

Summary
State-of-the-art proteomics software routinely quantifies thousands of peptides per experiment with minimal need for manual validation or processing of data. For the emerging field of discovery lipidomics via liquid chromatography tandem mass spectrometry (LC-MS/MS), comparably mature informatics tools do not exist. Here we introduce LipiDex, a freely available software suite which unifies and automates all stages of lipid identification, reducing hands-on processing time from hours to minutes for even the most expansive data sets. LipiDex utilizes flexible in-silico fragmentation templates and lipid-optimized MS/MS spectral matching routines to confidently identify and track hundreds of lipid species and unknown compounds from diverse sample matrices. Unique spectral and chromatographic peak purity algorithms accurately quantify co-isolation and co-elution of isobaric lipids, generating identifications which match the structural resolution afforded by the LC-MS/MS experiment. During final data filtering, ionization artifacts are removed to significantly reduce dataset redundancy. LipiDex interfaces with several LC-MS/MS software packages making robust lipid identification readily incorporated into preexisting data workflows.
Keywords: Lipidomics, mass spectrometry, open-source software
Graphical abstract
Hutchins et al. introduce an open-source software suite which automates lipid identification and significantly reduces hands-on processing time for large LC-MS/MS lipidomic data sets. They demonstrate the confident identification of hundreds of diverse lipids from complex lipid extracts using optimized spectral matching techniques and multiple identification filters.
Introduction
When coupled with chromatographic separation, the high resolution, mass accuracy, and speed of modern MS systems render them the premier tool for unbiased analysis of diverse biomolecules (Cajka and Fiehn, 2016; Hebert et al., 2014; Patti et al., 2012). Although less mature relative to other -omic technologies (Figure S1), discovery lipidomic analysis by LC-MS/MS is poised to make significant contributions to biomedical research. Already, this technology has uncovered novel lipids which improve glucose tolerance in mice (Yore et al., 2014), mapped lipid remodeling during platelet activation (Slatter et al., 2016), and revealed lipid dysregulation antecedent to islet autoimmunity in type 1 diabetes (Orešič et al., 2008). However, on account of lipid structural heterogeneity, co-fragmentation of isobaric lipids, and chromatographic peak (feature) degeneracy, significant data analysis challenges remain (Cajka and Fiehn, 2014; Kuhl et al., 2012; Mahieu and Patti, 2017).
To date, numerous informatics approaches exist to address one or more of these challenges including mzMine 2 (Pluskal et al., 2010), LipidBlast (Kind et al., 2013), LipidSearch (Taguchi and Ishikawa, 2010), MS-Dial (Tsugawa et al., 2015), LipidMatch (Koelmel et al., 2017), and Lipid Data Analyzer 2 (Hartler et al., 2017). Accurate lipid identification requires customizable in-silico spectral libraries, lipid-specific MS/MS matching, accurate assignment of MS/MS identifications to chromatographic peaks, and multi-dimensional downstream data filtration (Tsugawa et al., 2017a). To promote broad usability and acceptance, these elements should be vendor neutral and integrated in graphical user interface-based (GUI) software suites. At present, realizing this ideal workflow for global feature profiling often requires either significant manual validation or integration of multiple software packages. We developed LipiDex to automate and unite these crucial lipid identification steps into a single intuitive software package capable of efficiently processing expansive lipid data sets.
Results
Lipid Identification and Quantitation Workflow
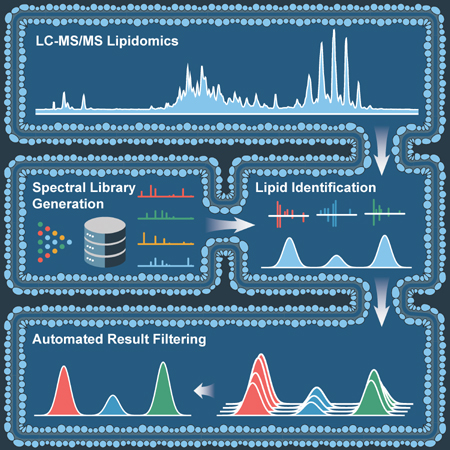
LipiDex comprises a set of lipid spectral library management and data processing tools optimized for high-resolution MS analysis and linked together in an intuitive graphical user interface (Figure 1A). Developed in Java (JRE v.1.8.0), LipiDex runs efficiently on multiple operating systems, supports vendor neutral and open-source MS file types, and accepts chromatographic feature tables from several commercial and open-source LC-MS/MS software packages. Here we describe LipiDex's major lipid identification algorithms and demonstrate their performance with multiple data sets. A detailed explanation of the algorithms is provided in the STAR Methods section, a feature comparison to other software packages is provided in Figure S2, and the software suite and accompanying multimedia tutorials describing the use of LipiDex's major functions are freely available at http://www.ncqbcs.com/resources/software/.
