

SUMMARY
Approximately 30% of triple-negative breast cancers (TNBCs) exhibit functional loss of the RB tumor suppressor, suggesting a target for precision intervention. Here, we use drug screens to identify agents specifically antagonized by the retinoblastoma tumor suppressor (RB) using CDK4/6 inhibitors. A number of candidate RB-synthetic lethal small molecules were identified, including anti-helmenthics, chemotherapeutic agents, and small-molecule inhibitors targeting DNA-damage checkpoints (e.g., CHK) and chromosome segregation (e.g., PLK1). Counter-screens using isogenic TNBC tumor cell lines and cell panels with varying endogenous RB statuses confirmed that therapeutic effects were robust and selective for RB loss of function. By analyzing TNBC clinical specimens, RB-deficient tumors were found to express high levels of CHK1 and PLK1. Loss of RB specifically resulted in loss of checkpoint functions governing DNA replication, yielding increased drug sensitivity. Xenograft models demonstrated RB-selective efficacy of CHK inhibitors. This study supports the possibility of selectively targeting RB loss in the treatment of TNBC.
In Brief
Witkiewicz et al. demonstrate that the activation state of the RB tumor suppressor is a critical determinant for selected targeted therapies in models of TNBC. Loss of RB yields a selective vulnerability to replication and chromosome segregation stress. Drugs targeting CHK and PLK have increased efficacy in RB-deficient tumors.
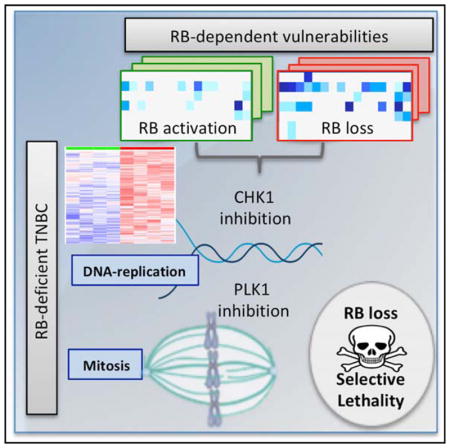
INTRODUCTION
In general, triple-negative breast cancer (TNBC) harbors a poor prognosis relative to other breast cancer subtypes (Foulkes et al., 2010; Reis-Filho and Tutt, 2008). This poor outcome is due to the heterogeneous and aggressive nature of the disease, coupled with the lack of highly recurrent and actionable biomarkers that can be used to direct therapy (Pareja et al., 2016; Turner and Reis-Filho, 2013). Almost all patients with a TNBC diagnosis are treated with chemotherapy regimens with varying efficacy (Anders et al., 2013). Despite an overall poor prognosis, a subset of TNBC tumors is responsive to conventional chemotherapy (Anders et al., 2013; Carey et al., 2007; Turner and Reis-Filho, 2013). However, for patients who have recurrent disease after treatment or progressive disease during chemotherapy, treatment options are limited.
One of the key clinical challenges in TNBC is to define actionable means for patient stratification and to delineate targeted approaches to treatment. Genetic analyses have shown that TNBC tumors carry a wide array of mutations (Cancer Genome Atlas, 2012); however, many of the mutated genes represent tumor suppressors that currently cannot be targeted (e.g., TP53 or RB1). Despite the intrinsic difficulty in targeting tumor suppressor loss, the ability to target BRCA1 or BRCA2 deficiency via a synthetic lethal approach with PARP inhibitors suggests that, in principle, selective therapeutic sensitivities for tumor suppressor loss can be developed (Farmer et al., 2005; Lord and Ashworth, 2017; McCabe et al., 2006).
The retinoblastoma tumor suppressor (RB) plays a key role in coordinating cell-cycle transitions and is frequently lost in TNBC (Burkhart and Sage, 2008; Witkiewicz et al., 2012; Witkiewicz and Knudsen, 2014). Functionally, RB acts downstream of CDK4/6 kinases to control cell-cycle progression into S phase (Asghar et al., 2015; Knudsen and Knudsen, 2008; Sherr et al., 2016). This action is largely dependent on RB-mediated repression of the E2F-family of transcriptional regulators that coordinate the expression of multiple genes required for DNA replication, mitosis, and DNA repair (Markey et al., 2007; Nevins, 2001). In the absence of RB, there is deregulated cell-cycle progression, which can contribute to tumor development. Additionally RB deficiency has been associated with multiple forms of chromosomal instability and, therefore, could also fuel tumorigenesis by removing safeguards that limit oncogenic transformation (Dyson, 2016; Manning and Dyson, 2012). In TNBC, the RB gene (RB1) is typically lost via homozygous deletion (Cancer Genome Atlas, 2012) but has also been reported to be the subject of epigenetic silencing. Depending on the TNBC cohort studied, RB dysfunction is estimated to occur in ~30% of cases (Witkiewicz and Knudsen, 2014). Thus, due to its frequency, the RB loss in TNBC represents a potentially relevant event for treatment stratification.
Prior studies have suggested that RB can play multiple distinct roles in the context of therapeutic interventions (Asghar et al., 2015; Knudsen and Knudsen, 2008; Knudsen and Wang, 2010). RB is downstream of CDK4/6-cyclin D complexes and is critical for the cytostatic activity of CDK4/6 inhibitors (Knudsen and Witkiewicz, 2017; Sherr et al., 2016). In this context, the induction of RB function has been associated with resistance to specific chemotherapeutic agents (e.g., carboplatin) (Johnson et al., 2010; Roberts et al., 2012). As a result, RB activation has been proposed to ameliorate toxic side effects of chemotherapeutic agents. In contrast, RB deficiency has been associated with sensitivity to chemotherapeutic agents in preclinical models and retrospective analysis of breast cancer clinical cases (Robinson et al., 2013; Stengel et al., 2008; Witkiewicz et al., 2012). In addition, RB loss has been associated with resistance to multiple targeted agents, including endocrine therapy, EGFR, and BRAF inhibitors (Bosco and Knudsen, 2007; Bosco et al., 2007; Niederst et al., 2015; Xing et al., 2012). The goal of this study was to delineate functional sensitivities related to RB activation status that could be actionable for the treatment of TNBC.
