

Abstract
Transforming growth factor-β (TGF-β) is a potent inhibitor of growth and proliferation of breast epithelial cells, and loss of sensitivity to its effects has been associated with malignant transformation and tumorigenesis. The biological effects of TGF-β are mediated by the TGF-β receptor complex, a multimer composed of TGF-β receptor type I (TβR-I) and TGF-β receptor type II (TβR-II) subunits. Evidence suggests that loss of expression of TβR-II is implicated in the loss of sensitivity of tumorigenic breast cell lines to TGF-β-mediated growth inhibition. A panel of human breast cell lines, including the immortalized MCF-10F and tumorigenic MCF-7, ZR75-1, BT474, T47-D, MDA-MB231, BT20, and SKBR-3 cell lines, was characterized for responsiveness to TGF-β-induced G1 growth arrest. Only the nontumorigenic MCF-10F and the tumorigenic MDA-MB231 cell lines demonstrated a significant inhibitory response to TGF-β1 and a significant binding of 125I-labeled TGF-β ligand. While expression of TβR-I mRNA was similar across the panel of cell lines, TβR-II mRNA expression was decreased significantly in all seven tumorigenic cell lines in comparison with the nontumorigenic MCF-10F cell line. When total cellular protein was fractionated by centrifugation, TβR-I protein was observed in both the cytosolic and membrane fractions at similar levels in all cell lines; however, TβR-II protein was present in the cytosolic fraction in all cell lines, but was observed in the membrane fraction of only the TGF-β-responsive MCF-10F and MDA-MB231 cells. Thus, lack of membrane-bound TβR-II protein appears to be an important determinant of resistance to TGF-β-mediated growth inhibition in this group of breast cell lines.
Keywords: Breast cancer, Transforming growth factor-β, Transforming growth factor-β receptors, Growth inhibition
BREAST cancer is the second most common malignancy in women in the United States (10). In 2000, an estimated 175,000 new cases of invasive breast cancer and 40,000 additional cases of in situ breast cancer will be diagnosed, and 43,300 women will die of the disease (10). Known risk factors for the development of breast cancer implicate lifetime estrogen exposure in breast cancer etiology (13); estrogens regulate breast epithelial cell growth, stimulating proliferation of both normal and breast tumor cells (12). However, other locally acting growth factors are also important regulators of breast epithelial cell growth and differentiation. The role of these growth factors in breast carcinogenesis, individually and in relationship to estrogen, remains an important area of research.
Transforming growth factor-β (TGF-β) is a family of multifunctional polypeptides that serve to promote differentiation and to inhibit growth and proliferation of most epithelial cell types in vitro as well as in vivo. TGF-βs are involved in the regulation of tissue morphogenesis, the production of extracellular matrix, and in the promotion of angiogenesis (32). Recent data suggest that TGF-βs also function in the cell microenvironment to suppress immune function, including natural killer cell function (3). Three homologous mammalian TGF-β isoforms have been identified, TGF-β1, -β2, and -β3, with similar biologic effects in vitro, although their differential expression in human breast cancer cell lines and tumor tissues is complex and not fully understood (22). Studies of TGF-β effects on normal and transformed breast epithelial cells in vitro provide strong evidence for its role as an inhibitor of breast epithelial cell growth (21). Initial in vitro studies of transformed breast epithelial cell lines suggest that estrogen may function as a negative regulator of TGF-β expression in estrogen responsive cell lines (16), while antiestrogens, including tamoxifen, effectively block estrogen-induced downregulation of TGF-β expression, resulting in increased cellular TGF-β production and induction of G1 cell cycle arrest (41).
The biological effects of TGF-β are mediated by the TGF-β receptor complex, which is composed of two essential independent and interacting subunits, TGF-β receptor type I (TβR-I) and TGF-β receptor type II (TβR-II), and an associated TGF-β receptor type III (TβR-III) (54). TβR-I and TβR-II are membrane-bound serine/threonine kinases, with an extracellular cysteine-rich ligand binding domain, a hydrophobic transmembrane domain, and an intracellular kinase domain (31). Ligand binding to TβR-II results in the recruitment and phosphorylation of TβR-I and the formation of the heteromeric signaling complex (5). Intracellular signaling from the TGF-β receptor complex is mediated by the SMAD family of proteins. Formation of the activated heteromeric complex initiates the recruitment and phosphorylation of the cytoplasmic signaling proteins SMAD2 or SMAD3, formation of the SMAD2/SMAD4 or SMAD3/SMAD4 heterodimer, and the subsequent translocation of the SMAD heterodimer to the cell nucleus (11). Within the cell nucleus, formation of the SMAD2/SMAD4/FAST-1 transcription factor complex induces transcription of key genes involved in TGF-β-mediated G1 cell cycle arrest, including the cyclin-dependent kinase inhibitors p15 and p21 (Fig. 1) (25).
Figure 1.
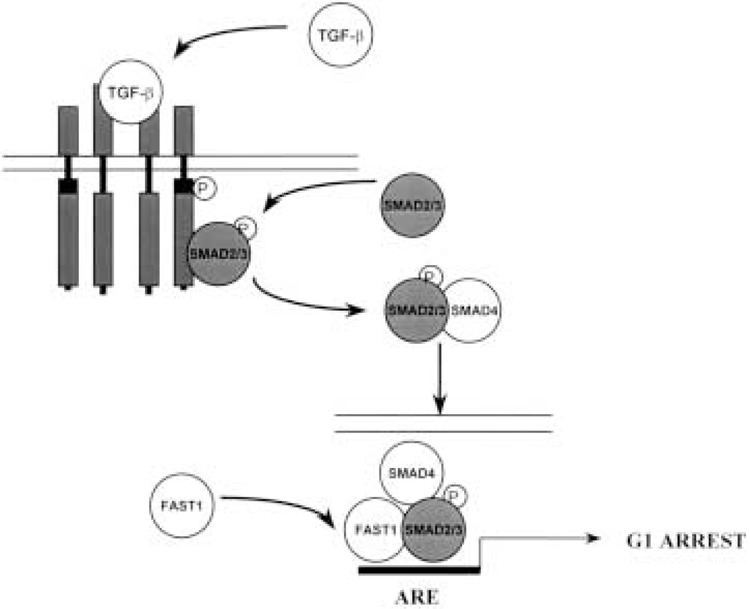
Heteromeric model of the TGF-β receptor signal transduction pathway. The TGF-β receptor complex is composed of two essential independent and interacting subunits, TβR-I and TβR-II, and an associated TβR-III. TβR-I and TβR-II are membrane-bound serine/threonine kinases, with an extracellular cysteine-rich ligand binding domain, a hydrophobic transmembrane domain, and an intracellular kinase domain. Ligand binding to TβR-II results in the recruitment and phosphorylation of TβR-I and the formation of the heteromeric signaling complex. Intracellular signaling is mediated by the SMAD family of proteins. Formation of the activated TGF-β receptor complex initiates the recruitment and phosphorylation of the cytoplasmic signaling proteins SMAD2 or SMAD3, formation of the SMAD2/SMAD4, or SMAD3/ SMAD4 heterodimer and the subsequent translocation of the SMAD heterodimer to the cell nucleus. Within the cell nucleus, formation of the SMAD2/SMAD4/FAST-1 transcription factor complex induces transcription of key genes involved in TGF-β-mediated G1 cell cycle arrest.
Loss of sensitivity to TGF-β-mediated inhibition of epithelial cell proliferation has been implicated in malignant transformation of several human epithelial cell types (9). Resistance to the growth inhibitory effects of TGF-β has been attributed to loss of expression of TβR-II in several human tumor cell lines including retinoblastoma (20), small-cell lung cancer (36), hepatoma (14), gastric cancer (39), esophageal cancer (38), breast cancer (2,17), and colon cancer (28). Studies in human colon cancer cell lines with replication error defects and an associated high incidence of genomic microsatellite instability have demonstrated mutations in TβR-II, resulting in loss of expression of TβR-II message and protein and subsequent resistance to TGF-β growth inhibition (29,51). In the MCF-7 breast cancer cell line, lack of binding of TGF-β ligand and loss of expression of TβR-II message correlated with resistance to TGF-β growth inhibition, suggesting that loss of TβR-II expression occurs at the transcriptional level. This finding was further supported by reintroduction of sensitivity to TGF-β upon stable transfection of a TβR-II message expression vector (48). A subsequent study in MCF-7 breast cells found TβR-II message to be significantly reduced in MCF-7 cells, but TβR-II protein to be present, although only in the cell cytoplasm (23). This study raises the question of posttranslational defects in trafficking of TβR-II to the cell membrane as a mechanism of resistance. These in vitro studies demonstrate that loss of expression of TβR-II is a key mechanism by which transformed cells develop resistance to TGF-β-mediated growth inhibition. The mechanisms by which tumor cells achieve independence from TGF-β-mediated growth inhibition range from mutations in TβR-II, transcriptional or posttranscriptional events leading to decreased levels of TβR-II message, or posttranslational defects in trafficking of TβR-II protein to the cell membrane.