Figure 1. Lipid Identification and Quantitation Workflow.

(A) Overview of library construction (Library Generator), lipid identification (Spectrum Searcher), and result filtering (Peak Finder) in LipiDex.
(B) Extraction and alignment of lipid chromatographic peaks using external software packages.
(C) Identification of MS/MS spectra (grey circles and red spectrum) using in-silico lipid databases and a modified dot-product scoring algorithm which ranks identifications according to spectral similarity to library entries (blue spectrum).
(D) Spectral deconvolution and relative quantitation (colored circles) of co-fragmented isobaric lipid species by sequential matching against and subtracting from the experimental spectra.
(E) Accurate association of MS/MS identifications to chromatographic features via Gaussian peak modeling (shaded color).
(F) Calculation of total peak purity for each chromatographic peak across all samples using the Gaussian peak profile to modify spectral purity measurements.
(G) Final identification of each chromatographic peak using either the sum composition (i.e. PC40:1) or molecular composition (i.e. PC 16:1_24:0) if the weighted peak purity rises above a user-supplied threshold.
To achieve confident lipid identification at the sum or molecular composition level (Liebisch et al., 2013), LipiDex integrates precursor mass-to-charge (m/z), fragmentation spectra, chromatographic retention time, and chromatographic elution profile (Figure 1B). In-silico lipid spectral libraries for MS/MS identification are first created by Library Generator which utilizes a set of customizable lipid metadata tables and fragmentation rules to accurately model the m/z and relative intensity of diverse fragment types (Figure S2) (Kind et al., 2014). To facilitate utilization, LipiDex includes extensive pre-built spectral libraries based on numerous authentic reference standards, experimental identifications, and published databases (refer to Table S1, Table S2 for library composition) (Kind et al., 2014; Tsugawa et al., 2017b). Next, Spectrum Searcher generates lipid spectral matches by searching experimental MS/MS spectra against these libraries using a two-step searching algorithm. Experimental MS/MS spectra are first assigned putative identifications via a modified dot-product score (Figure 1C) and are then deconvoluted to quantify lipid co-fragmentation (Figure 1D). Peak Finder links these MS/MS identifications with externally-generated feature tables using a Gaussian peak model to accurately assign quantitative abundance values to unique lipid species (Figure 1E). Final result filtering, lipid grouping, and retention time filtering remove degenerate peaks and collapse redundant identifications (Figure 1F), automating the manual data validation normally required for robust large-scale discovery lipidomics.
Generating Confident Lipid Identifications
Although many powerful LC-MS/MS analysis packages identify chromatographic features from user-supplied or public spectral libraries, these packages are not specifically optimized for lipid data analysis. Accordingly, we present several in-silico and experimental datasets for the optimization and validation of LipiDex's core functions. To generate candidate spectral matches, MS/MS spectra are searched against user-selected spectral libraries leveraging a lipid-optimized dot-product spectral similarity score and the intact precursor m/z as a search constraint. The dot-product score represents the scaled cosine of the angle between the in-silico and experimental spectral vectors (Stein and Scott, 1994) and contains mass (n) and intensity (m) weighting factors which can be modulated to tailor the equation to specific applications (Kim et al., 2012). In Figure 2A, we demonstrate the systematic optimization of these terms using a structurally diverse set of 82 lipid reference standard spectra fragmented via higher-energy collisional dissociation (HCD) from the NIST Tandem Mass Spectral Library (National Institute of Standards and Technology, 2012). The optimal weighting factor set (m:1.2, n:0.9) returns a median top match similarity score of 955, a median first and second match score difference of 887, 82 correct identifications (Figure 2A), and performs similarly well for collision-induced dissociation (CID) spectra (Figure S5) and LC-MS/MS analysis of pure reference standards (Figure S6). Importantly, this large difference between the first and second match scores greatly increases spectral match confidence in the absence of a false discovery rate (FDR) estimate as commonly used in spectral matching for discovery proteomics.