RESULTS
Functional Antagonism Screen to Define RB-Pathway-Dependent Therapeutic Agents
In recognition that RB has multiple roles in cell biology and therapeutic response, we initially queried how activation of RB with CDK4/6 inhibition impinges on therapeutic sensitivities. RB-positive MDA-MB231 cells (MB231) were treated with palbociclib (1 μM) or DMSO for 24 hr and then exposed to the Prestwick library of 1,280 compounds, and viability was assessed. Palbociclib treatment resulted in substantial cell-cycle inhibition in MB231 cells (Figure S1). In total, 56 drugs from the Prestwick library were identified as causing significant cell killing from the primary drug screen (Figures 1A and S1). Of these agents, 21 were antagonized by palbociclib pretreatment. The drug sensitivity was reduced for select chemotherapeutic agents (e.g., docetaxel and gemcitabine) as well as anti-helmenthics (e.g., albendazole and mebandazole) (Figures 1A and S1). Evaluation of these drugs relative to their principle targets suggested that in this library there was potent selection for drugs that target microtubule dynamics or nucleotide metabolism. This finding is consistent with the concept that CDK4/6 inhibition could serve to ameliorate the cytotoxic effects of specific chemotherapies (Roberts et al., 2012). The primary screen results with anti-helmenthics were confirmed by dose-response analysis (Figure 1B). Since the targets of anti-helmenthics are parasitic worms/larva, we could evaluate the potential for CDK4/6 inhibition to differentiate between human and lower organisms. This is particularly relevant, as in basal organisms, there is either no CDK4/6 ortholog, or it is not associated with cell-cycle progression (Cao et al., 2014; Meyer et al., 2000). Treatment with palbociclib prevented colchicine-induced G2/M accumulation and cell death due to cell-cycle arrest in the human tumor cell line MDA-MB231 (Figure 1C). In contrast, in D. melanogaster S2 cells, palbociclib had no effect on cell-cycle control or sensitivity to colchicine. Thus, in principle, CDK4/6 inhibitors could be used to expand a therapeutic index relative to lower eukaryotes, based on cell-cycle regulatory differences. The selective antagonism related to specific chemotherapies was confirmed by dose response, wherein docetaxel and gemcitabine were antagonized by palbociclib treatment, but sensitivity to epigenetic agents 5-azacytidine and vorinostat was independent of CDK4/6 inhibition (Figure 1D; data not shown). To determine whether the sensitivities were intrinsically dependent on the presence of RB in tumor cells, we used the RB-deficient TNBC cell line MB468 (Figures 1E and S1). In this model, CDK4/6 inhibition did not antagonize the response to docetaxel and gemcitabine, and the MB468 cell line exhibited increased sensitivity to these chemotherapeutics (Figure 1E). These findings indicate that CDK4/6 inhibition and RB-activation status controls the sensitivity to a significant subset of agents present within the Prestwick library.
Figure 1. CDK4/6 Inhibition Antagonizes Sensitivity to Select Agents.

(A) Sensitivity of agents within the Prestwick chemical library is defined as individual dots. The sensitivity in the presence (red) or absence (blue) of CDK4/6 inhibitor palbociclib (PD) is plotted. Select agents, for which pretreatment with CDK4/6 inhibitor antagonized initial cytotoxicity (robust Z score less than −3.0), are shown in the table. The score shown is the percent effect on viability, where −100 represents complete loss of viability.
(B) Dose response of cells treated with the indicated agents, either alone (black bars) or with prior treatment with a CDK4/6 inhibitor (gray bars). The mean and SD are shown (***p < 0.001, as determined by t test).
(C) Flow cytometry showing the DNA-content of either MB231 cells or S2 cells treated with colchicine, palbociclib (PD), or pretreated with palbociclib and then treated with colchicine. Representative histograms are shown.
(D) Dose response of cells treated with the indicated agents, either alone (black bars) or with prior treatment with a CDK4/6 inhibitor (gray bars). The mean and SD are shown (***p < 0.001, as determined by t test).
(E) MB231 and MB468 cells were treated with the indicated agents at increasing dose. The mean and SD are shown (***p < 0.001, as determined by t test).
Multiple Drugs Targeting PLK1 and CHK Are Antagonized by CDK4/6 Inhibition
Since the Prestwick library has only a limited number of targeted cancer therapeutics, the screen was repeated with a library of 305 cancer drugs (Figure 2A) that are in preclinical or clinical development (Witkiewicz et al., 2016). Consistent with the data from the Prestwick library, palbociclib elicited significant protection against the cytotoxicity mediated by chemotherapeutic agents, including gemcitabine and taxanes (Figure 2B). Because the drug library includes multiple drugs for a given target, we focused on hits that occurred with multiple drugs targeting the same protein/pathway. Using these criteria, we observed significant protection against the toxicity induced by polo-like kinase 1 (PLK1), aurora kinase (AURK), and checkpoint kinase (CHK) inhibitors (Figures 2C, 2D, and S2). In measuring the biochemical response to AZD7762 and CHIR124 inhibitors, we observed the expected increase in replication stress associated with CHK1 inhibition that is measured by the phosphorylation of replication protein A (RPA). Conversely, treatment with volasertib induced the phosphorylation of PLK1 on threonine 210 indicative of pharmacological kinase inhibition (Figure 2E). To determine the relevance of RB for these responses, we employed engineered MB231 models in which the RB protein was depleted as a consequence of a synthetic microRNA (miRNA) targeting the RB transcript or CRISPR/Cas9 gene editing. Immunofluorescence and immunoblotting were used to confirm homogeneous depletion of RB in cell cultures (Figure 2F). Since pharmaceutical agents can have off-target effects, we used RNAi to deplete CHK1 and PLK1. With this approach, CDK4/6 inhibition also resulted in protection from the RNAi-mediated toxicities in RB dependent fashion, as the CRISPR-RB derivatives were refractory to the effects of palbociclib. RB-deficient derivatives also exhibited increased sensitivity to CHK1 or PLK1 depletion (Figure 2G). Similar effects were observed through the acute depletion of RB using RNAi transfection (Figure S2). Therefore, the sensitivity to PLK1 and CHK1 depletion is coordinated by the activation status of RB, and RB deficiency may be associated with increased sensitivity.
Figure 2. CDK4/6 Inhibition Selectively Inhibits Sensitivity to Cell-Cycle Active Agents.

(A) Sensitivities of agents within the SelleckChem chemical library are defined as individual dots. The sensitivity in the presence (red) or absence (blue) of CDK4/6 inhibitor palbociclib (PD) is plotted.
(B) Dose-response relationship between naive and palbociclib-pretreated MB231 cultures with the indicated chemotherapy agents. The mean and SD are shown (*p < 0.05, **p < 0.01, ***p < 0.001, as determined by t test).
(C) Dose-response relationship between naive and palbociclib-pretreated MB231 cultures with the indicated PLK1 inhibitors. The mean and SD are shown (*p < 0.05, **p < 0.01, ***p < 0.001, as determined by t test).
(D) Dose-response relationship between naive and palbociclib pretreated MB231 cultures with the indicated CHK inhibitors. The mean and SD are shown (*p < 0.05, **p < 0.01, ***p < 0.001, as determined by t test).
(E) Immunoblot analysis of the indicated proteins from MB231 cells treated with the CHK (AZD7762 and CHIR124) and PLK1 inhibitors.
(F) Immunoblot analysis of RB-proficient and deficient models developed with a synthetic miRNA (miRB) or CRISPR/CAS9 mediated deletion. Immunofluorescence was used to confirm that less than 1% of the cells contained detectable nuclear RB staining (scale bar, 100 μm).
(G) Knockdown of PLK1 and CHK1 was performed in either RB-positive or RB-deficient cell populations by RNAi transfection. The efficacy of the knockdown was confirmed by immunoblot analysis of PLK1 and CHK1 in MB231 cells. Treatment with palbociclib was protective only in cell lines that contain RB, and RB-deficient models were more sensitive to the depletion of CHK1 and PLK1. The mean and SD are shown (**p < 0.01, ***p < 0.001, as determined by t test).