In order to further elucidate the mechanisms by which breast cancer cells develop resistance to TGF-β growth inhibition, we have characterized the responsiveness of a panel of breast cell lines, both estrogen responsive-estrogen receptor positive (ER+) and estrogen receptor negative (ER−), to the growth inhibitory effects of TGF-β1. Next, we screened these breast cell lines for mutations in TβR-I and TβR-II. We then correlated TGF-β responsiveness to expression of TβR-I and TβR-II message and protein. Our data suggest that lack of membrane-bound TβR-II protein is the key determinant in the development of resistance to TGF-β-mediated growth inhibition in this panel of breast cancer cell lines.
MATERIALS AND METHODS
Cell Culture
The Mv1Lu and MCF-10F cell lines were obtained from the American Type Culture Collection (Rockville, MD). Mv1Lu cells, derived from mink lung epithelium, are well-characterized TGF-β1-responsive cells (26). Mv1Lu cells were used as a positive control for growth studies. The MCF-10F cell line was derived from normal breast epithelium and is nontumorigenic in nude mice (46). The estrogen receptor-positive and estrogen-responsive MCF-7, ZR75-1, BT474, and T47-D cell lines, as well as the estrogen receptor-negative MDA-MB231, BT20, and SKBR-3 cell lines, were obtained from frozen stocks of the laboratory of R. Brueggemeier. All cell lines, except for MCF-10F, were cultured in Media B (Gibco BRL, Gaithersburg, MD), a modified Eagle’s medium with Earle’s salts, 1.5× essential amino acids, 2× nonessential amino acids, and 1.5× vitamins without sodium bicarbonate and without phenol red, supplemented with 10% FBS (Gibco BRL), 2 mM l-glutamine (Gibco BRL), and 20 mg/L gentamycin (Gibco BRL). MCF-10F cells were cultured in DMEM/F12 supplemented with 5% horse serum treated with Chelex 100 (BioRad, Hercules, CA) to remove all calcium, 0.04 mM CaCl2, 20 ng/ml epidermal growth factor, 100 ng/ml cholera toxin, 10 μg/ ml bovine insulin, and 50 ng/ml hydrocortisone (Sigma, St. Louis, MO). Defined medium (DM) used for the cell proliferation assay consisted of Media B supplemented with 2 mM l-glutamine and ITS+ premix (Collaborative Biomedical Products, Bedford, MA) for all cell lines except MCF-10F. DM for the MCF-10F cells also contained 20 ng/ml epidermal growth factor, 100 ng/ml cholera toxin, and 50 ng/ml hydrocortisone. Cells were cultured in 5% CO2 in a humidified incubator at 37°C.
Cell Proliferation Assay
Effects on cell proliferation, in response to TGF-β1 treatment, were quantified by measuring BrdU incorporation into synthesizing cellular DNA. Approximately 0.5–1.0 × 104 cells were plated per well and six wells were plated for each observation in a 96-well multiwell plate (MTP) in DM and incubated for 24 h. DM was removed and cells were treated with TGF-β1 in DM (R&D Systems, Minneapolis, MN) at concentrations of 0 (control), 0.01, 0.10, and 1.0 nM, for 12 h. Measurement of BrdU incorporation was performed using the Cell Proliferation ELISA Colorimetric assay (Boehringer Mannheim, Germany). Control and treated cells were incubated with 10 μM BrdU for 6 h, fixed and denatured, then labeled with mouse monoclonal anti-BrdU peroxidase-conjugated antibody for 90 min. BrdU–antibody complexes were detected by reaction of the conjugate with tetramethylbenzidine substrate. The reaction product was quantified by measuring absorbance at 372 nm using a scanning multiwell spectrophotometer (Molecular Devices, Sunnyvale, CA). Each data point is the average OD for six observations. Data were analyzed using Excel (Microsoft, Redmond, WA) and GraphPad (GraphPad Software Incorporated).
[125I]TGF-β Labeling of Breast Cells Cultures
Binding of 125I-labeled TGF-β1 to Mv1Lu control and each breast cell line was determined using a standard protocol (30). Cell monolayers, plated in six-well tissue culture plates and grown to subconfluence in their standard growth media, were washed in cold binding buffer (50 mM HEPES, pH 7.5, 128 mM NaCl, 1.2 mM CaCl2, 5 mM MgSO4, 5 mM KCl, 0.2% FA-free BSA), equilibrated in binding buffer at 4°C, and labeled with 5 ng (154 μCi/μg) [125I]TGF-β1 or 5 ng [125I]TGF-β1 with 250 ng unlabeled TGF-β1 in 5 ml cold binding buffer and incubated at 4°C for 4 h. Labeling media were removed and cells washed with cold binding buffer. Bound labeled ligand was crosslinked to the cell surface by incubating monolayers with 25 μl 27 mM DSS (Pierce, Rockford, IL) in 5 ml binding buffer without BSA with agitation at 4°C for 15 min. Cells were washed, scraped into detachment buffer (0.25 M sucrose, 10 mM Tris, pH 7.4, 1 mM EDTA, 0.3 mM PMSF), pelleted, and lysed in cell fractionation buffer with 0.8% Triton X-100. Cell lysates were cleared by centrifugation at 12,000 rpm for 15 min and analyzed by SDS-PAGE electrophoresis through 0.8% Tris-Glycine precast gels (Novex, San Diego, CA). Gels were then dried and developed by autoradiography (Amersham, Chicago, IL).
RNA and DNA Isolation
Cells cultured in 100-mm plates to subconfluence were scraped into 1 ml Trizol reagent per plate and RNA was isolated according to the manufacturer’s protocol. All samples were analyzed for integrity of 18S and 28S rRNA by ethidium bromide staining of 1% agarose/formaldehyde gels. DNA was isolated using a modification of the manufacturer’s protocol developed in our laboratory (D. Ramljak). Briefly, DNA was precipitated from the Trizol organic phase and interphase with 50 mM Tris (pH 7.4), 1 mM EDTA, washed twice with CHCl3 and 70% ethanol, and the associated protein digested in a buffer containing 50 mM Tris, 5 mM CaCl2, 10% SDS, and 10 mg/ml proteinase K with incubation at 50°C overnight. DNA was extracted 1× with phenol/CHCl3/isoamyl alcohol (25:24:1) and 2× with CHCl3/isoamyl alcohol (24:1) and precipitated with 7.5 M ammonium acetate.
Semiquantitative RT-PCR
Reverse transcription was performed using 2 μg total RNA in a final reaction volume of 20 μl containing 1× PCR buffer (200 mM Tris-HCl, pH 8.4, 500 mM KCl), 5 mM MgCl2, 1 mM each dNTP, 100 pmol random primers, 1 U/μl RNAsin (Clontech, Palo Alto, CA), and 2.5 U/μl Superscript Reverse Transcriptase (Gibco BRL). RNA in DEPC H2O was preincubated at 60°C for 5 min and the reaction mix was added. Reverse transcription was performed at 25°C for 10 min and 37°C for 60 min, followed by 99°C for 5 min to denature the enzymes and terminate the reaction. PCR was performed with 1 μl cDNA in a reaction volume of 25 μl containing 1× PCR buffer (200 mM Tris-HCl, pH 8.4, 500 mM KCl), 2 mM MgCl2, 0.2 mM each dNTP, 0.2 μM each PCR primer, and 1.25 μl Taq DNA polymerase (Gibco BRL) incubated with TaqStart Antibody (Clontech). Primers for PCR were designed according to GenBank Accession Numbers: L11695 (TβR-I), L07594 (TβR-II), and M26434 (HPRT). Optimal annealing temperature (T a) and the linear range of amplification was determined empirically for each set of PCR primers. Primers used for semiquantitative RT-PCR are shown in Table 1.
TABLE 1.
PRIMERS USED FOR SEMIQUANTITATIVE RT-PCR
| mRNA | Primer Sequence | T a | Cycle Number |
|---|---|---|---|
| TβR-I | 5′-GGAACTGGCAGCTGTCATTG 5′-TTCTTCTCCCCGCCACTTTC |
61°C | 28 |
| TβR-II | 5′-GGTCAGAAGTCGGTTAATAA 5′-TGCACTCATCAGAGCTACAG |
58°C | 28 |
| HPRT | 5′-GTAATGACCAGTCAACAGGGGAC 5′-CCAGCAAGCTTGCGACCTTGACCA |
60°C | 28 |
PCR products were analyzed on precast 10% polyacrylamide gels (Novex) and stained with ethidium bromide. The intensity of staining of PCR fragments was determined using a digital imaging system (Alpha Innotech Co., San Leandro, CA). The ratio of TβR-I and TβR-II to HPRT mRNA expression was determined for each cell line.
