., Kaluzhny D., Shchyolkina A., Borisova O., Smirnov I., Pozmogova G.. Conformation and thermostability of oligonucleotide d(GGTTGGTGTGGTTGG) containing thiophosphoryl internucleotide bonds at different positions. Biophys. Chem. 2010; 146:1–6. [DOI] [PubMed] [Google Scholar]
- 88. Peng C.G., Damha M.J.. G-quadruplex induced stabilization by 2′-deoxy-2′-fluoro-D-arabinonucleic acids (2′F-ANA). Nucleic. Acids. Res. 2007; 35:4977–4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lokesh G.L., Wang H., Lam C.H., Thiviyanathan V., Ward N., Gorenstein D.G., Volk D.E.. X-aptamer selection and validation. Methods Mol. Biol. 2017; 1632:151–174. [DOI] [PubMed] [Google Scholar]
- 90. Zeidan E., Shivaji R., Henrich V.C., Sandros M.G.. Nano-SPRi Aptasensor for the detection of progesterone in buffer. Sci. Rep. 2016; 6:doi:1038/srep26714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lu E., Elizondo-Riojas M.A., Chang J.T., Volk D.E.. Aptaligner: automated software for aligning pseudorandom DNA X-aptamers from next-generation sequencing data. Biochemistry. 2014; 53:3523–3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pallan P., Yang X., Sierant M., Abeydeera N., Hassell T., martinez C., Janicka M., Nawrot B., Egli M.. Crystal structure, stability and Ago2 affinity of phosphorodithioate-modified RNAs. RSC Adv. 2014; 4:64901–64904. [Google Scholar]
- 93. Gupta S., Hirota M., Waugh S.M., Murakami I., Suzuki T., Muraguchi M., Shibamori M., Ishikawa Y., Jarvis T.C., Carter J.D. et al. . Chemically modified DNA aptamers bind interleukin-6 with high affinity and inhibit signaling by blocking its interaction with interleukin-6 receptor. J. Biol. Chem. 2014; 289:8706–8719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Gawande B.N., Rohloff J.C., Carter J.D., von Carlowitz I., Zhang C., Schneider D.J., Janjic N.. Selection of DNA aptamers with two modified bases. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:2898–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Davies D.R., Gelinas A.D., Zhang C., Rohloff J.C., Carter J.D., O’Connell D., Waugh S.M., Wolk S.K., Mayfield W.S., Burgin A.B. et al. . Unique motifs and hydrophobic interactions shape the binding of modified DNA ligands to protein targets. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:19971–19976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mueller U., Förster R., Hellmig M., Huschmann F.U., Kastner A., Malecki P., Pühringer S., Röwer M., Sparta K., Steffien M. et al. . The macromolecular crystallography beamlines at BESSY II of the Helmholtz-Zentrum Berlin: Current status and perspectives. Eur. Phys. J. Plus. 2015; 130:141–150. [Google Scholar]
- 97. Cianci M., Bourenkov G., Pompidor G., Karpics I., Kallio J., Bento I., Roessle M., Cipriani F., Fiedler S., Schneider T.R.. P13, the EMBL macromolecular crystallography beamline at the low-emittance PETRA III ring for high- and low-energy phasing with variable beam focusing. J. Synchr. Rad. 2017; 24:323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers PDB IDs: 6EO6 (α-thrombin:T4W complex) and 6EO7 (α-thrombin:T4K complex).