RB Loss Is Associated with Increased Sensitivity to PLK1 and CHK Inhibitors
To specifically define vulnerabilities occurring as a result of RB status, drug screening was performed with a number of cell lines that exhibit endogenous RB expression (BT20, MB453, MCF7, and MB231), intrinsically lack RB (MB468, BT549, AW23), or harbor RB depletion via specific manipulation (MB231 miRB). These data revealed a diversity of responses. RB-deficient models were consistently sensitive to CHK and PLK1 inhibitors (Figures 3A, 3B, and S3). Similarly RB-deficient models were more sensitive to select chemotherapies (taxanes and gemcitabine) (Figure 3C). Composite analysis of all agents and the impact of RB loss or activation with palbociclib indicated that there were several select response classes (Figure 3D): (1) agents that killed all cells irrespective of RB status (e.g., dinaciclib); (2) agents that had no therapeutic effect at the concentrations employed (e.g., AMG.208); (3) classes of drugs where RB loss was associated with drug resistance, including a variety of drugs that impact on signaling pathways (e.g., the phosphatidylinositol 3-kinase [PI3K] inhibitor GDC0980); and (4) PLK1 inhibitors, CHK inhibitors, and chemotherapy agents, where RB loss was associated with enhanced sensitivity while palbociclib caused drug resistance. As a complimentary approach and to define the effect of acute RB loss, RNAi was used to deplete RB protein from the MB453 model (Figure 3E). In this context, RB deficiency was associated with increased sensitivity to the CHK inhibitor AZD7762 and the PLK1 inhibitor volasertib.
Figure 3. RB Loss Is Associated with Increased Sensitivity to PLK1 and CHK Inhibitors.

(A) Relative sensitivity of the indicated RB-proficient (blue) and deficient (green) cell lines is shown for the indicated CHK inhibitors at 1 μM. The mean and SD are shown. Dose-response analysis is shown for the indicated RB-proficient and deficient cell lines to AZD7762. Statistical analysis of the aggregated data for RB-proficient and deficient models was determined by t test (**p < 0.01, ***p < 0.001). Rhe analysis of dose responses was determined by ANOVA analysis (p < 0.001) for the comparison across RB statuses.
(B) Relative sensitivity of RB-proficient (blue) and deficient (green) cell lines is shown for the indicated PLK1 inhibitors at 1 μM. The mean and SD are shown. Dose-response analysis is shown for the indicated RB-proficient and deficient cell lines to volasertib. Statistical analysis of the aggregated data for RB-proficient and deficient models was determined by t test (**p < 0.01, ***p < 0.001). The analysis of dose responses was determined by ANOVA analysis (p < 0.001) for the comparison across RB status.
(C) Relative sensitivity of RB-proficient (blue) and deficient (green) cell lines is shown for the indicated chemotherapies at 1 μM. The mean and SD are shown. Statistical analysis of the aggregated data for RB-proficient and deficient models was determined by t test (**p < 0.01, ***p < 0.001).
(D) Heatmap illustrating different drug responses in RB-proficient, RB-deficient, and palbociclib-treated cell lines. The color bar denotes the log fold change in cell viability, with blue indicating sensitivity.
(E) MB453 cells were transfected with either control non-targeting (NT) or RB1-directed RNAi. The efficacy of the knockdown was confirmed by immunofluorescence staining for RB protein (scale bar, 50 μm). Cells were treated with the indicated dose of AZD7762 or volasertib, and cell viability was determined. The mean and SD are shown. RB deficiency was associated with increased sensitivity (**p < 0.01, ***p < 0.001, as determined by t test).
(F) Representative immunofluorescence staining of mixed cultures of RB-proficient (MB231) and RB-deficient cell lines (AW23) (scale bar, 50 μm).
(G) Two independent mixed cultures were developed (MB231/AW23 and MB453/MB468). The relative fraction of RB-positive and RB-negative cells was determined for more than 250 cells in random fields from three independent experiments. Data for untreated cultures versus those treated with CHIR124 (500 nM) or volasertib (500 nM) are shown. The statistical difference in the percentage of RB-deficient cells was determined by t test (**p < 0.01, ***p < 0.001).
To interrogate the selectivity of the observed therapeutic sensitivities for RB-deficient TNBC models, heterogeneous cultures of RB-proficient and deficient cells were developed. This approach could be viewed as a model for intrinsic intra-tumor heterogeneity. Upon vehicle treatment, RB-deficient tumor cells dominated the heterogeneous cultures. In contrast, CHK and PLK1 inhibitors limited growth of the RB-deficient tumor population and resulted in cultures dominated by RB-proficient cells (Figures 3F and 3G), indicating that both drugs were selectively lethal in tumor cells with RB defects.
To further confirm these findings, a number of additional studies specifically focused on CHK1 inhibition were performed. Isogenic MCF10A and CALU1 subclones with RB defects were developed by small hairpin RNA (shRNA) and CRISPR technologies, respectively (Figure 4A). As observed in other models, depletion of RB caused increased sensitivity to CHK1 gene silencing (Figure 4B; p < 0.05 in both model systems). Selective sensitivity to the clinical CHK1 inhibitor PF-0047736 was then assessed in a larger panel of TNBC cell lines, and we could show that loss of RB correlated with enhanced sensitivity (Figures 4C and S4). To determine a specific role for RB in modulating responses to CHK inhibitors, we restored functional RB using ectopic expression of the constitutively active RB allele (7LP) by adenoviral transduction (Knudsen et al., 1998) (Figures 4D and S4). The expression of this RB mutant was confirmed in the RB-deficient MB436 and MB468 cell lines, and it reduced toxicity observed with the CHK inhibitor AZD7762 (Figure 4D). Restored RB activity also limited sensitivity to volasertib (Figure S4).
Figure 4. RB Deficiency Is Associated with Increased Sensitivity to CHK Inhibitors in Multiple Models of TNBC.

(A) Immunoblot analysis of CALU1 and MCF10A cells with RB gene CRISPR-mediated deletion and shRNA-mediated knockdown respectively.
(B) MCF10A or CAL51 cells and RB-deficient derivatives were reverse transfected with a CHK1 containing RNAi library in 384-well plates. Cell viability was determined by the use of CellTiter-Glo reagent (Promega). Luminescence values were log transformed and converted into Z scores compared to the median effect of all siRNAs within the 384-well plate. Negative Z scores indicate cell inhibition. Data show the mean effect from three replica experiments. Error bars represent SEM; significance was determined using the t test.
(C) The indicated cell lines were plated when growing in log phase in 384-well plates and exposed to a titration of PF-00477736 (1, 0.5, 0.1, 0.05, 0.01, 0.005, 0.001, 0.0005, and 0 μM) for five continuous days, at which point cell viability was determined. Values were normalized to the median of the per-plate DMSO-vehicle-only wells, and area under the dose-response curve (AUC) measurements were calculated. Each cell line was assessed in triplicate, with median AUC values shown. Compared to RB wild-type tumor cell lines, RB-defective tumor cell lines exhibited enhanced sensitivity to PF-00477736 (p = 0.0061, t test). The heatmap provides a complementary visualization of the data.
(D) RB-deficient TNBC cell lines (MB436 and MB468) were transduced with adenoviruses encoding LacZ or a constitutively active allele of RB (7LP). The degree of transduction was evaluated by immunofluorescence staining, with >90% of cells expressing RB. A representative image of MB436 cells is shown (scale bare is 50 μm). 24 hr post-transduction, cells were treated with increasing concentrations of AZD7762, and cell viability was evaluated after 72 hr. The mean and SD are shown. Expression of the constitutively active RB was associated with resistance to AZD7762 (t test, ***p < 0.001).