RβR-I and TβR-II DNA Sequence Analysis
TβR-I and TβR-II exons were amplified using intron-based primers. TβR-I primers were designed according to genomic structure and intron sequence determined by intron capture PCR experiments performed in this laboratory (data unpublished) and reported to GenBank, accession numbers AF035662–AF035670. TβR-I exon 1 primers were generously provided by M. Reiss (Yale University). TβR-II primers were designed according to GenBank accession numbers U37070, U52240-U52246. PCR was performed with 2 μg DNA in a reaction volume of 50 μl containing 1× PCR buffer (200 mM Tris-HCl, pH 8.4, 500 mM KCl), 2 mM MgCl2, 0.2 mM each dNTP, 0.2 μM each PCR primer, and 1.25 μl Taq DNA polymerase (Gibco BRL) incubated with Taq-Start Antibody (Clontech). Optimal primer annealing temperature (T a) was determined empirically for each set of PCR primers. PCR products were purified and directly sequenced using the Perkin Elmer ABI377 Prism Automated DNA Sequencer. TβR-I and TβR-II primers are shown in Table 2.
TABLE 2.
TβR-I AND TβR-II PRIMERS
| Exon | Primers | Size | T a |
|---|---|---|---|
| TβR-I | |||
| 1 | 5′-GAG GCG AGG TTT GCT GGG GTG AGG CA 5′-CAT GTT TGA GAA AGA GCA GGA GCG AG |
244 bp | 55°C |
| 2 | 5′-CTG TTA ACC TTG AGA TTT TT 5′-ATG AAG AGT TTT TCT TGT AG |
344 bp | 53°C |
| 3 | 5′-TGT CGT TGT TGA TGT TTA TT 5′-AGC AAG TTG GGT TAT TAG AA |
361 bp | 56°C |
| 4 | 5′-ATA TTG TTG ATT GTG TTG AG 5′-CTG TAA AGA CTT AAA GAG AT |
333 bp | 56°C |
| 5 | 5′-ATG CAG CCC AAC CGA AAT GT 5′-CTC AGC CTC CCA AAG TGA TG |
281 bp | 53°C |
| 6 | 5′-TGT GAG TTG TGA TTG GTA TT 5′-TAT GAA AGA GAA GGG AAA AA |
224 bp | 50°C |
| 7 | 5′-AAA GGA GGT TCA TCC AAA TA 5′-CAA CTT CTG CTC ATG ACA AA |
241 bp | 56°C |
| 8 | 5′-CTC TGT TCC ACA TAC CTA CT 5′-AAT TGC CTA ATA TCA AAA AG |
283 bp | 52°C |
| 9 | 5′-TAT CCA GAC CAA TGG AAA AT 5′-GGA GCA GAT CTG AAG AAA AA |
231 bp | 56°C |
| TβR-II | |||
| 1 | 5′-TTG CGA GCG GGC GCC ACA TC 5′-GGA CCA CTC ACC CGA CTT CT |
266 bp | 62°C |
| 2 | 5′-AAA TTG CAT AAC ATC TTC AG 5′-CAC TGA CTG TGT GTA CTA TG |
330 bp | 58°C |
| 3 | 5′-CCA ATG AAT CTC TTC ACT CT 5′-TCA GGT CCC ACA CCC TTT AG |
247 bp | 62°C |
| 4a | 5′-CCT TCT CTC CTT GTT TTG TT 5′-GCT CTG TGT TGT GGT TGA TG |
297 bp | 62°C |
| 4b | 5′-AGC GAG CAC TGT GCC ATC AC 5′-TGT CTT CCA AGA GGC ATA CT |
231 bp | 61°C |
| 4c | 5′-AGT CAA GAT CTT TCC CTA TG 5′-CAC TGT GGA GGT GAG CAA TC |
266 bp | 61°C |
| 4d | 5′-ACG CCA AGG GCA ACC TAC AG 5′-TTC CCA GGC TCA AGG TAA AG |
318 bp | 62°C |
| 5 | 5′-GGC CTC ACT GTC TGT TTT TG 5′-TCC ACA CCT ACC TCC CAC TG |
179 bp | 62°C |
| 6 | 5′-GGC TGC ACA TGC CAT TCT CA 5′-GGG ACC TTC CCT CAN ATT TA |
281 bp | 61°C |
| 7 | 5′-TGT CCC TTT GGA TCT CTT TC 5′-GGG GCA GCT TCC TGC TCT CT |
284 bp | 58°C |
Protein Isolation
Cells that were grown on three 100-mm culture dishes were washed with cold PBS, scraped into 1 ml cold PBS, pelleted at 2000 × g, resuspended in cold fractionation buffer (20 mM Tris HCl, pH 7.4, 2 mM EDTA, 25 mM NaF, 1 mM DTT, 2 mM NaMO4, 2 mM NaVO4, 1 μ/ml aprotinin, and 10 mM PMSF), and homogenized by passing through a 26-gauge needle. Of the total cell homogenate, 250 μl of total lysate was aliquotted and brought to a final Triton X-100 concentration of 0.8% and 750 μl was centrifuged at 100,000 × g for 1 h at 4°C. The cytosolic fraction was decanted and brought to a final concentration of Triton X-100 of 0.8% and the pellet was solubilized in cold fractionation buffer with 0.8% Triton X-100, incubated on ice for 1 h, vortexed repeatedly, and centrifuged at 12,000 × g for 15 min to remove insoluble cell debris.
Western Blotting
Aliquots of protein (10 μg) were heated to 100°C in Tris-Glycine SDS sample buffer (63 mM Tris HCl, pH 7.4, 10% glycerol, 2% SDS, 0.0025% bromphenol blue) (Novex) and 5% β-mercaptoethanol (Sigma) and separated by SDS-PAGE electrophoresis on precast 8% Tris-Glycine gels (Novex) and transferred to Immobilon P membranes (Millipore, Bedford, MA). Membranes were stained with Ponceau stain (Sigma) to ensure homogenous transfer of proteins and to allow for accurate marking of the transferred 10-κDa ladder (Gibco BRL) for estimation of protein molecular weight. Ponceau-stained membranes were washed 3 in PBS-T (0.1% Tween 20 in PBS) and were blocked with 5% nonfat dry milk in PBS-T for 1 h at room temperature. Blocked membranes were incubated with TβR-I (V-22) or TβR-II (L-21) antibodies (Santa Cruz Biotechnology, Palo Alto, CA) at 400 ng/ml in PBS-T overnight at 4°C. Blocking peptide controls using SC-398 (TβR-I, V-22 antibody) and SC-400 (TβR-II, L-21 antibody) to demonstrate specificity of antibody binding were also performed. Membranes were then washed 3 in PBS-T. Bound antibodies were detected by incubation with peroxidase-conjugated anti-rabbit immunogloblins (Amersham, Arlington Heights, IL) at 1:5000 dilution in PBS-T for 1 h at room temperature, washing 3 in PBS-T, and enhanced chemiluminscent autoradiography (ECL: Amersham).
RESULTS
Resistance of Tumorigenic Breast Cell Lines to TGF-β-Induced Cell Cycle Arrest
Sensitivity and resistance of breast cell lines to TGF-β1-mediated cell cycle arrest have been reported in previous studies (4,33,55). Results have varied from study to study, based upon passage number of the cell line and culture conditions unique to each laboratory. Due to this variability in published data, we undertook a determination of the responsiveness of eight breast cell lines to TGF-β1-mediated growth inhibition. Using incorporation of BrdU as a marker of DNA synthesis, we found that TGF-β1 had no inhibitory effect on DNA synthesis in subconfluent cultures of four of the tumorigenic cell lines, ZR75-1, BT474, T47-D, and SKBR-3 (Fig. 2) and only a minimal inhibitory effect on the MCF-7 and BT20 cell lines at the highest concentration of 1.0 nM. TGF-β1 significantly inhibited DNA synthesis in subconfluent cultures of Mv1Lu (positive control) and the nontumorigenic MCF-10F cell lines in a dose-dependent manner, whereas the MDA-MB231 cells demonstrated an intermediate sensitivity to TGF-β1, with 75% growth inhibition at 10 pM and greater than 50% growth inhibition at 1.0 nM.
Figure 2.
