

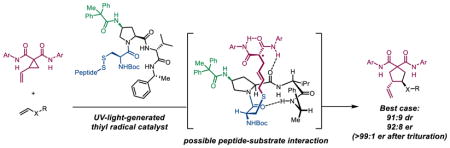
Abstract
An enantioselective vinylcyclopropane ring-opening/cycloaddition cascade. The active thiyl radical catalysts are generated in situ via UV light-promoted homolysis of cystine-based dimers. Amide-functionalization of the peptide at the 4-proline position is essential for effective asymmetric induction. Stereochemical communication is dependent on steric interactions with this substituent that are enforced by H-bonding to the peptide backbone.
Graphical Abstract
As a catalytic residue in enzymatic systems, cysteine mediates myriad transformations proceeding through both polar and radical mechanisms. Ubiquitous cysteine proteases and other enzymes crucial to DNA biosynthesis operate via two-electron, nucleophilic mechanisms (Scheme 1a).1 Equally essential are cysteine-mediated biochemical processes in which a sulfur-based radical is generated within the active site and directly participates in a chemical reaction, as in ribonucleotide reductase and pyruvate formate lyase (Scheme 1b).2
Scheme 1.

Polar and radical cysteine catalysts in biology and synthesis
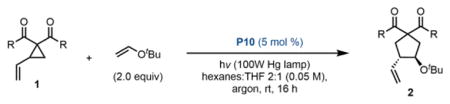
Our group has previously reported a cysteine catalyst for the polar Rauhut-Currier reaction,3 which has been suggested to have a biosynthetic counterpart (Scheme 1c).4 In addition, mimics of protein disulfide isomerase have been developed to catalyze disulfide bridge interchanges.5 However, unlike the polar chemistry of cysteine-based systems, single-electron chemistry with cysteine thiyl radicals has yet to be employed in organic synthesis. Inspired by the mechanistic versatility of cysteine in biological processes, and in line with our interest in the convergence of biological motifs with stereoselective catalysis, we explored the potential of cysteine-based thiyl radicals to catalyze asymmetric transformations. These studies culminate in the design of a cysteine thiyl radical catalyst embedded within a peptide framework. We demonstrate the effectiveness of these catalysts in an enantioselective vinylcyclopropane (VCP) ring-opening/cycloaddition cascade (Scheme 1d).
As a context to explore these ideas, we were inspired by the enantioselective, aryl thiyl radical-catalyzed VCP ring-opening/cycloaddition cascade recently disclosed by Maruoka and coworkers.6 This report, which constitutes the singular example of asymmetric catalysis of these processes, achieves asymmetric induction through a steric blocking strategy. In contrast, we speculated that enantiocontrol could be achieved through attractive noncovalent interactions provided by a peptidic scaffold. Thus, photolysis of a disulfide-bridged peptide pre-catalyst was projected to yield the derived alkyl thiyl radical; addition to the VCP substrate delivers the stabilized radical intermediate (I) (Scheme 1e). Olefin addition to I provides intermediate II, which can cyclize to the product and regenerate the active thiyl radical catalyst.
Our first challenge in implementing this proposal involved the in situ generation of cysteine thiyl radicals, which are less accessible than aryl thiyl radicals due to differences in bond dissociation energies.7 Alkyl thiyl radicals can be generated via H-atom transfer from thiols, but such an approach requires external initiators, oxidants, and/or bases, which can give rise to undesired side reactions. Alternatively, UV light-promoted homolysis of disulfide bonds can directly produce two equivalents of the corresponding thiyl radical.8 We chose to implement this strategy using cysteine-disulfide dimers of the active peptide catalysts, with continuous exposure of the reaction mixture to unfiltered UV light.9
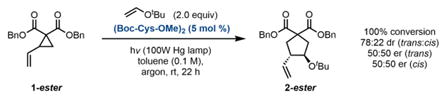
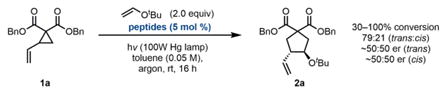
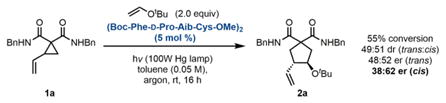
Under these conditions, excellent reactivity is observed using diester substrates of the type studied by Maruoka, with (Boc-Cys-OMe)2 as the catalyst (Eq. A). High levels of reactivity are maintained with the use of more complex peptides that were anticipated to provide some level of selectivity (Eq. B). However, nearly racemic product is obtained with these catalysts, indicating that the determinants of selectivity in Maruoka’s system are quite dissimilar from ours. To further take advantage of peptide-substrate H-bonding interactions, we converted to amide- functionalized VCP substrates (Eq. C). With this different substrate class, promising levels of enantioselectivity are obtained, indicating that the H-bond-donating ability of the substrate is important for achieving selectivity. To further take advantage of this presumed interaction, subsequent screens were performed on the more electron-deficient bis(amide) substrate 1b.