RB Loss Deregulates Mitotic and Replication Control in TNBC
To define how RB loss impacts the biology of TNBC tumors, immunohistochemically defined RB-positive and RB-deficient clinical cases were selected (Figure 5A). As previously reported, RB-negative cases showed upregulation of p16ink4a and a high proliferation index (Witkiewicz et al., 2011). RB-proficient and RB-deficient TNBC cases were matched for histologic grade and clinical stage (Figure S5), and both tumor tissue and associated normal tissue were utilized for RNA sequencing. In these specimens, RB loss was associated with a statistically significant change in 1,353 genes (p < 0.05). As expected, RB expression was diminished in the immunohistochemically negative tumors, and the compensatory upregulation of CDKN2A encoding p16ink4a was also evident (Figure 5B). While we observed a large number of additional genes that were downregulated in RB-deficient tumors, there was no strong enriched for a specific pathway or ontology (data not shown). In contrast, there was a significant upregulation of a large collection of genes associated with DNA replication and mitosis. The upregulated genes included CHK1 and PLK1 (Figure 5C), suggesting that high levels of these genes play an important role in sustaining viability upon RB loss. The upregulated genes were highly enriched for E2F-target genes, confirming a direct role for RB in modulating their expression (Figure S5). Additional breast cancer cases from The Cancer Genome Atlas (TCGA) Data Portal were analyzed and also illustrated that RB genetic status was associated with the expression of multiple factors involved in DNA replication and mitosis (Figure S5). Because these genes could be reflective of cell-cycle stress, we evaluated an established DNA replication stress signature in the RB-deficient versus RB-proficient tumors (Allera-Moreau et al., 2012). These analyses showed overall enrichment of the replication stress signature in RB-deficient tumors (Figure 5D).
Figure 5. RB-Deficient TNBC Harbor High Levels of Genes Associated with Replication and Mitosis.

(A) Representative immunostaining for RB, p16ink4a, and Ki67 to define RB-deficient TNBC cases.
(B) Heatmap of significantly altered genes as determined by RNA sequencing of RB-proficient and RB-deficient cases. Gene ontology analysis of upregulated genes shows highly significant enrichment for genes associated with DNA replication and mitosis. Transcript levels of RB1 and CDKN2A support the immunohistochemical analysis (t test, **p < 0.01, ***p < 0.001).
(C) Transcript levels of PLK1 and CHEK1 in normal, RB-positive, and RB-negative TNBC cases. The mean and specific transcript levels are plotted (t test, *p < 0.05).
(D) Heatmap depicting the Z score in the expression of genes in a replication stress signature. Red and green color bars denote RB-negative and RB-positive cases, respectively.
Mechanistic Impact of RB Loss on Replication and Mitotic Stress
To decipher the impact of RB loss on sensitivity to CHK and PLK1 inhibitors, detailed cell-cycle analysis was performed (Figures 6A and 6B). While both RB-proficient and deficient TNBC cell lines had similar cell-cycle profiles and proliferation rates in the absence of cellular stress, the response to therapeutic stresses was significantly different. With PLK1 inhibition RB-proficient MB231 cells progressed into G2/M, but further cell-cycle progression was attenuated as noted by a reduction in bromodeoxyuridine (BrdU) incorporation (Figure 6C). In contrast, RB-deficient cells maintained a high-level of DNA replication and progressed to higher 8N ploidy (Figure 6D). These data indicate that RB plays a critical role in coordinating G2/M arrest and preventing aberrant DNA replication. These general findings were recapitulated in other RB-proficient and deficient cell lines (Figure S6). In the case of CHK inhibition, RB-deficient cells experienced S phase collapse that is indicative of irreparable damage occurring in S phase (Figures 6A and 6B). Treatment with CHK inhibitors demonstrated substantial PCNA engagement with chromatin and the appearance of fragmented nuclei (Figure 6E) that was accompanied by the selective induction of cell death as indicated by PARP cleavage (Figures 6F and S6). Conversely, restoration of RB expression in MB436 cells resulted in cell-cycle inhibition that limited mitotic entry and nuclear fragmentation/mitotic catastrophe with volasertib (Figure 6G). In the case of CHK inhibition, RB activation resulted in the suppression of DNA replication and associated DNA damage as determined by γ-H2AX reactivity (Figure 6H). Similar results were observed with the protective effect of CDK4/6 inhibition on sensitivity to CHK and PLK1 inhibitors in MB231 cells (Figure S6). Together, these data suggest that deficits in DNA replication control are likely a key determinant of therapeutic sensitivities imparted by RB loss.
Figure 6. RB-Deficient Cells Are Prone to Replicative Catastrophe and Mitotic Dysregulation.

(A) Flow-cytometry traces of MB231 and MB231 RB CRISPR cells treated with the indicated agents (x axis, DNA content; y axis, BrdU incorporation). The positions of 2N, 4N, and 8N ploidy are indicated.
(B) Quantitation of BrdU intensity in control (red) or CHK inhibitor (CHIR124)-treated (blue) cells. The mean and SD are shown. The BrdU collapse is indicative of increased S phase DNA damage and failure or replication.
(C) Quantitation of the percentage of BrdU-positive cells treated with CHIR124. The mean and SD are shown. Significant differences between RB-proficient and deficient cells were determined by t test (**p < 0.01).
(D) Quantitation of cell ploidy in the RB-proficient and deficient cells with the indicated treatments. The mean and SD are shown. Significant differences between RB-proficient and deficient cells were determined by t test (**p < 0.01).
(E) Quantitation of chromatin-associated PCNA following treatment with AZD7762. The mean and SD are shown (**p < 0.01). Representative immunofluorescence images of RB-proficient and deficient cells following treatment with the CHK inhibitor AZD7762 (scale bar, 25 μm).
(F) Immunoblotting of the indicated cell lines treated with increasing doses of CHK inhibitor (AZD7762). PARP cleavage is a marker for apoptotic cell death.
(G) MB436 cells either transduced with control virus (LacZ) or expressing constitutively active RB (7LP) were treated with volasertib, and the percentage of cells exhibiting mitotic entry was determined by staining for phosphorylated serine 10 of histone H3. The mean and SD are shown. Significant effect of constitutively active RB on the response to volasertib was determined by t test (***p < 0.001). Representative images showing combined staining for EdU and phosphorylated serine 10 histone H3 and DAPI (scale bar, 20 μm).
(H) MB436 cells either transduced with control virus (LacZ) or expressing constitutively active RB (7LP) were treated with CHIR124, and the percentage of cells exhibiting γ-H2AX reactivity was determined by immunofluorescent staining. The mean and SD are shown. Significant effect of constitutively active RB on the response to CHIR124 was determined by t test (***p < 0.001). Representative images showing combined staining for EdU and phosphorylated γ-H2AX and DAPI (scale bar, 20 μm).
Selective Efficacy of CHK Inhibitors in RB-Deficient Xenografts
To define the selective vulnerability imparted by RB loss in vivo, xenograft studies were performed. For this work, isogenic MB231 xenografts were established in NSG mice (Figure 7A). Mice were randomized to vehicle, AZD7762 or AZD7762 + gemcitabine treatment arms on the indicated schedule (Figure 7B). To ensure that drugs were not overtly toxic at the delivered doses, mouse weight was recorded throughout the study. There was minimal effect on weight, and animals appeared healthy on treatment (Figure 7C). Mice were treated for 21 days, with tumor volume measured daily. In MB231 xenografts, there was no significant tumor growth inhibition with CHK inhibitor alone and only a minor reduction with the gemcibabine + AZD7762 combination (Figures 7D and 7E). In contrast, the MB231 RB CRISPR model was markedly sensitive to both single and combination treatments (Figures 7D and 7E). The same treatments were also performed in the RB-deficient MB436 model that was also highly responsive to the single agent and combination therapy (Figure S7). Analysis of tumor histology demonstrated significant areas of necrosis and evidence of mitotic catastrophe in the RB-deficient tumors (Figure 7F and not shown). Similarly the RB-deficient tumors harbored more DNA damage as indicated by staining for γ-H2AX (Figure S7). These data indicate that RB loss translates into increased sensitivity to CHK inhibition in vivo.