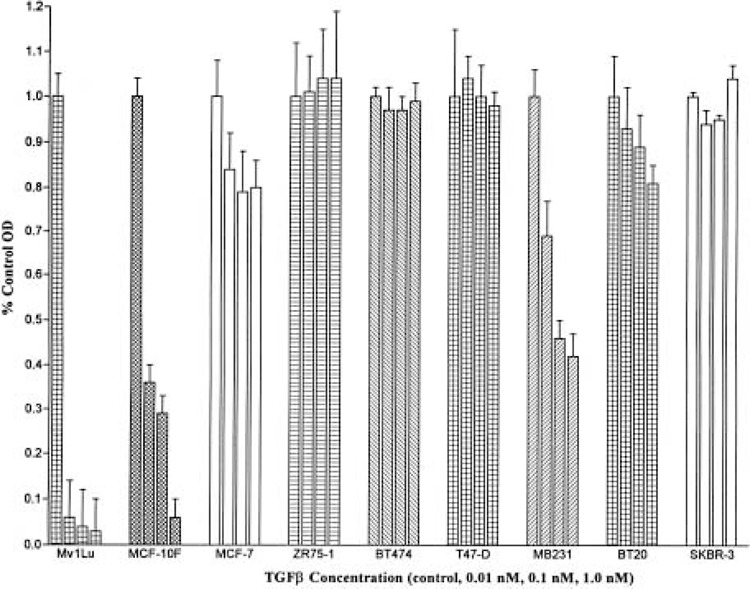
Responsiveness of human breast cell lines to TGF-β1-mediated growth inhibition. Cell DNA synthesis in response to TGF-β was quantified by measuring BrdU incorporation into synthesizing cellular DNA. Cells (0.5–1.0 × 104) cells were plated per well and six wells were plated for each observation in a 96-well multiwell plate (MTP) in defined media and incubated for 24 h. Cells were treated with TGF-β at concentrations of 0 (control), 0.01, 0.10, and 1.0 nM for 12 h. Measurement of BrdU incorporation was performed using the Cell Proliferation ELISA, BrdU, Colorimetric assay (Boehringer Mannheim, Germany). Control and treated cells were incubated with BrdU, fixed and denatured, and labeled with mouse monoclonal anti-BrdU peroxidase-conjugated antibody. BrdU–antibody complexes were detected with substrate and the reaction product was quantified by measuring absorbance at 372 nm using a scanning multiwell spectrophotometer. Each data point is the average OD for six observations. Data were analyzed using Excel and GraphPad and are expressed as percent control OD.
Binding of 125I-Labeled Ligand to TGF-β-Responsive Breast Cell Lines
Previous studies have demonstrated that a lack of responsiveness of tumorigenic breast cell lines to TGF-β1 is associated with reduced membrane binding of TGF-β1 (17). To determine if our observed unresponsiveness to TGF-β1 followed a similar pattern, binding of 125I-labeled TGF-β1 to each of the cell lines was assayed. Only the TGF-β1-responsive Mv1Lu, MCF-10F, and MDA-MB231 cell lines demonstrated a significant binding of [125I]TGF-β1 to TβR-I and TβR-II (Fig. 3). This binding was successfully competed against by unlabeled TGF-β1 (data not shown). The TβR-II receptor ligand complex was revealed at ∼95 kDa and TβR-I at ∼68 kDa, which have been previously identified as the appropriate sizes for the TBR-II–ligand complex and TβR-I (29). Only those cell lines, Mv1Lu, MDF-10F, and MDA-MB231, that demonstrated a significant growth inhibitory response to TGF-β1 showed significant binding of 125I-labeled TGF-β1 to TβR-II and associated TβR-I. Thus, lack of significant membrane binding of 125I-labeled TGF-β1 under these culture conditions correlated with the observed lack of responsiveness of the MCF-7, ZR75-1, BT474, T47-D, BT20, and SKBR-3 tumorigenic cell lines to TGF-β1-mediated growth inhibition.
Figure 3.
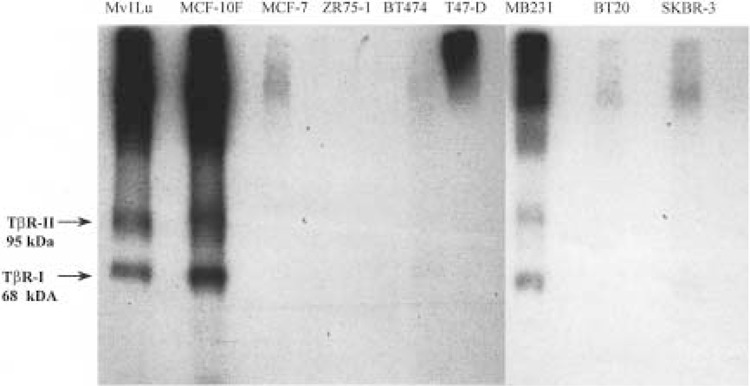
125I-labeled TGF-β1 binding to human breast cells. Binding of 125I-labeled TGF-β1 to MvlLu control and each breast cell line was determined in cell monolayers, plated in six-well tissue culture plates. Cells were labeled with 5 ng (154 μCi/μg) [125I]TGF-β1 or 5 ng [125I]TGF-β1 with 250 ng unlabeled TGF-β1. Bound labeled ligand was crosslinked to the cell surface. Cells were lysed and lysates were cleared by centrifugation and analyzed by SDS-PAGE electrophoresis and autoradiography. Radioactive labeling of TβR-I at 68 kDa and the TβR-II–ligand complex at 95 kDa was observed in the TGF-β-sensitive Mv1Lu (positive control), MCF-10F (nontumorigenic breast), and MDA-MB231 (tumorigenic breast) cell lines.
Mutation Assays for TβR-I and TβR-II in Human Breast Cell Lines
Mutations in TβR-I and TβR-II have been associated with loss of expression of functional TβR-I or TβR-II protein and resulting resistance to TGF-β1-mediated growth inhibition (37). Each exon of TβR-I and TβR-II was PCR amplified from high-quality DNA isolated from each of the cell lines using intron-based primers. PCR fragments containing each exon were purified and sequenced to determine if mutations in the coding region of TβR-I or TβR-II were present. The only coding change identified was at codon 35 in exon 2 of the BT474 cell line where an ATG→ATC change is associated with an isoleucine to methionine amino acid substitution (Table 3). The significance of this change is unclear. No other mutations in the coding regions of TβR-I and TβR-II were identified. Thus, mutational inactivation of TβR-I and TβR-II does not appear to contribute to the apparent loss of binding of TGF-β1 ligand and lack of response to TGF-β1-mediated growth inhibition in the breast cancer cell lines.
TABLE 3.
SEQUENCE ANALYSIS OF TβR-I AND TβR-II IN HUMAN BREAST CELL LINES*
| Cell Line | TβR-I Sequence | TβR-II Sequence |
|---|---|---|
| MCF-10F | wild type | wild type |
| MCF-7 | wild type | wild type |
| ZR75-1 | wild type | wild type |
| BT474 | wild type | codon 35:ATG→ATC† |
| T47-D | wild type | wild type |
| MDA-MB231 | wild type | wild type |
| BT20 | wild type | wild type |
| SKBR-3 | wild type | wild type |
TβR-I and TβR-II exons were PCR amplified using intron-based primers. PCR fragments were purified and directly sequenced using the Perkin Elmer ABI377 Prism Automated DNA Sequencer.
The BT474 cell line demonstrated a point mutation in codon 35 of exon 2 resulting in the substitution of an isoleucine for a methionine. No other sequence changes in TβR-I or TβR-II were observed.
Expression of TβR-I and TβR-II mRNA in Resistant and Sensitive Cell Lines
Lack of responsiveness to TGF-β1-mediated growth inhibition demonstrated by six of seven of the tumorigenic breast cell lines could be a manifestation of decreased expression of TβR-I or TβR-II message with a resulting decrease in protein expression. A previous study in human breast cell lines demonstrated that a lack of responsiveness to TGF-β1 growth inhibition correlated well with decreased levels of TβR-II message expression (17). In order to examine the fidelity of this relationship, relative levels of message expression among the panel of TGF-β1-responsive and -resistant breast cell lines was determined by semiquantitative RT-PCR (8) using the nontumorigenic MCF-10F cells as a standard. Using normalized values for mRNA expression, TβR-I message was consistently expressed among all cell lines, while TβR-II message expression was significantly decreased in all TGF-β1-resistant tumorigenic cell lines (Table 4 and Fig. 4). TβR-II message was decreased by fivefold in MCF-7, by 50-fold in ZR75-1, and to an undetectable level in T47-D cells (using ethidium bromide staining and keeping within the linear range of amplification) (Table 4 and Fig. 4). MDA-MB231 cells, which demonstrated an intermediate sensitivity to TGF-β1 and binding of 125I-labeled TGF-β1, showed decreased TβR-II message expression, although not as pronounced as in the other tumor cell lines. Thus, decreased TβR-II message expression and/or loss of stability of TβR-II message in the TGF-β1-resistant tumorigenic cell lines was associated with the lack of binding of labeled TGF-β1 and the resistance of these cell lines to TGF-β1-mediated growth inhibition.