Figure 2. Generating Confident Lipid Identifications.

(A) Color coded values for the median score, median score difference to next match, and correct identifications for the top-scoring spectral matches from 82 lipid reference standard spectra from the NIST Tandem Mass Spectral Library searched using various combinations of intensity and mass weighting factors.
(B) Sequential matching (colored lines) and subtraction (light grey lines) from experimental triacylglycerol MS/MS spectrum and representative fragment intensities used to quantify co-fragmentation of isobaric lipids. Note the displayed spectrum is enlarged to highlight the fatty acid-identifying peaks (m/z 530-600).
(C) Violin plot showing the measured (blue) and expected (dashed line) spectral purities for 98 isobaric lipid pairs mixed in-silico at varying ratios. Boxplots within each violin demarcate the median (white stripe), the 25th to 75th percentile (interquartile range; box), and 1.5 times the interquartile range (whiskers).
(D) Number of identified and unidentified chromatographic feature groups from Hap1 cells, human plasma, mouse liver, and yeast cells analyzed in triplicate after sequential filtering for common LC-MS/MS artifacts and lipid identification filtering.
Spectral similarity scoring's accuracy markedly improves upon addition of orthogonal identification filters (Kwiecien et al., 2015), particularly for spectra containing multiple co-fragmented lipid regioisomers (Hartler et al., 2017; Koelmel et al., 2017). Spectrum Searcher utilizes a unique spectral deconvolution algorithm to quantify the co-fragmentation of isobaric lipids (Figure S6). In Figure 2B, we demonstrate spectral deconvolution for a complex experimental triacylglycerol (TG) spectrum where multiple TGs share common chain-identifying fragments. By sequentially subtracting select matched fragments, the deconvolution algorithm avoids assigning the full intensity of fragments shared by multiple isobaric precursors to each species, an inaccuracy inherent to methods which sum the intensity of all matched fragments (Hartler et al., 2017; Koelmel et al., 2017). The accuracy of the algorithm is globally demonstrated using a set of pure isobaric phospho-, glycero-, and sphingo-lipid spectral pairs extracted from experimental LC-MS/MS data and mixed in-silico at various ratios to simulate co-fragmentation (Figure 2C). The deconvolution algorithm accurately returns the correct mixing ratio for the vast majority of in-silico pairs (average spectral purity error of 3.5). Note the returned values represent a metric of spectral purity and are not used to modify the peak area values for associated features, rendering small inaccuracies in the purity value insignificant.
Removing redundant features which arise from the same analyte (degenerate features) and data artifacts generated from the discovery LC-MS/MS lipidomic workflow is a critical step for robust downstream statistical analysis which often requires significant manual data validation (Collins et al., 2016; Kuhl et al., 2012; Mahieu and Patti, 2017). To minimize this laborious, but essential step, LipiDex automatically detects and removes degenerate identified and unidentified features. In Figure 2D, we present the LC-MS/MS analysis and data filtration of complex lipid extracts of human Hap1 cells, human plasma, mouse liver, and yeast cells. After removal of features present in a blank injection by the peak picking software, each dataset contains thousands of features (8,276, 9,287, 8,494, and 4,921 respectively). LipiDex automatically removes spurious feature detections, adducts, dimers, in-source fragments, and misidentified isotopes resulting in an average feature reduction of 59%.