 |
(A) |
 |
(B) |
 |
(C) |
A screen of catalyst motifs that exhibit high enantioselectivity in other peptide-catalyzed reactions10 did not lead to a highly selective catalyst for this cascade reaction; representative peptides are shown in Scheme 2a. Modifications of these sequences at all positions, using natural or commercially available synthetic amino acids, afforded no significant improvement in enantioselectivity. These observations made clear that our previously developed catalyst scaffolds are not able to engage effectively with the amide substrate. We recognized that the reactive centers of the putative radical intermediates are likely distant from any chiral information in the catalyst, and hypothesized that additional functionality orthogonal to the peptide backbone would provide an expanded chiral pocket and enhance stereochemical communication (Scheme 2b).11 Incorporation of a substituent at the 4-position of the proline can achieve this extension of chiral information with minimal disruption to the secondary structure of the peptide catalyst (Scheme 2c). Indeed, this strategy was successful as phenylacetamide-bearing catalyst P3 yields both diastereomers with significantly improved enantioselectivity. A survey of other functional groups at this position revealed that amide substituents are the most promising for achieving high enantioselectivity (Scheme 2d). Extensive investigation to identify a suitable amide at the 4-proline position led us to catalyst P10, which bears a diphenylmethylacetamide substituent. Modifications of non-prolyl residues revealed that no other position impacts enantioselectivity as significantly as functionalization at the 4-proline position. Such changes, including to the C-terminal capping group and the N-terminal protecting group, did not provide any increase in selectivity.12
Scheme 2. Peptide optimization studiesa.

aReactions were performed on a 0.05 mmol scale and were quenched by exposure to air. Conversions, diastereomeric ratios, and enantiomeric excesses were determined by CSP-HPLC (254 nm, uncorrected). Ar = 4-CF3Ph.
Under our standard reaction conditions13 using catalyst P10, the diester substrates examined by Maruoka and coworkers still afford nearly racemic product (Table 1, entry 1). However, our amide-substituted VCPs, which can presumably interact with the peptide through H-bonding interactions, are more successful substrates for this transformation. Specifically, several electron-deficient amide substrates deliver the cycloaddition products with high enantioselectivity and yield (entries 3, 8–11). Further support for the proposed VCP–peptide H-bond is provided by the result obtained in methanol (entry 5). This reaction proceeds with significantly diminished enantioselectivity, potentially due to disruption of the H-bonding interaction. Consistent with our H-bonding hypothesis, electron-rich amide substrates furnish the products with marginally diminished enantioselectivity (entries 6 and 7). With this class of bis(amide) substrates, diastereoselectivity is unperturbed by the peptide and is intrinsic to each substrate. The trans diastereomer predominates and is consistently formed with higher enantioselectivity than the cis diastereomer.
Table 1.
Exploration of VCP substrate scope
| ||||||
|---|---|---|---|---|---|---|
| entrya | R = | solventb | yield (%)c | dr [trans:cis)d | er (trans)e | er (cis)e |
| 1f,g |
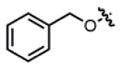 2-ester |
THF | 44 | 78:22 | 47:53 | ND |
| 2 |
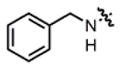 2a |
THF | 70 | 40:60 | 83:17 | 72:28 |
| 3 |
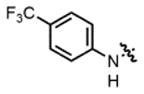 2b |
hex/THF | 84 | 79:21 | 90:10 | 75:25 |
| 4 | THF | 82 | 77:23 | 88:12 | 76:24 | |
| 5f | MeOH | 82 | 76:22 | 70:30 | 65:35 | |
| 6 |
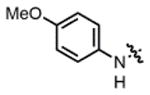 2c |
THF | 79 | 68:32 | 85:15 | 75:25 |
| 7 |
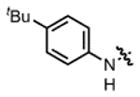 2d |
hex/THF | 32 | 68:32 | 86:14 | 75:25 |
| 8 |
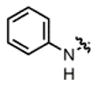 2e |
hex/THF | 77 | 73:27 | 90:10 | 79:21 |
| 9 |
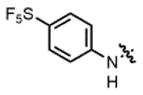 2f |
hex/THF | 83 | 80:20 | 90:10 | 76:24 |
| 10 |
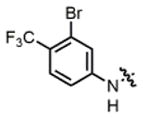 2g |
hex/THF | 81 | 82:18 | 90:10 | 73:27 |
| 11 |
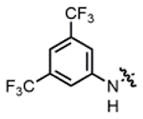 2h |
hex/THF | 94 | 82:18 | 90:10 | 73:27 |
Reactions were performed on a 0.1 mmol scale and were quenched by exposure to air. Reported data is the average of 2 separate experiments, unless noted.