TABLE 4.
SEMIQUANTITATIVE ANALYSIS OF TβR-I AND TβR-II mRNA EXPRESSION IN BREAST CELL LINES
| Cell Line | TβR-I Expression | TβR-II Expression |
|---|---|---|
| MCF-10F | 1.0 | 1.0 |
| MCF-7 | 0.83 | 0.22 |
| ZR75-1 | 1.32 | 0.02 |
| BT474 | 0.89 | 0.10 |
| T47-D | 0.94 | 0 |
| MDA-MB231 | 0.88 | 0.66 |
| BT20 | 0.62 | 0.08 |
| SKBR-3 | 1.1 | 0.13 |
Total RNA extracted from subconfluent cell monolayers was reverse transcribed. A constant amount of cDNA from each cell line was used as template for PCR amplification of mRNA sequence of TβR-I, TβR-II, and HPRT fragments. PCR products were quantified by gel electrophoresis, ethidium bromide staining, and digital imaging. TβR-I and TβR-II expression was normalized to HPRT expression for each cell line. The nontumorigenic MCF-10F cell line value was set at 1.0 and all other cell line values were normalized to MCF-10F.
Figure 4.
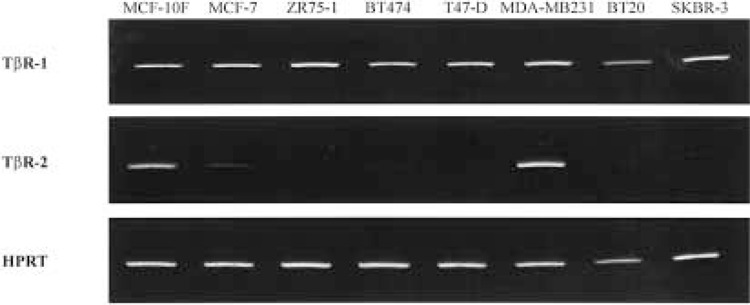
TβR-I and TβR-II mRNA expression in human breast cell lines. Reverse transcription was performed using 2 μg total RNA. Primers for PCR were designed according to GenBank accession numbers: L11695 (TβR-I), L07594 (TβR-II), and M26434 (HPRT). Optimal annealing temperature (T a) and the linear range of amplification were determined empirically for each set of PCR primers. PCR products were analyzed on 10% polyacrylamide gels and the intensity of ethidium bromide staining of PCR fragments was determined using a digital imaging system.
Expression of TβR-I and TβR-II Protein in Breast Cell Lines
To determine if the decreased levels of TβR-II mRNA also resulted in a decrease of TβR-II protein, total cellular protein was isolated from subconfluent cell cultures and Western blotting was performed. Conditions for Western blotting were optimized and specificity of binding of the antibodies was confirmed by use of the blocking peptide provided by the antibody manufacturer. Aliquots of protein used for the TβR-I and TβR-II experiments were derived from the same sample and the blots for TβR-I and TβR-II protein were performed in parallel as an additional control.
TβR-I protein was recognized at 55 kDa and TβR-II protein at 70 kDa, as predicted by the manufacturer of the antibodies and as demonstrated in previous published studies (23). Both TβR-I (Fig. 5) and TβR-II protein (Fig. 6) were observed in total cellular protein at relatively consistent levels across all cell lines. The apparent discrepancy between mRNA and protein expression in T47-D cells (Figs. 4 and 6) is most likely due to the insensitive visualization method (ethidium bromide staining) and limiting the number of PCR cycles in order to stay within the linear range of amplification for semiquantitative evaluation. Expression of T47-D mRNA was obtained when the PCR was performed through 40 cycles of amplification.
Figure 5.
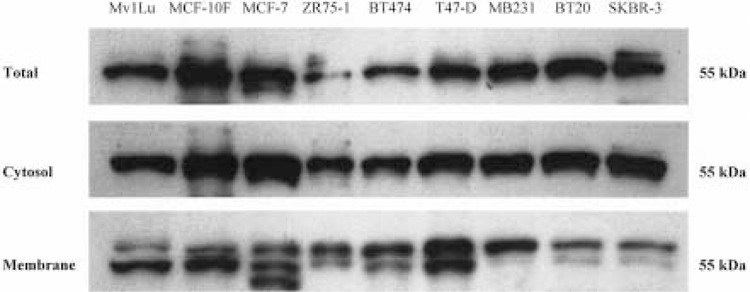
TβR-I protein expression in total lysate, cytosol, and membrane fractions from human breast cell lines. Aliquots (10 μg) of total cell lysate and fractionated protein were analyzed by SDS-PAGE electrophoresis, transferred to PVDF membranes, blocked with 5% nonfat dry milk in PBS-T, and incubated with TβR-I antibody. Bound antibodies were detected by incubation with peroxidase-conjugated antirabbit immunoglobulin and enhanced chemiluminscent autoradiography. A 55-kDa band corresponding to the presence of TβR-I protein in total lysate, cytosolic, and membrane-bound protein fractions was observed. Membrane-bound fractions (and to a lesser extent the total cellular lysate) contain two (or three in the case of MCF-7) bands of close molecular weight, the significance of which remains to be determined.
Figure 6.
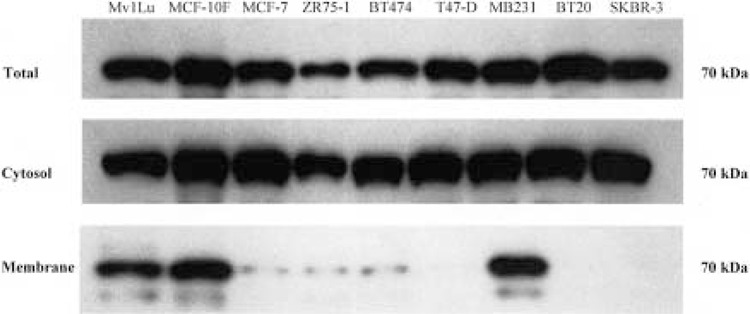
TβR-II protein expression in total lysate, cytosol, and membrane fractions from human breast cell lines. Aliquots (10 μg) of total cell lysate and fractionated protein were analyzed by SDS-PAGE electrophoresis, transferred to PVDF membranes, blocked with 5% nonfat dry milk in PBS-T, and incubated with TβR-II antibody. Bound antibodies were detected by incubation with peroxidase-conjugated antirabbit immunoglobulin and enhanced chemiluminscent autoradiography. A 70-kDa band corresponding to the size expected for TβR-II protein was observed in all cell lines in total lysate and the cytosolic fraction. The membrane fractions demonstrate the clear presence of TβR-II protein in the Mv1Lu positive control, the MCF-10F nontumorigenic, and the tumorigenic MDA-MB231 cell lines. The other cell lines have little or no TβR-II protein present in the membrane fraction.
Levels of Membrane-Bound TβR-II Protein in Resistant Tumorigenic Breast Cell Lines
Because similar levels of total TβR-II protein were found in all of the cell lines, we chose to investigate the relative levels of TβR-II protein in the cytosolic and membrane-bound protein fractions from these same cell lines, speculating that trafficking of TβR-II protein to the cell membrane may be one of the mechanisms of resistance to TGF-β1 growth inhibition. Total cellular protein was fractionated by velocity differential centrifugation and Western blotting was performed. As before, TβR-I protein was found at similar levels in both the cytosolic and membrane fractions across all cell lines (Fig. 5). However, Western blotting for TβR-II in the cytosolic and membrane fractions showed bands at 70 kDa in only the cytosolic fraction, with membrane-bound TβR-II protein fractions found only in the TGF-β1-responsive cell lines, Mv1Lu, MCF-10F, and MDA-MB231. The weak bands that were observed in the membrane fraction of the MCF-7, ZR75-1, BT474, and BT20 cell lines were likely due to contamination of the membrane fractions by incomplete separation from the cytosolic fraction. No TβR-II bands were observed in the membrane fraction of the T47-D and the SKBR-3 cell lines. These data strongly suggest that lack of significant levels of membrane TβR-II protein is the primary mechanism by which the resistant cell lines escape TGF-β1-mediated growth control.
