
Figure 6. Changes in mitochondrial morphology lead to tubular damage in acute kidney injury.
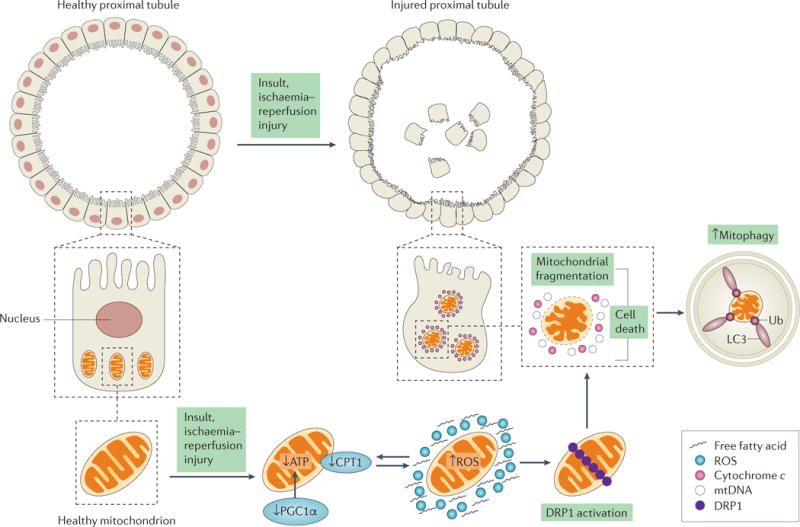
A healthy proximal tubule consists of an intact brush border with tight junctions and contains a network of mitochondria to maintain its function. After ischaemia–reperfusion injury (IRI), changes in mitochondrial function and morphology lead to mitochondrial dysfunction, and eventually to injured proximal tubules. In the early stages of acute kidney injury (AKI), a number of events may happen concurrently to cause a decrease in ATP production. These events include a decrease in the expression of carnitine O-palmitoyltransferase 1 (CPT1) (causing fatty acid accumulation and decreasing β-oxidation for ATP production), a decrease in the expression of peroxisome proliferator-activated receptor-γ co-activator 1α (PGC1α) and an increase in the production of reactive oxygen species (ROS) (bidirectional arrows). Together, these events can trigger the activation and accumulation of dynamin 1-like protein (DRP1) on the mitochondrial outer membrane, promoting mitochondrial fragmentation and eventually cell death. The release of cytochrome c and mitochondrial DNA (mtDNA) from dysfunctional mitochondria causes an increase in mitophagy. Mitochondrial dysfunction can induce cell death in injured proximal tubules, resulting in the loss of nuclei and tight junctions and in disrupted brush borders. Apoptotic or necrotic tubules can lead to cell sloughing, as seen in the centre of the tubule.