After removal of artifacts, LipiDex infers the minimum number of lipid identifications which accurately explain all identified lipid features and their associated MS/MS identifications (Figure S7) in a process roughly analogous to protein inference (Nesvizhskii and Aebersold, 2005). Although these filters remove the vast majority of incorrect lipid identifications, false positives can remain. Lipid retention time (RT) represents a highly specific and orthogonal filter which can greatly improve identification confidence (Aicheler et al., 2015; Hartler et al., 2017; Tsugawa et al., 2015, 2017b). Many approaches predict lipid RT by correlating chemical identifiers to experimentally measured RTs which inherently requires a static chromatographic setup. LipiDex circumvents this problem by implementing an optional outlier detection algorithm to remove lipid identifications falling outside of the retention time cluster elution pattern of lipid reversed-phase analysis (Figure S8). While these filters do not exhaustively remove degenerate features or false identifications in a sample, the tremendous reduction in candidate features significantly expedites data validation and makes LipiDex a flexible tool for discovery lipidomic analysis.
Discussion
Highly scalable, automated, and robust lipid identification is readily achievable with LipiDex. Already, LipiDex has been used in multiple lipidomics studies to analyze a wide array of sample types (Lapointe et al., 2017; Reidenbach et al., 2017; Rhoads et al., 2018; Stefely et al., 2016; Veling et al., 2017; Velsko et al., 2017). To extend this identification capability to new biological systems of interest and novel fragmentation techniques, LipiDex's extensive curated lipid databases are easily expanded using its intuitive fragmentation templates. Beyond library generation, the detailed fragmentation metadata provided for each predicted MS/MS fragment empowers accurate spectral matching and deconvolution. Unlike many general small-molecule data analysis packages, LipiDex dynamically modifies the structural resolution encoded within each lipid identification to match the experimental MS/MS data. Moreover, these and other unidentified features are extensively filtered to remove degenerate peaks, redundant lipid identifications, and spurious lipid features. In the absence of a method to calculate a false discovery rate, LipiDex applies multi-dimensional filtering to increase lipid identification confidence. Recently published methods for small molecule FDR estimation via target-decoy methods raise the tantalizing possibility of statistically rigorous lipid identification (Palmer et al., 2016; Scheubert et al., 2017). We note that LipiDex's detailed fragmentation rule sets may prove valuable for automatically generating realistic and expandable decoy databases, a vital step for the rapid evolution of discovery lipidomic analysis into an essential systems biology tool.
Star Methods
Contact For Reagent And Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joshua J. Coon (jcoon@chem.wisc.edu).
Method Details
Supported lipidomic workflows
LipiDex bases lipid identifications on accurate precursor m/z and matching of high or low resolution MS/MS spectra against theoretical in-silico libraries. These identifications are then extensively filtered to ensure datasets with minimal artefactual and redundant entries. Although the software was initially developed and benchmarked using RP-LC-MS/MS data collected on the Q-Exactive platform, it supports numerous common lipidomic workflows from diverse LC-MS/MS setups including acetate and formate mobile phase systems, reversed phase and HILIC separations, HCD and CID fragmentation spectra, and fast polarity switching and separate polarity data acquisition. Additionally, the flexible spectral library generation tools allow the software package to be readily expanded to cover novel lipid classes and fragmentation templates. Currently, LipiDex does not support data collected using data-independent acquisition (DIA), all ion fragmentation (AIF), or infusion-based methods.
Spectral similarity matching
Candidate lipid identifications are assigned to experimental MS/MS spectra using a modified dot-product spectral similarity score. First, user-selected in-silico lipid spectra are extracted from generated .msp files and binned by precursor m/z for efficient retrieval prior to spectral matching. Experimental MS/MS spectra in the Mascot Generic Format (MGF) or mzXML format are then extracted and the fragment intensities normalized to 1000. All library spectra whose intact precursor m/z falls within a user-defined tolerance of the experimental MS/MS precursor m/z are then compared to the experimental MS/MS using the dot-product score. The following dot-produce equation is used to measure spectral similarity:
The optimized intensity (m = 1.2) and mass (n = 0.9) weighting factors give greater emphasis to higher m/z and intensity fragments which often identify individual fatty acid moieties of the lipid. To reduce the deleterious effect of co-fragmented lipid species on spectral scoring, the intensities of peaks present in the experimental spectrum but not in the library entry are reduced by 50% (Tsugawa et al., 2015). For the reverse dot-product score, only peaks present in both experimental and library spectra (within the user-defined m/z tolerance) are considered for spectral similarity scoring. Peak Finder utilizes both the standard and reverse dot-product scores to filter out potential false identifications whose spectral similarity scores fall below a user-supplied threshold.