DISCUSSION
Investigations into the role of TGF-β in malignant progression have contributed to a growing understanding of the biphasic nature of the effects of this growth factor in tumorigenesis. In normal epithelium and among transformed cells, activated TGF-β within the microenvironment functions to inhibit cell cycle progression, providing a mechanism to inhibit clonal expansion of mitogenically activated cells. A critical event in tumor progression is the point at which a population of transformed cells escape growth inhibition mediated by TGF-β. At this point, the malignant tumor cells become an important source of TGF-β in the microenvironment, where activated TGF-β functions to promote tumor development by promoting angiogenesis, facilitating tumor invasion and metastasis, and contributing to local immune suppression (42). Thus, a better understanding of the mechanisms by which human breast tumor cells escape from TGF-β-mediated growth control will provide important insights into breast tumorigenesis and possible targets for future chemopreventive and therapeutic strategies.
In vitro studies of TGF-β-mediated effects on normal and transformed breast epithelium suggest that the development of resistance to TGF-β-mediated growth inhibition is a prevalent event among tumorigenic breast epithelial cell cultures. Using a human mammary epithelial cell culture system (HMEC), developed specifically for the investigation of cell cycle control and signal transduction, Stampfer et al. (47) have demonstrated that normal HMEC are growth inhibited by TGF-β, arresting in late G1 phase of the cell cycle. In contrast, transformed HMEC showed variable resistance to TGF-β-mediated growth inhibition; however, all transformed cultures developed a population of cells that were resistant to the effects TGF-β. The heterogeneity of responses to TGF-β among transformed HMEC and the persistence of a population of resistant cells suggests an epigenetic, and possibly a later genetic, mechanism involved in the development of resistance to TGF-β growth inhibition.
Assays for responsiveness of widely used established breast cancer cell lines to TGF-β-mediated growth inhibition have demonstrated differing results between laboratories. Initial studies of MCF-7 cells, using inhibition of anchorage independent growth as the marker of TGF-β sensitivity, showed marked responsiveness of MCF-7 cells to TGF-β1 effects (43). However, later studies, using MCF-7 cells that had been in culture for several years, demonstrated resistance of MCF-7 cells to TGF-β-mediated growth inhibition (4,55). Our studies utilized established cell lines passaged in our laboratory (R. Brueggemeier) for several years, except for the MCF-10F and the Mv1Lu cells, which were obtained from ATCC. In our experiments, the estrogen receptor-positive tumorigenic MCF-7, ZR75-1, BT474, and T47-D cell lines and the estrogen receptor-negative tumorigenic BT20 and SKBR-3 cell lines were resistant to TGF-β1 growth inhibition. The positive control, Mv1Lu, and the nontumorigenic breast epithelial MCF-10F cell lines showed similar responses to TGF-β1, with greater than 50% inhibition of DNA synthesis at 10 pM TGF-β1. Our MDA-MB231 cells showed a significant response to TGF-β1, although not as marked as the MCF-10F cells. Variation in responsiveness of established breast cell lines to TGF-β among laboratories suggests that selection of TGF-β-resistant clones occurs under routine culture conditions, indicating a mechanism for genetic selection for TGF-β resistance, or possibly an epigenetic response of tumorigenic cells in culture to their microenvironment.
Binding of 125I-labeled TGF-β1 to the cell membrane in our cell lines parallels the inhibition of DNA synthesis by TGF-β1, with TGF-β-responsive Mv1Lu, MCF-10F, and MDA-MB231 cells demonstrating binding of 125I-labeled TGF-β1 and the TGF-β-resistant cells showing no membrane binding of labeled ligand. One hypothesis to explain this correlation is the lack of TβR-II receptor protein in the cell membrane. However, lack of membrane binding of TGF-β ligand in TGF-β-resistant cell lines may be due to other causes. For example, Chen et al. (7) have shown that introduction of a TβR-III expression vector into MCF-7 induces sensitivity to TGF-β. Furthermore, binding of 125I-labeled TGF-β1 to TβR-I and TβR-II in the parent MCF-7 cells was undetectable, but binding of labeled ligand to TβR-I and TβR-II protein was shown in cells that contained the TβR-III expression vector (7). These data suggest that TβR-III also plays an essential role in ligand binding and presentation to the TβR-I/TβR-II complex or in the stability of the TβR type I and/or type II receptors. Further studies are needed to elucidate the mechanism by which TβR-III functions to facilitate binding of TGF-β ligand to TβR-I and TβR-II.
A significant mechanism by which tumor cells develop resistance to TGF-β involves mutational inactivation of TβR-I or TβR-II. Mutations in TβR-I have been identified in the human prostate cell line, LNCaP (19), as well as in human breast tumors (6). Mutations in TβR-II are a common cause of receptor inactivation and TGF-β resistance in tumor cell lines that demonstrate microsatellite instability, including colon cancer (29,51), gastric cancer (39), and malignant gliomas (15). In human clinical tumor specimens, characteristic inactivating TβR-II mutations associated with microsatellite instability have also been identified in replication error-positive human colon cancer (40), gastric cancer (35), and ovarian cancer (27), but not in endometrial carcinomas (35). Furthermore, inactivating TβR-II mutations, unrelated to microsatellite instability, have been identified in sporadic colon cancer (40), squamous cell carcinoma of the head and neck (52), and ovarian cancers (40), but not in human breast tumors (49). Using TβR-I intron sequences determined by a novel PCR technique, each exon of TβR-I was PCR amplified and directly sequenced, but no sequence changes were found in this panel of breast cell lines. Using recently published intron sequences for TβR-II (50), a similar approach was used to screen for mutations in TβR-II. Only in the BT474 cell was a sequence alteration identified at codon 35, resulting in an isoleucine→methionine amino acid substitution. The effect of this mutation on receptor function is unknown at this time. In summary, mutations in TβR-I and TβR-II do not appear to play an important role in the inactivation of the TβR complex in this panel of breast cell lines.
Decreased expression of TβR-II mRNA among the TGF-β-resistant MCF-7, ZR75-1, BT474, T47-D, BT20, and SKBR-3 human breast cell lines in our studies reflects previously published experiments using these same cells (17,23). In the studies by Kalkhoven et al. (17), a similar reduction in relative levels of TβR-II mRNA was observed even though the MCF-7 cells in their laboratory were responsive to TGF-β1 growth inhibition and demonstrated binding of 125I-labeled TGF-β1 ligand. These data and our results suggest that the observed relative reduction in levels of TβR-II message is not the primary mechanism by which breast cancer cells escape growth inhibition by TGF-β1.
Our data on expression and trafficking of TβR-I and TβR-II protein provide further evidence that TβR-II protein is a key target for the development of resistance of tumor cells to TGF-β-mediated growth inhibition as evidenced by the lack of TβR-II protein in the membrane fraction in the TGF-β1-resistant breast cell lines. Schindler et al. (45) have suggested that the trafficking of TβR-II protein in breast tumor cells (MCF-7) is affected by lack of pH control of acidic compartments, thus causing the cells to be defective in recycling and secretory pathways. Evidence for this supposition was provided by Koli and Arteaga (23), who found that drug-resistant cells (after adriamycin treatment) were normal in compartmental acidification, expressed membrane TβR-II protein, and were inhibited by TGF-β. Recent work on the processing of TβR-I and TβR-II protein has shown differential processing and turnover of the two receptor types and may help to explain the differences in membrane binding of TβR-I and TβR-II observed in our studies. While the half-life of TβR-II protein is quite short (less than 60 min), the half-life of TβR-I protein is 3 h or longer (53). In addition, the half-life of TβR-II is further reduced by the presence of TGF-β1 (24). Thus, TβR-II protein has a rapid turnover and is subject to negative feedback regulation by the TGF-β ligand, which would make TβR-II protein a potential target for critical regulatory events in cell responsiveness to TGF-β ligand. Moreover, studies in hamster ovary cells have identified a cytoplasmic pool of TβR-II protein that is hormonally regulated, suggesting a mechanism for crosstalk between TGF-β and other cellular signal transduction pathways (44).
Recent observations regarding TβR-II protein expression in clinical human tumor specimens reflect our observations in human breast cell lines in vitro. In our studies of human ovarian carcinomas, we found loss or decreased TβR-II protein expression in 15 of 22 ovarian carcinomas by immunohistochemistry (35). Similar decreases in expression of TβR-II protein have been observed in adenocarcinomas of the lung (18) and squamous cell carcinomas of the head and neck (34). Interestingly, a recent study of women with mammary epithelial hyperplasia revealed that decreased expression of TβR-II protein was associated with a 1.98 odds ratio of future invasive breast cancer and loss of expression of TβR-II protein was associated with a 3.41 odds ratio of invasive breast cancer, suggesting that loss of TβR-II protein expression may be an important marker of malignant progression in human breast cancer. These data from human tumors further support the hypothesis that loss of TβR-II protein expression is a common event among human tumors and may be the key event by which human tumor cells escape TGF-β-mediated growth inhibition, contributing to malignant progression.