Figure 7. Selective Efficacy of CHK Inhibitors in RB-Deficient Xenografts.

(A) Representative RB immunohistochemistry for MB231 and MB231 RB CRISPR xenografts. Scale bar, 50 μm.
(B) Treatment schedules for xenograft models. Mice were randomized to the treatments when tumors were ~250 mm3, and treatment proceeded for 21 days.
(C) Mouse weight was determined daily. No significant differences in mouse body weights were observed through the treatment course.
(D) Relative tumor volume was determined by digital calipers daily for RB-proficient tumors treated with vehicle (n = 11), AZD7762 (n = 11), or AZD7762 + gemcitabine (n = 11) and RB CRISPR tumors treated with vehicle (n = 7), AZD7762 (n = 8), or AZD7762 + gemcitabine (n = 8). The mean and SD of tumor size are plotted. In the case of MB231 tumors, a statistically significant difference in tumor volume was observed only with gemcitabine + AZD7762 treatment. In the case of MB231 RB CRISPR tumors, statistically significant differences were observed with all treatments.
(E) Representative images of excised tumors at the end of 21 days of treatment. Scale bar, 1 cm.
(F) Representative images of H&E-stained sections showing large areas of tumor cell necrosis in the MB231 RB CRISPR tumors treated with drug. Scale bar, 100 μm.
DISCUSSION
Despite substantial genetic analysis, few targeted therapies have emerged for the treatment of TNBC (Herold and Anders, 2013; Turner and Reis-Filho, 2013). Here, we explored whether loss of the RB tumor suppressor could represent a specific vulnerability for therapeutic intervention. Unlike other breast cancer subtypes, TNBC does not have a clear oncogenic driver, although there are defined subclasses of TNBC that harbor distinct prognoses (Geyer et al., 2017; Lehmann et al., 2011). RB loss occurs in ~30% of TNBC cases, and therefore, selectively targeting this event could have a substantial impact on patients. Several groups have evaluated how RB deficiency impinges on therapeutic sensitivity in breast cancer models (Jones et al., 2016; Knudsen and Wang, 2010; Robinson et al., 2013). These studies have indicated that RB loss can sensitize to treatment with select chemotherapies, radiation therapy, or agents that impact on mitochondrial translation. Both preclinical analysis and evaluation of RB-deficient tumors support the premise that RB loss is associated with increased sensitivity to select chemotherapies in breast cancer (Bosco et al., 2007; Witkiewicz et al., 2012). However, the spectrum of agents for which therapeutic responses are modulated by RB status has not been clearly defined, and the RB-targeted therapeutic strategy in TNBC has yet to emerge.
By using a CDK4/6 inhibitor, we observed that activated RB antagonized a surprisingly narrow spectrum of agents in TNBC cell lines. This included microtubule poisons that are used in chemotherapy and as anti-helmenthic agents. Additionally, CDK4/6 inhibition antagonized the action of agents that would be expected to function through replication stress and represent both historical (e.g., mercaptopurine) and currently used (e.g., gemcitabine) therapeutic agents. However, CDK4/6 inhibition did not antagonize a myriad of additional compounds, including antibiotics, HDAC inhibitors, and targeted drugs against multiple signaling pathways (e.g., MEK, PI3K, and MTOR inhibitors). Among molecularly targeted agents, CDK4/6 inhibition consistently antagonized PLK, AURK, and CHK inhibitors. Significantly, these were not single agent-specific effects, as similar results were observed across multiple agents within a drug class. Because PLK (PLK1, PLK2, and PLK3), AURK (AURKA and AURKB), and CHK (CHK1 and CHK2) represent kinase families, it is difficult to ascribe sensitivity to the specific inhibition of one member. Agents targeting PLK are generally directed at PLK1 but can also inhibit other PLK family members. Among CHK inhibitors utilized in this study, AZD7762 equivalently inhibits CHK1 and CHK2, while other agents have more specificity toward CHK1. Biochemical analysis as well as knockdown approaches also suggest that CHK1 and PLK1 are the important targets for RB-dependent lethality. While the functional antagonism scored in these assays is important for assessing a functional role of the RB pathway in the drug response, it is important to note that short-term antagonism may not be representative of long-term therapeutic responses with CDK4/6 combinations that are being tested in multiple clinical trials (Knudsen and Witkiewicz, 2017). Carefully designed preclinical studies with appropriate scheduling will be required to determine whether long-term combination therapeutic approaches with CDK4/6 inhibitors could have a positive clinical effect in spite of short-term antagonism.
Counter-screens using panels of RB-deficient cell lines and isogenic models confirmed a number of the sensitivities elucidated by CDK4/6 antagonism. Using panels of cells lines with intrinsically varying RB status is important, as it reflects genetic features of TNBC that developed with or without RB tumor suppressor loss. However, isogenic models (e.g., shRNA knockdowns and CRISPR-mediated deletions) are also critical in confirming specificity of the observed effects relative to RB status. These approaches were employed in parallel to define specificity of RB status mediated sensitivities to CHK and PLK inhibitors. Efficacy was further confirmed in xenografts, which demonstrated a significant increase in the therapeutic index in models with RB loss. Importantly, the treatments were very well tolerated in animals, suggesting that targeting CHK in RB-deficient tumors would be clinically feasible.
That RB loss sensitizes to drugs that impact cell-cycle checkpoints provides an important extension in understanding biological functions of this tumor suppressor. TNBC is a highly aggressive tumor irrespective of RB status and, in fact, depleting RB has no discernible effect on proliferation rate. However, analysis of clinical samples from RB-deficient TNBC demonstrated that these tumor cells express high levels of DNA replication and G2/M factors and overexpress the specific targets of both CHK1 and PLK1 inhibition. Additionally, RB loss is associated with inherent DNA-replication stress and deficits in G2/M progression that ostensibly cooperate with the inhibition of CHK1 and PLK1, respectively (Manning and Dyson, 2012; Tort et al., 2006). Multiple agents that induce replication stress were highly effective in RB-deficient tumor cells, suggesting that a greater dependence on CHK1 is an intrinsic feature associated with RB loss. Presumably the activity of these parallel cell-cycle regulatory mechanisms allows for the survival of RB-deficient TNBC and therefore served as a synthetic vulnerability. While CHK1 and PLK1 have been the targets of robust clinical development, to date, clinical success has been modest (Gutteridge et al., 2016; Thompson and Eastman, 2013). In part, this could reflect the lack of biomarkers upon which to direct treatment. Clinical trials with CHK1 and PLK1 inhibitors have largely been open to all patients with a specific malignancy diagnosis and not directed with any biomarker. The one exception is a clinical trial of CHK1 inhibitors under conditions of replicative stress or homologous repair deficiency (NCT02873975). Importantly, only through rigorous prospective clinical trials will it be possible to fully leverage preclinical findings to potentially advance TNBC treatment approaches. Given the frequency of RB deficiency in TNBC, it would be possible to evaluate efficacy of CHK1 or PLK1 inhibition in tumors exhibiting RB. In xenograft models, we observed strong disease control with single-agent CHK1 inhibition over a 3-week period. Gemcitabine modestly enhanced the response, suggesting potential for synergistic interactions. Together, the data herein support selective targeting of RB loss in TNBC and indicate several different strategies for a precision intervention in TNBC based on tumor suppressor loss.