Standard conditions: 2:1 hexanes:THF.
Isolated yield of a mixture of trans and cis diastereomers.
Determined by 1H NMR.
Determined by CSP-HPLC (254 nm, uncorrected).
Single experiment.
dr is crude before purification to remove cis, yield is pure trans.
Multiple olefins are successful olefin coupling partners, with an evident requirement for an electron-rich olefin bearing an α-heteroatom (Scheme 3). Other vinylogous functional groups, including styrenes, acrylamides, and unactivated olefins, are unreactive, likely due to polarity matching requirements between the electron-deficient α-dicarbonyl radical and the olefin.14 The system developed by Maruoka and coworkers also suffers from the same restriction. Diastereoselectivity remains intrinsic to each VCP/olefin combination, again favoring the trans diastereomer. The diminished enantioselectivity observed when linear n-propyl vinyl ether (3) is used suggests that enhanced bulk on the olefin partner is beneficial for stereochemical communication. This is also consistent with the results obtained from N-vinyl acetamide (5), in contrast to the bulkier vinyl carbamate (4). Olefins lacking an N–H bond furnish the corresponding products with higher enantioselectivity, suggesting that the free N–H bond may interfere with enantioinduction. Of these products, trans-7 could be enantioenriched through filtration to remove an insoluble product, which is assumed to be an aggregate of minor stereoisomers [enriched to 92:8 dr (trans:cis), >99:1 er (trans)].
Scheme 3. Breadth of olefin coupling partnersa.

aReactions were performed on a 0.1 mmol scale and were quenched by exposure to air. Isolated yield of a mixture of trans and cis diastereomers. Diastereomeric ratios determined by 1H NMR, enantiomeric ratios determined by CSP-HPLC (254 nm, uncorrected). Average of 2 separate experiments. Ar = 4-CF3Ph. c10 mol % catalyst. dDiastereomeric ratio reported after purification, crude dr 80:20 (trans:cis)
1,1-Disubstituted olefins are considerably less reactive, but afford similar enantiomeric excess as other olefins. The internal olefin of a cyclic vinyl carbamate also undergoes the transformation, delivering 5,5-bicyclic pyrrolidine (10). However, our optimal catalyst is evidently mismatched to the intrinsic diastereoselectivity of this olefin (>95:5 cis:trans with phenyl disulfide), and the product is instead obtained in lower dr, and the minor trans diastereomer formed with higher enantioselectivity. This is the only case in which diastereoselectivity is perturbed in the peptide-catalyzed reaction relative to the phenyl disulfide-catalyzed reaction.
Given the importance of the 4-proline substituent in achieving enantioinduction in this system and its uniqueness relative to our previously reported catalysts, we wanted to better understand its function within this peptide catalyst and its effect on enantio-induction. A brief study of electronically varied benzamide catalysts revealed that the 4-proline substituent appears to act neither as an H-bond donor nor H-bond acceptor (Table 2a). Electron-deficient amides provide the least enantioselective outcomes; however, other Lewis basic substituents, such as a urea (P6), are also poorly selective. However, the steric profile of this substituent is clearly critical to enantioinduction—a small acetamide catalyst (P15) is less selective than a bulkier pivalamide (P4) (Table 2b). Enantioselectivity generally improves with increased steric bulk, although excessive bulk is detrimental—after increasing the substituent size to an adamantyl group, enantioselectivity decreases (P17). Our attempts to find the optimal degree of bulk on this substituent led us to catalyst P10. Catalyst P18, which is as bulky as P4 but ostensibly a better H-bond donor, gives approximately the same results as catalyst P4, reinforcing our hypothesis that the H-bonding capability of the amide is secondary to its steric profile in terms of governing enantioinduction.
Table 2.
Catalyst structure-selectivity investigationa

|
Reactions were performed on a 0.05 mmol scale and were quenched by exposure to air. Conversions, diastereomeric ratios, and enantiomeric ratios were determined by CSP-HPLC (254 nm, uncorrected). Ar = 4-CF3Ph.
In our catalyst optimization studies, we observed that changes to other parts of the catalyst have minimal impact on enantioselectivity. This prompted us to examine whether the catalyst could be abbreviated at the proline residue and still perform effectively (Table 2c). Enantioselectivity is largely retained with these catalysts when the proline residue is capped as an N,N-disubstituted amide (P19); a larger decrease is observed when the amide bears a free N–H (P20). The observation that enhanced Lewis basicity at this position is beneficial, combined with the results obtained with steric perturbations, leads us to advance a hypothetical stereo-chemical model as shown in Table 4d. We propose that the VCP substrate engages with the peptide backbone through an H-bonding interaction with the proline amide, with a chiral pocket defined by the steric profile of the 4-proline substituent. The absolute configuration of 2g-trans was determined by X-ray crystallography15 and is consistent with orientation of the tert-butyl moiety away from the 4-proline substituent, further supporting a steric role. Studies toward a mechanistic understanding of the effect of this substituent on the enantioselectivity-determining step are underway.