An important question to answer is the possibility of pharmacologic manipulation of TβR-II expression as a potential chemopreventive (1) or chemotherapeutic strategy. The effects of estrogens and antiestrogens on trafficking of TβR-II protein to the cell membrane is an important area of research. Using these breast cancer cell lines as a model, hormonal and other chemopreventive strategies for the reintroduction of responsiveness to TGF-β can be explored in vitro, transferred to animal models of breast carcinogensis, and hopefully translated to clinical trials. Studies using human breast tissues to identify the extent to which decreased TβR-II protein expression correlates with malignancy and to identify the point in malignant progression at which this occurs would also contribute to the use of expression of TβR-II as a marker of malignant progression and as a potential endpoint in chemoprevention trials.
ACKNOWLEDGMENTS
This study was supported in part by an award from the V Foundation, Cary, NC, and NIH-NIDCR grant 1PO1 DE12704 (C.M.W.) and NIH-NCI grants R21 CA66193 (R.W.B.) and P30 CA16058 (OSUCCC).
REFERENCES
- 1. Ariazi E. A.; Satomi Y.; Ellis M. J.; Haag J. D.; Shi W.; Sattler C. A.; Gould M. N. Activation of the transforming growth factor beta signaling pathway and induction of cytostasis and apoptosis in mammary carcinomas treated with the anticancer agent perillyl alcohol. Cancer Res. 59:1917–1928; 1999. [PubMed] [Google Scholar]
- 2. Arteaga C. L.; Carty-Dugger T.; Moses H. L.; Hurd S. D.; Pientenpoi J. A. Transforming growth factor beta 1 can induce estrogen-independent tumorigenicity of human breast cancer cells in athymic mice. Cell Growth Differ. 4:367–374; 1993. [PubMed] [Google Scholar]
- 3. Arteaga C. L.; Koli K. M.; Dugger T. C.; Clarke R. Reversal of tamoxifen resistance of human breast carcinomas in vitro by neutralizing antibodies to transforming growth factor-β. J. Natl. Cancer Inst. 91:4653; 1999. [DOI] [PubMed] [Google Scholar]
- 4. Arteaga C. L.; Tandon A. K.; VonHoff D. D.; Osborne C. K. Transforming growth factor β: Potential autocrine growth inhibitor of estrogen receptor-negative human breast cancer cells. Cancer Res. 48:3898–3904; 1988. [PubMed] [Google Scholar]
- 5. Carcamo J.; Zentella A.; Massague J. Disruption of transforming growth factor β signalling by a mutation that prevents transphosphorylation within the receptor complex. Mol. Cell. Biol. 15:1573–1581; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen T.; Carter D.; Garrigue-Antar L.; Reiss M. Transforming growth factor-β type I receptor kinase mutant associated with metastatic breast cancer. Cancer Res. 58:4805–4810; 1998. [PubMed] [Google Scholar]
- 7. Chen C.; Wang W. F.; Sun L. Z. Expression of transforming growth factor β (TGFβ) type III receptor restores autocrine TGFβ1 activity in human breast cancer MCF-7 cells. J. Biol. Chem. 272:12862–12867; 1997. [DOI] [PubMed] [Google Scholar]
- 8. Freeman W. M.; Walker S. J.; Vrana K. E. Quantitative RT-PCR: Pitfalls and potential. Biotechniques 26: 124–125; 1999. [DOI] [PubMed] [Google Scholar]
- 9. Fynan T. M.; Reiss M. Resistance to inhibition of cell growth by transforming growth factor-β and its role in oncogenesis. Crit. Rev. Oncol. 4:493–540; 1993. [PubMed] [Google Scholar]
- 10. Greenlee R. T.; Murray T.; Bolden S.; Wingo P. A. Cancer statistics, 2000. CA Cancer J. Clin. 50:7–33; 2000. [DOI] [PubMed] [Google Scholar]
- 11. Heldin C. H.; Miyazono K.; ten Dijke P. TGF-beta signaling from cell membrane to nucleus through SMAD proteins. Nature 390:465–471; 1997. [DOI] [PubMed] [Google Scholar]
- 12. Henderson B. E.; Ross R. K.; Bernstein L. Estrogen as a cause of human cancer: The Richard and Hinda Rosenthal Foundation award lecture. Cancer Res. 48: 246–253; 1988. [PubMed] [Google Scholar]
- 13. Henderson B. E.; Ross R. K.; Pike M. C. Hormonal chemoprevention of cancer in women. Science 259: 633–638; 1993. [DOI] [PubMed] [Google Scholar]
- 14. Inagaki M.; Moustakas A.; Lin H. Y.; Lodish H. F.; Carr B. I. Growth inhibition by transforming growth factor β (TGF-β) type I is restored in TGF-β resistant hepatoma cells after expression of TGF-β receptor type II cDNA. Proc. Natl. Acad. Sci. USA 90:5359–5363; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Izumoto S.; Arita N.; Ohnishi T.; Hiraga S.; Taki T.; Tomita N.; Ohue M.; Hayakawa T. Microsatellite instability and mutated type II transforming growth factor receptor gene in gliomas. Cancer Lett. 112:251–256;1997. [DOI] [PubMed] [Google Scholar]
- 16. Jeng M. H.; ten Dijke P.; Iwata K. K.; Jordan V. C. Regulation of the levels of three transforming growth factor β mRNAs by estrogen and their effects on the proliferation of human breast cancer cells. Mol. Cell. Endocrinol. 97:115–123; 1993. [DOI] [PubMed] [Google Scholar]
- 17. Kalkhoven E.; Roelen B. A. J.; de Winter J. P.; Mummery C. L.; van den Eijnden-van Raaij A. J. M.; van der Saag P. T.; van der Burg B. Resistance to transforming growth factor beta and activin due to reduced receptor expression in human breast tumor cell lines. Cell Growth Differ. 6:1151–1161; 1995. [PubMed] [Google Scholar]
- 18. Kim W. S.; Park C.; Jung Y. S.; Kim H. S.; Han J.; Park C. H.; Kim K.; Kim J.; Shim Y. M.; Park K. Reduced transforming growth factor-β type II receptor (TGF-β RII) expression in adenocarcinoma of the lung. Anticancer Res. 19:301–306; 1999. [PubMed] [Google Scholar]
- 19. Kim I. Y.; Zelner D. J.; Lee C. Genetic change in transforming growth factor beta (TGF-beta) receptor type I gene correlates with insensitivity to TGF-betal in human prostate cancer cells. Cancer Res. 56:44–48; 1996. [PubMed] [Google Scholar]
- 20. Kimchi A.; Wang X. F.; Weinberg R. A.; Cheifetz S.; Massague J. Absence of TGF-beta receptors and growth inhibitory responses in retinoblastoma cells. Science 240:196–199; 1988. [DOI] [PubMed] [Google Scholar]
- 21. Knabbe C.; Lippman M. E.; Kasid A.; Flanders R.; Wakefield L. M.; Derynck R.; Dickson R. B. Evidence that transforming growth factor β is a hormonally regulated negative growth factor in human breast cancer cells. Cell 48:417–428; 1987. [DOI] [PubMed] [Google Scholar]
- 22. Koli K. M.; Arteaga C. L. Complex role of tumor cell TGF-βs on breast cancer progression. J. Mammary Gland Biol. Neopl. 1:373–380; 1996. [DOI] [PubMed] [Google Scholar]
- 23. Koli K. M.; Arteaga C. L. Predominant cytosolic localization of type II transforming growth factor β receptors in human breast carcinoma cells. Cancer Res. 57:970-977; 1997. [PubMed] [Google Scholar]
- 24. Koli K. M.; Arteaga C. L. Processing of the transforming growth factor β type I and II receptors. J. Biol. Chem. 272:6423–6427; 1997. [DOI] [PubMed] [Google Scholar]
- 25. Kretzschmar M.; Massague J. SMADs: Mediators and regulator of TGF-beta signaling. Curr. Opin. Genet. Dev. 8:103–111; 1998. [DOI] [PubMed] [Google Scholar]
- 26. Like B.; Massague J. The antiproliferative effect of type beta transforming growth factor occurs at a level distal from receptors for growth activating factors. J. Biol. Chem. 261:13426–134269; 1986. [PubMed] [Google Scholar]
- 27. Lynch M. A.; Nakashima R.; Song H.; Degroff V. L.; Wang D.; Enomoto T.; Weghorst C. M. Mutational analysis of the transforming growth factor β receptor type II gene in human ovarian cancer. Cancer Res. 58:4227–4232; 1998. [PubMed] [Google Scholar]