EXPERIMENTAL PROCEDURES
Cell Culture
The cell lines used in the study were obtained from the ATCC, with the exception of the AW23 cell line that was developed from a patient with RB-deficient triple-negative breast cancer. Cell lines were cultured as denoted by the ATCC. The AW23 cell line was cultured in DMEM + 10% fetal bovine serum (FBS). The identity of cell lines was confirmed by short tandem repeat (STR) typing using the StemElite Kit (Promega). Profiles were confirmed using the Children’s Oncology Group (COG) Cell Culture and Xenograft Repository bank (http://strdb.cogcell.org) or JCRB cell bank (http://cellbank.nibiohn.go.jp/english). In addition, cell lines were tested monthly throughout experimentation for mycoplasma infection using the MycoAlert kit (Lonza).
Drug Screening
The Prestwick library was applied to MB231 cells growing in standard media or pretreated with 1 μM palbociclib for 24 hr. Cell survival was evaluated after 72 hr by CellTiter-Glo (CTG). Selection of hits for validation was determined by multi-point rich Z (RZ) scores (Birminghamet al., 2009). Validation dose response analysis was performed in MCF7, MB231, and MB468 cells. The oncology drug panel that contains 305 drugs covering multiple distinct drug classes was previously described (Witkiewicz et al., 2016). This panel was delivered at 250 nM and 1 μM to the indicated cell lines either alone or following pretreatment with the CDK4/6 inhibitor palbociclib. Cell viability was determined by CTG analysis at 72 hr following delivery of the library. Drug screening was performed as previously published (Witkiewicz et al., 2016). In determining the sensitivity of multiple breast cancer lines to PF-00477736, the indicated cell lines were cultured at different drug doses (1, 0.5, 0.1, 0.05, 0.01, 0.005, 0.001, 0.0005, and 0 μM) for five continuous days. Cell viability was determined using CellTiter-Glo reagent (Promega). Luminescence values were normalized to the median of the per-plate DMSO vehicle only control wells and the dose-response relationships modeled using 3-parameter logistic regression provided by the DRC R-package. Area under the dose-response curve (AUC) measurements calculated using this package were used as a readout of drug sensitivity. Each cell line was assessed in triplicate, with median AUC values shown.
RNAi Transfection and Adenovirus Transduction
TNBC cell lines were reverse transfected when growing in log phase in a 384-well plate format with a CHK1 siRNA as previously described (Campbell et al., 2016). Alternatively, cells were forward or reverse transfected with RNAi to PLK1, RB1, or CHK1 using standard procedures (Dharmacon). After 3–4 days of culture, cell viability was determined using CellTiter-Glo reagent or cells were utilized for western blotting or immunofluorescence microscopy. Adenovirus encoding LacZ or PSM.7-LP have been previously described (Braden et al., 2006). Titration was performed by immunofluorescence staining of infected cultures to define a transduction efficiency of 90%–100%. Cells were employed 24 hr post-infection for response to drug treatment.
Cell-Cycle Analysis
Cells were treated with drug (CHIR124 or volasertib) for 48 hr. Prior to harvesting, cells were pulsed with BrdU or 5-ethynyl-2′-deoxyuridine (EdU) for 1 hr. Cells were trypsinized after BrdU pulse and fixed in 70% ethanol overnight in 4°C. Cell pellets were washed once with IFA buffer (1× HEPES, 4% FBS and 0.1% NaN3) and then with IFA + 0.5% Tween20. Pellets were incubated in fluorescein isothiocyanate (FITC)-conjugated anti-BrdU (BD Pharmingen) for 1 hr at room temperature. Cells were suspended in propidium iodide and RNase before analysis in FASCanto II flow cytometer. Cell-cycle analysis was performed with FlowJo software. EdU incorporation was detected using Click-iT EdU imaging kits, and percent incorporation was determined by counting random fields (Thermo Fisher).
Immunoblotting
Cells were seeded in 60-mm plates at 1.5 × 105 density and were treated the next. After 48 hr, cells were collected and subject to RIPA lysis buffer (50 mM Tris, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, sodium orthovanadate, sodium fluoride, EDTA, and leupeptin). Protein concentration was determined by Bradford Protein Assay dye (Bio-Rad) and 30 μg of protein per sample was resolved by SDS-PAGE. Gel was transferred to Immobilon-P membrane (Millipore). Blots were blocked with 5% milk for 1 hr at room temperature and incubated with primary antibody overnight at 4°C. Incubation in secondary antibody was at room temperature for 1 hr. Antibodies for specific proteins detected were PARP (9542, Cell Signaling), RB (4H1, Cell Signaling), PLK1 (208G4, Cell Signaling), pRPA32 (T21, Abcam), and phospho-PLK1 (Thr210, Cell Signaling).
Immunofluorescence
Cells were seeded on glass coverslips and treated with drug (AZD7762, CHIR124, and volasertib) for 48 hr. For the staining of γ-H2AX (2572, Cell Signaling), PCNA (PC10, Santa Cruz), and phospho-serine 10 histone H3 (06-570, Millipore), cells were washed in PBS and fixed in 4% paraformaldehyde. They were then permeabilized in 0.5% Triton X-100. For PCNA staining, cells were pre-extracted with CSK buffer (10 mM HEPES, 300 mM sucrose, 100 mM NaCl, and 3 mM MgCl2) and extraction buffer (50 mM NaF, 0.1 mM sodium orthovanadate, 1 mM PMSF, 0.5% Triton X-100, and protease inhibitor). After permeabilization, cells were blocked in immunofluorescence (IF) buffer (5% BSA, 0.4% NP40 in PBS) and incubated with primary antibody diluted in IF buffer for 1 hr in 37°C. Coverslips were washed in PBS and secondary antibody diluted in IF buffer was applied for 1 hr in 37°C. Coverslips were washed after staining after staining with the secondary antibody and mounted on slides. Images were taken with a Leica confocal microscope at 63×magnification. For the staining of RB, cells were fixed in methanol and stained with 4H1 antibody (Cell Signaling) as previously described (Franco et al., 2016).
Mixed Culture Analysis
The indicated RB-proficient and deficient cells were mixed at a 1:3 ratio and plated onto collagen-coated coverslips. Cells were treated for 72 hr and fixed in methanol, and the fraction of RB-positive cells was determined by immunofluorescence microscopy. Experiments were performed in triplicate, with over 200 cells counted per condition from random fields. The percentage of cells that were RB positive or negative was determined and the average and SD determined. Differences in the percentage of RB-negative cells were assessed for statistical significance using the Student’s t test.