The VCP-ring opening cascade described herein represents a new mode of peptide-based catalysis, in which asymmetric induction on reactive and dynamic radical intermediates using flexible catalysts can be achieved. Furthermore, this class of catalyst is newly developed and may have broad application in other thiyl radical- and other peptide-catalyzed reactions. The reactivity of cysteine-derived thiyl radicals with VCPs demonstrated herein may not necessarily be biologically relevant. However, given the extant roles for thiyl radicals in biological systems, perhaps biochemical discovery of related carbon–carbon bond-forming processes may yet emerge.
Supplementary Material
Acknowledgments
This work is supported by the National Institute of General Medical Sciences of the United States National Institutes of Health (R01-GM096403). J.M.R. acknowledges the Department of Defense (DoD) for funding through the National Defense Science & Engineering Graduate Fellowship (NDSEG) Program. We also thank Dr. Brandon Q. Mercado for X-ray crystallographic analyses and Dr. Eric Paulson for NMR assistance.
Footnotes
Notes
The authors declare no competing financial interests.
The Supporting Information is available free of charge on the ACS Publications website. Experimental methods, characterization of all compounds, and copies of spectra can be found in the Supporting Information.
References
- 1.(a) Otto HH, Schirmeister T. Chem Rev. 1997;97:133–171. doi: 10.1021/cr950025u. [DOI] [PubMed] [Google Scholar]; (b) Carreras CW, Santi DV. Annu Rev Biochem. 1995;64:721–762. doi: 10.1146/annurev.bi.64.070195.003445. [DOI] [PubMed] [Google Scholar]
- 2.(a) Stubbe J, van der Donk WA. Chem Rev. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]; (b) Licht S, Gerfen GJ, Stubbe J. Science. 1996;271:477–481. doi: 10.1126/science.271.5248.477. [DOI] [PubMed] [Google Scholar]; (c) Knappe J, Elbert S, Frey M, Wagner AFV. Biochem Soc T. 1993;21:731–734. doi: 10.1042/bst0210731. [DOI] [PubMed] [Google Scholar]
- 3.(a) Aroyan CE, Miller SJ. J Am Chem Soc. 2007;129:256–257. doi: 10.1021/ja067139f. [DOI] [PubMed] [Google Scholar]; (b) Aroyan CE, Dermenci A, Miller SJ. J Org Chem. 2010;75:5784–5796. doi: 10.1021/jo101018t. [DOI] [PubMed] [Google Scholar]; (c) Dermenci A, Selig PS, Domaoal RA, Spasov KA, Anderson KS, Miller SJ. Chem Sci. 2011;2:1568–1572. doi: 10.1039/C1SC00221J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim HJ, Ruszczycky MW, Choi S-h, Liu Y-n, Liu H-w. Nature. 2011;473:109–112. doi: 10.1038/nature09981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kersteen EA, Barrows SR, Raines RT. Biochemistry. 2005;44:12168–12178. doi: 10.1021/bi0507985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hashimoto T, Kawamata Y, Maruoka K. Nature Chem. 2014;6:702–705. doi: 10.1038/nchem.1998. [DOI] [PubMed] [Google Scholar]
- 7.Dénès F, Pichowicz M, Povie G, Renaud P. Chem Rev. 2014;114:2587–2693. doi: 10.1021/cr400441m. [DOI] [PubMed] [Google Scholar]
- 8.Singleton DA, Church KM. J Org Chem. 1990;55:4780. [Google Scholar]
- 9.Activation with a 365 nm-filtered UV lamp provided appreciable but incomplete conversion, possibly due to poor overlap of the alkyl disulfide bond absorbance. UV-vis spectra of (Boc-Cys-OH)2 and catalyst P10 are provided in the supporting information.
- 10.For a detailed study of peptide structure, see Metrano AJ, Abascal NC, Mercado BQ, Paulson EK, Hurtley AE, Miller SJ. J Am Chem Soc. 2017;139:492–516. doi: 10.1021/jacs.6b11348.
- 11.Peptide model is based on the three-dimensional structure elucidated in: Jakobsche CE, Peris G, Miller SJ. Angew Chem Int Ed Engl. 2008;47:6707. doi: 10.1002/anie.200802223.
- 12.Additional optimization data can be found in the supporting information.
- 13.While the reaction proceeds in a variety of solvents (EtOAc, PhMe, MeOH, MEK), non-polar, ethereal solvent mixtures provided the highest reactivity and enantioselectivity.
- 14.Roberts BP. Chem Soc Rev. 1999;28:25–35. [Google Scholar]
- 15.The x-ray crystallographic data is presented in the supporting information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.