- 28. Mackay S. L.; Auffenberg T.; Tannahill C. L.; Ksontini R.; Josephs M. D.; Nowak M.; Moldawer L. L.; Copeland E. M. Transfection of the type II TGF-beta receptor into colon cancer cells increases receptor expression, inhibits cell growth, and reduces the malignant phenotype. Ann. Surg. 227:781–789; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Markowitz S.; Wang J.; Myeroff L.; Parsons R.; Sun L.; Lutterbaugh J.; Fan R. S.; Zborowska E.; Kinzler K. W.; Vogelstein B.; Brattain M.; Wilson J. K. V. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 268:1336–1338; 1995. [DOI] [PubMed] [Google Scholar]
- 30. Massague J. Identification of receptors for type-β transforming growth factor. Methods Enzymol. 146: 174–195; 1987. [DOI] [PubMed] [Google Scholar]
- 31. Massague J. Receptors for the TGF-β Family. Cell 69: 1067–1070; 1992. [DOI] [PubMed] [Google Scholar]
- 32. Massague J.; Cheifetz S.; Laiho M.; Ralph D. A.; Weis F. M.; Zentella A. Transforming growth factor β. Cancer Surveys 12:1989–2003; 1992. [PubMed] [Google Scholar]
- 33. Mazars P.; Barboule N.; Baldin V.; Vidal S.; Ducommum B.; Valette A. Effects of TGF-β1 (transforming growth factor-β1) on the cell cycle regulation of human breast adenocarcinoma (MCF-7) cells. FEBS Lett. 362:295–300; 1995. [DOI] [PubMed] [Google Scholar]
- 34. Muro-Cacho C. A.; Anderson M.; Cordero J.; Munoz-Antonia T. Expression of transforming growth factor β type II receptors in head and neck squamous cell carcinomas. Clin. Cancer Res. 5:1243–1248; 1999. [PubMed] [Google Scholar]
- 35. Myeroff L. L.; Parsons R.; Kim S. J.; Hedrick L.; Cho K. R.; Orth K.; Mathis M.; Kinzler K. W.; Lutterbaugh J.; Park K.; Bang Y. J.; Lee H. Y.; Park J. G.; Lynch H. T.; Roberts A. B.; Vogelstein B.; Markowitz S. D. A transforming growth factor β receptor type II gene mutation common in colon and gastric but rare in endometrial cancers with microsatellite instability. Cancer Res. 55:5545–5547; 1995. [PubMed] [Google Scholar]
- 36. Norgaard P.; Damstrup L.; Rygaard K.; Spang-Thomsen M.; Poulsen H. S. Growth suppression by transforming growth factor-β1 of human small-cell lung cancer cell lines is associated with expression of type II receptor. Br. J. Cancer 69:802–808; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Norgaard P.; Hougaard S.; Poulsen H. S.; Spang-Thomsen M. Transforming growth factor β and cancer. Cancer Treat. Rev. 21:367–403; 1995. [DOI] [PubMed] [Google Scholar]
- 38. Okamoto A.; Jiang W.; Kim S. J.; Spillare E. A.; Stoner G. D.; Weinstein I. B.; Harris C. C. Overexpression of cyclin D1 reduces transforming growth factor beta (TGF-beta) type II receptor and growth inhibition by TGF-beta 1 in an immortalized human esophageal cell line. Proc. Natl. Acad. Sci. USA 91: 11576–80; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Park K.; Kim S. J.; Bang Y. J.; Park J. G.; Kim N. K.; Roberts A. B.; Sporn M. B. Genetic changes in the transforming growth factor beta (TGF-β) type II receptor gene in human gastric cancer cells correlates with sensitivity to growth inhibition by TGF-beta. Proc. Natl. Acad. Sci. USA 91:8772–8776; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Parsons R.; Myeroff L. L.; Liu B.; Willson J. K. V.; Markowitz S. D.; Kinzler K. W.; Vogelstein B. Microsatellite instability and mutations of the transforming growth factor β type II receptor gene in colorectal cancer. Cancer Res. 55:5548–5550; 1995. [PubMed] [Google Scholar]
- 41. Perry R. R.; Kang Y.; Greaves B. R. Relationship between tamoxifen-induced transforming growth factor β expression, cytostasis and apoptosis in human breast cancer cells. Br. J. Cancer 61:612–617; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reiss M. TGF-β and cancer. Microbes Infection 1: 1327–1347; 1999. [DOI] [PubMed] [Google Scholar]
- 43. Roberts A. B.; Anzano M. A.; Wakefield L. M.; Roche N. S.; Stern D. F.; Sporn M. B. Type β transforming growth factor: A bifunctional regulator of cellular growth. Proc. Natl. Acad. Sci. USA 82:119–123; 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roy S. K.; Kole A. R. Transforming growth factor-β receptor type II expression in the hamster ovary: Cellular sites, biochemical properties, and hormonal regulation. Endocrinology 136:4610–4620; 1995. [DOI] [PubMed] [Google Scholar]
- 45. Schindler M.; Grabski S.; Hoff E.; Simon S. M. Defective pH regulation of acidic compartments in human breast cancer cells (MCF-7) is normalized in adriamycin-resistant cells (MCF-7adr). Biochemistry 35:2811–2817; 1996. [DOI] [PubMed] [Google Scholar]
- 46. Soule H. D.; Maloney T. M.; Wolman S. R.; Peterson W. D. Jr.; Brenz R.; McGrath C. M.; Russo J.; Pauley R. J.; Jones R. F.; Brooks S. C. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 50:6075–6086; 1990. [PubMed] [Google Scholar]
- 47. Stampfer M. R.; Yaswen P. Culture systems for study of human mammary epithelial cell proliferation, differentiation and transformation. Cancer Surveys 18:7–34; 1993. [PubMed] [Google Scholar]
- 48. Sun L.; Wu G.; Willson J. K. V.; Zborowska E.; Yang J.; Rajkarunanayake I.; Wang J.; Gentry L. E.; Wang X. F.; Brattain M. G. Expression of transforming growth factor beta type II receptor leads to reduced malignancy in human breast cancer MCF-7 cells. J. Biol. Chem. 263:26449–26455; 1994. [PubMed] [Google Scholar]
- 49. Takenoshita S.; Mogi A.; Tani M.; Osawa H.; Sunaga H.; Kakegawa H.; Yanagita Y.; Koida T.; Kimura M.; Fujita K. I.; Kato R.; Nagamachi Y. Absence of mutations in the analysis of coding sequences of the entire transforming growth factor-beta type II receptor gene in sporadic human breast cancers. Oncol. Rep. 5:367–371; 1998. [PubMed] [Google Scholar]
- 50. Takenoshita S.; Tani M.; Nagashima M.; Hagiwara K.; Bennett W. P.; Yokota J.; Harris C. C. Mutation analysis of coding sequences of the entire transforming growth factor beta type II receptor gene in sporadic human colon cancer using genomic DNA and intron primers. Oncogene 15:117–122; 1997. [DOI] [PubMed] [Google Scholar]
- 51. Wang J.; Sun L.; Myeroff L.; Wang X.; Gentry L. E.; Yang J.; Liang J.; Zborowska E.; Markowitz S.; Willson J. K.; Brattain M. G. Demonstration that mutations of the type II transforming growth factor beta receptor inactivates its tumor suppressor activity in replication error-positive colon carcinoma cells. J. Biol. Chem. 270:22044–22049; 1995. [DOI] [PubMed] [Google Scholar]
- 52. Wang D.; Song H.; Evans J. A.; Lang J. C.; Schuller D. E.; Weghorst C. M. Mutation and downregulation of the transforming growth factor beta type II receptor gene in primary squamous cell carcinomas of the head and neck. Carcinogenesis 18:2285–2290; 1997. [DOI] [PubMed] [Google Scholar]
- 53. Wells R. G.; Yankelev H.; Lin H. Y.; Lodish H. F. Biosynthesis of type I and type II TGF-β receptors. J. Biol. Chem. 27:11444–11451; 1997. [DOI] [PubMed] [Google Scholar]
- 54. Wrana J. L.; Attisano L.; Carcamo J.; Zentella A.; Doody J.; Laiho M.; Wang X. F.; Massague J. TGF-β signals through a heteromeric protein kinase receptor complex. Cell 71:1003–1014; 1992. [DOI] [PubMed] [Google Scholar]
- 55. Zugmaier G.; Ennis B. W.; Deschauer B.; Katz D.; Knabbe C.; Wilding G.; Daly P.; Lippman M. E.; Dickson R. B. Transforming growth factors type β1 and β2 are equipotent growth inhibitors of human breast cancer cell lines. J. Cell. Physiol. 141:353–361; 1989. [DOI] [PubMed] [Google Scholar]