Immunohistochemistry and RNA-Sequencing Analysis of TNBC Cases
Excess tissue from TNBC surgical resections was collected under an institutional review board (IRB)-approved protocol. The immunohistochemistry for RB and p16ink4a was performed on formalin-fixed paraffin-embedded specimens as previously reported (Knudsen et al., 2012). Total RNA was prepared from frozen tissue for cases that were either RB+/p16ink4alow or RB−/p16ink4ahigh as determined by immunohistochemistry. Samples were sequenced with Thermo Fisher’s SOLiD whole-transcriptome sequencing with 50-bp paired ends. RNA reads were aligned using Thermo Fisher’s LifeScope Genomic Analysis software v.2.5.1 and counts generated from aligned BAM files using HTSeq. The accession number for the RNA sequencing data reported in this paper is GEO: GSE108757, deposited at the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/), and can be accessed with the manuscript title “Targeting the vulnerability of RB tumor suppressor loss in triple negative breast cancer.” Counts were log-normalized using the edgeR R package. p values were calculated between RB-positive and RB-negative samples using a Student’s t test, assuming unequal variance. Genes were determined to be differentially expressed between the two groups if p < 0.05. Z scores were calculated for each gene and heatmaps were generated in R. Gene ontology analysis was run through PANTHER. Boxplots were generated using log-normalized expression values for select genes in GraphPad Prism 7.
Analysis of TCGA Data
RB1 mutation status was determined for triple-negative breast cancer cases from TCGA (Cancer Genome Atlas, 2012). Cases were considered RB negative if a truncating variant or homozygous deletion was called in the sample. Log-fold changes and p values were calculated for genes with a p value less than 0.1 in our triple-negative breast cancer samples. The volcano plot was created for these genes using R.
In Vivo Xenograft Studies
Female NSG mice were injected with MB231, MB231 RB CRISPR, or MB436 cells (2–3 × 106 cells per mouse). Mice were randomized to the control, AZD7762, and combination (AZD7762 and gemcitabine) groups when tumor volumes reached 150–200 mm3. In the control group, mice were administered with vehicle. AZD7762 group was administered AZD7762 (25 mg/kg) via intra-peritoneal (IP) injections daily. Combination group was injected with gemcitabine (50 mg/kg, IP) on day 1, followed by AZD7762 (25 mg/kg, IP) on days 2 and 3. The therapy lasted for 21 days, unless tumor volumes reached 2,000 mm3 at the earlier time point. AZD7762 was prepared in 11.3% 2-hydroxyproply-β-cyclodextrin (Sigma, St Louis, MO) and sterile saline. Gemcitabine was dissolved in sterile saline. Tumor size was measured every day, and volume was calculated using the following equation: V = 0.5 × ([greatest diameter] × [shortest diameter]2).
Immunohistochemistry on Xenograft Tumors
Staining for RB1 and γ-H2AX was performed on harvested formalin-fixed paraffin-embedded tumor tissue as previously described (Knudsen et al., 2012; Witkiewicz et al., 2016). Sections stained with for γ-H2AX were scored manually by the same operator (A.K.W.) using a Zeiss microscope with a 63× objective. γ-H2AX-positive cells were scored for nuclei with >5 γ-H2AX foci visible. Greater than 250 cells were scored per slide, and the average number of positive cells was determined. The counts were performed on three slides (corresponding to individual xenografts) for each treatment condition.
Supplementary Material
Highlights.
RB loss in TNBC drives increased DNA replication and mitotic gene expression programs
CDK4/6 inhibition yields RB-dependent protection against select chemotherapeutics
CHK and PLK inhibitors exploit a vulnerability conferred by RB loss in TNBC models
RB loss increases efficacy of select therapies in TNBC xenograft models
Acknowledgments
The authors thank all members of the Witkiewicz and Knudsen laboratories for technical support and discussion of the study results. This work was supported by NIH grants CA188650 and CA163863 (E.S.K. and A.K.W.) and Programme Funding from Breast Cancer Now (to C.J.L.).
Footnotes
Supplemental Information includes seven figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.01.022.
DECLARATION OF INTERESTS
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Conceptualization, A.K.W., E.S.K., and C.J.L.; Methodology Development, S.C., P.V., R.B., and A.K.W.; Formal Analysis of Data, S.C., P.V., R.B., J.F., E.S.K., A.K.W., and C.J.L.; Investigation, S.C., P.V., J.F., R.B., E.S.K., A.K.W., and C.L.; Writing and Editing, A.K.W., E.S.K., C.J.L., and S.C.; Supervision, A.K.W., E.S.K., and C.J.L.; Funding, E.S.K. and A.K.W.
References
- Allera-Moreau C, Rouquette I, Lepage B, Oumouhou N, Walschaerts M, Leconte E, Schilling V, Gordien K, Brouchet L, Delisle MB, et al. DNA replication stress response involving PLK1, CDC6, POLQ, RAD51 and CLASPIN upregulation prognoses the outcome of early/mid-stage non-small cell lung cancer patients. Oncogenesis. 2012;1:e30. doi: 10.1038/oncsis.2012.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders CK, Zagar TM, Carey LA. The management of early-stage and metastatic triple-negative breast cancer: a review. Hematol Oncol Clin North Am. 2013;27:737–749. viii. doi: 10.1016/j.hoc.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130–146. doi: 10.1038/nrd4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birmingham A, Selfors LM, Forster T, Wrobel D, Kennedy CJ, Shanks E, Santoyo-Lopez J, Dunican DJ, Long A, Kelleher D, et al. Statistical methods for analysis of high-throughput RNA interference screens. Nat Methods. 2009;6:569–575. doi: 10.1038/nmeth.1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco EE, Knudsen ES. RB in breast cancer: at the crossroads of tumorigenesis and treatment. Cell Cycle. 2007;6:667–671. doi: 10.4161/cc.6.6.3988. [DOI] [PubMed] [Google Scholar]
- Bosco EE, Wang Y, Xu H, Zilfou JT, Knudsen KE, Aronow BJ, Lowe SW, Knudsen ES. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J Clin Invest. 2007;117:218–228. doi: 10.1172/JCI28803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braden WA, Lenihan JM, Lan Z, Luce KS, Zagorski W, Bosco E, Reed MF, Cook JG, Knudsen ES. Distinct action of the retinoblastoma pathway on the DNA replication machinery defines specific roles for cyclin-dependent kinase complexes in prereplication complex assembly and S-phase progression. Mol Cell Biol. 2006;26:7667–7681. doi: 10.1128/MCB.00045-06. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Burkhart DL, Sage J. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer. 2008;8:671–682. doi: 10.1038/nrc2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell J, Ryan CJ, Brough R, Bajrami I, Pemberton HN, Chong IY, Costa-Cabral S, Frankum J, Gulati A, Holme H, et al. Large-scale profiling of kinase dependencies in cancer cell lines. Cell Rep. 2016;14:2490–2501. doi: 10.1016/j.celrep.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas; Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Chen F, Yang X, Xu W, Xie J, Yu L. Phylogenetic analysis of CDK and cyclin proteins in premetazoan lineages. BMC Evol Biol. 2014;14:10. doi: 10.1186/1471-2148-14-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, Ollila DW, Sartor CI, Graham ML, Perou CM. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007;13:2329–2334. doi: 10.1158/1078-0432.CCR-06-1109. [DOI] [PubMed] [Google Scholar]
- Dyson NJ. RB1: a prototype tumor suppressor and an enigma. Genes Dev. 2016;30:1492–1502. doi: 10.1101/gad.282145.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–1948. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- Franco J, Balaji U, Freinkman E, Witkiewicz AK, Knudsen ES. Metabolic reprogramming of pancreatic cancer mediated by CDK4/6 inhibition elicits unique vulnerabilities. Cell Rep. 2016;14:979–990. doi: 10.1016/j.celrep.2015.12.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer FC, Pareja F, Weigelt B, Rakha E, Ellis IO, Schnitt SJ, Reis-Filho JS. The spectrum of triple-negative breast disease: high- and low-grade lesions. Am J Pathol. 2017;187:2139–2151. doi: 10.1016/j.ajpath.2017.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutteridge RE, Ndiaye MA, Liu X, Ahmad N. Plk1 inhibitors in cancer therapy: from laboratory to clinics. Mol Cancer Ther. 2016;15:1427–1435. doi: 10.1158/1535-7163.MCT-15-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold CI, Anders CK. New targets for triple-negative breast cancer. Oncology (Williston Park) 2013;27:846–854. [PubMed] [Google Scholar]
- Johnson SM, Torrice CD, Bell JF, Monahan KB, Jiang Q, Wang Y, Ramsey MR, Jin J, Wong KK, Su L, et al. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J Clin Invest. 2010;120:2528–2536. doi: 10.1172/JCI41402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RA, Robinson TJ, Liu JC, Shrestha M, Voisin V, Ju Y, Chung PE, Pellecchia G, Fell VL, Bae S, et al. RB1 deficiency in triple-negative breast cancer induces mitochondrial protein translation. J Clin Invest. 2016;126:3739–3757. doi: 10.1172/JCI81568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen ES, Knudsen KE. Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer. 2008;8:714–724. doi: 10.1038/nrc2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen ES, Wang JY. Targeting the RB-pathway in cancer therapy. Clin Cancer Res. 2010;16:1094–1099. doi: 10.1158/1078-0432.CCR-09-0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen ES, Witkiewicz AK. The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer. 2017;3:39–55. doi: 10.1016/j.trecan.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen ES, Buckmaster C, Chen TT, Feramisco JR, Wang JY. Inhibition of DNA synthesis by RB: effects on G1/S transition and S-phase progression. Genes Dev. 1998;12:2278–2292. doi: 10.1101/gad.12.15.2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen ES, Pajak TF, Qeenan M, McClendon AK, Armon BD, Schwartz GF, Witkiewicz AK. Retinoblastoma and phosphate and tensin homolog tumor suppressors: impact on ductal carcinoma in situ progression. J Natl Cancer Inst. 2012;104:1825–1836. doi: 10.1093/jnci/djs446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355:1152–1158. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning AL, Dyson NJ. RB: mitotic implications of a tumour suppressor. Nat Rev Cancer. 2012;12:220–226. doi: 10.1038/nrc3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markey MP, Bergseid J, Bosco EE, Stengel K, Xu H, Mayhew CN, Schwemberger SJ, Braden WA, Jiang Y, Babcock GF, et al. Loss of the retinoblastoma tumor suppressor: differential action on transcriptional programs related to cell cycle control and immune function. Oncogene. 2007;26:6307–6318. doi: 10.1038/sj.onc.1210450. [DOI] [PubMed] [Google Scholar]
- McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O’Connor MJ, Tutt AN, Zdzienicka MZ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–8115. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- Meyer CA, Jacobs HW, Datar SA, Du W, Edgar BA, Lehner CF. Drosophila Cdk4 is required for normal growth and is dispensable for cell cycle progression. EMBO J. 2000;19:4533–4542. doi: 10.1093/emboj/19.17.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevins JR. The Rb/E2F pathway and cancer. Hum Mol Genet. 2001;10:699–703. doi: 10.1093/hmg/10.7.699. [DOI] [PubMed] [Google Scholar]
- Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, Katayama R, Costa C, Ross KN, Moran T, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun. 2015;6:6377. doi: 10.1038/ncomms7377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareja F, Geyer FC, Marchiò C, Burke KA, Weigelt B, Reis-Filho JS. Triple-negative breast cancer: the importance of molecular and histologic subtyping, and recognition of low-grade variants. NPJ Breast Cancer. 2016;2:16036. doi: 10.1038/npjbcancer.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis-Filho JS, Tutt AN. Triple negative tumours: a critical review. Histopathology. 2008;52:108–118. doi: 10.1111/j.1365-2559.2007.02889.x. [DOI] [PubMed] [Google Scholar]
- Roberts PJ, Bisi JE, Strum JC, Combest AJ, Darr DB, Usary JE, Zamboni WC, Wong KK, Perou CM, Sharpless NE. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J Natl Cancer Inst. 2012;104:476–487. doi: 10.1093/jnci/djs002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TJ, Liu JC, Vizeacoumar F, Sun T, Maclean N, Egan SE, Schimmer AD, Datti A, Zacksenhaus E. RB1 status in triple negative breast cancer cells dictates response to radiation treatment and selective therapeutic drugs. PLoS ONE. 2013;8:e78641. doi: 10.1371/journal.pone.0078641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ, Beach D, Shapiro GI. Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 2016;6:353–367. doi: 10.1158/2159-8290.CD-15-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stengel KR, Dean JL, Seeley SL, Mayhew CN, Knudsen ES. RB status governs differential sensitivity to cytotoxic and molecularly-targeted therapeutic agents. Cell Cycle. 2008;7:1095–1103. doi: 10.4161/cc.7.8.5737. [DOI] [PubMed] [Google Scholar]
- Thompson R, Eastman A. The cancer therapeutic potential of Chk1 inhibitors: how mechanistic studies impact on clinical trial design. Br J Clin Pharmacol. 2013;76:358–369. doi: 10.1111/bcp.12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tort F, Bartkova J, Sehested M, Orntoft T, Lukas J, Bartek J. Retinoblastoma pathway defects show differential ability to activate the constitutive DNA damage response in human tumorigenesis. Cancer Res. 2006;66:10258–10263. doi: 10.1158/0008-5472.CAN-06-2178. [DOI] [PubMed] [Google Scholar]
- Turner NC, Reis-Filho JS. Tackling the diversity of triple-negative breast cancer. Clin Cancer Res. 2013;19:6380–6388. doi: 10.1158/1078-0432.CCR-13-0915. [DOI] [PubMed] [Google Scholar]
- Witkiewicz AK, Knudsen ES. Retinoblastoma tumor suppressor pathway in breast cancer: prognosis, precision medicine, and therapeutic interventions. Breast Cancer Res. 2014;16:207. doi: 10.1186/bcr3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewicz AK, Knudsen KE, Dicker AP, Knudsen ES. The meaning of p16(ink4a) expression in tumors: functional significance, clinical associations and future developments. Cell Cycle. 2011;10:2497–2503. doi: 10.4161/cc.10.15.16776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewicz AK, Ertel A, McFalls J, Valsecchi ME, Schwartz G, Knudsen ES. RB-pathway disruption is associated with improved response to neoadjuvant chemotherapy in breast cancer. Clin Cancer Res. 2012;18:5110–5122. doi: 10.1158/1078-0432.CCR-12-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkiewicz AK, Balaji U, Eslinger C, McMillan E, Conway W, Posner B, Mills GB, O’Reilly EM, Knudsen ES. Integrated patient-derived models delineate individualized therapeutic vulnerabilities of pancreatic cancer. Cell Rep. 2016;16:2017–2031. doi: 10.1016/j.celrep.2016.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing F, Persaud Y, Pratilas CA, Taylor BS, Janakiraman M, She QB, Gallardo H, Liu C, Merghoub T, Hefter B, et al. Concurrent loss of the PTEN and RB1 tumor suppressors attenuates RAF dependence in melanomas harboring (V600E)BRAF. Oncogene. 2012;31:446–457. doi: 10.1038/onc.2011.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.