Spectral deconvolution
Accurate quantification of co-fragmentation of isobaric lipids and total spectral purity follows the logic flow detailed in Figure S2. For a given MS/MS spectrum, all spectral matches are ranked according to their dot-product score and added to the scan queue (SQ). Next, fragmentation template entry for each candidate lipid species is queried to find the fatty acid-identifying fragment type which is assigned the highest relative intensity. The experimental spectrum is then searched for fragments which correspond to this rule type. If all of the predicted highest intensity chain-identifying fragments are not found, the candidate identification is discarded. If all predicted highest intensity chain-identifying fragments are found, the relative intensity of matched fragments is corrected using any matching m/z values found in the correction list (CL). The corrected m/z values are then assigned the median intensity of the matched fatty acid-identifying fragments and added to the CL. Depending on the number of fatty acids for the identified lipid, either the median or maximum matched intensity is added to the intensity list (IL). This intensity value is used to quantify the relative abundance of the lipid in the MS/MS spectrum. This process is repeated until no candidate spectra remain. All intensities found in the IL are then scaled to the sum of all matched species' intensities.
Feature-MS/MS association
LipiDex accepts chromatographic feature tables generated by Compound Discoverer (Thermo Fisher Scientific) or MZmine 2 (v. 2.30) (Pluskal et al., 2010). To associate lipid spectral matches with their parent features, each feature is modeled using a Gaussian function generated from the feature metadata in the imported table. The following function is used for Gaussian modeling:
The parameter a is the apex height of the feature, b is the apex retention time, and FWHM is the full peak width at half of the maximum intensity. MS/MS identifications whose dot-product score is greater than a user-supplied threshold are then linked to chromatographic features if they stem from the same sample and if they share the same charge, precursor m/z (within user-supplied tolerance), and retention time. As MS/MS are not necessarily acquired at the feature apex, spectra are assigned to features if the retention time falls within a user-specified factor of the feature's peak width (FWHM). To accurately associate spectra for partially co-eluting isobaric peaks, each spectrum is assigned to the peak which has the highest Gaussian-modeled intensity at that retention time.
Weighted peak purity
To quantify co-elution of isobaric features, LipiDex combines the Gaussian feature profile and the deconvoluted spectral purity values to calculate a weighted purity value for the feature across all samples (feature group). All identified lipid spectral components generated from spectral deconvolution and assigned to a specific feature group are weighted according to their proximity to the peak apex using the Gaussian peak model. These weighted values are then used to calculate a weighted average for each component assigned to the feature group.
Spurious peak detection filtering
When analyzing complex LC-MS data, feature detection software routinely identifies chromatographic peaks which cannot be reliably quantified across many samples. These features are often of low abundance, suffer from poor chromatographic resolution, or are artifacts of the specific peak detection routine (Mahieu et al., 2015). To remove these features, LipiDex discards all feature groups which were not detected in at least a user-supplied number of LC-MS experiments from the dataset.
Degenerate peak filtering
Following assignment of MS/MS identifications to features, co-eluting feature groups are filtered to remove redundant adduct, multimers, misidentified isotopes, and in-source fragments. Feature groups are considered to co-elute if the apex retention time difference is less than half the average peak width (FWHM). In this way, small differences in apex retention time, which can arise from polarity-switching LC-MS/MS experiments, do not erroneously disqualify potential degenerate feature groups. Co-eluting peaks are considered adducts or dimers of the same parent compound if they satisfy the following equations within a user-defined m/z tolerance:
Here, m refers to the precursor m/z for feature groups 1 or 2 and a denotes the m/z of adducts x or y. All potential adduct combinations for the correct polarity are taken from a user-supplied list in Peak Finder. If two features are determined to stem from the same parent compound, the feature with the largest area is retained and the other discarded. If only one of the features is identified as a lipid from its MS/MS spectra, that feature is retained and the feature without an MS/MS spectral identification is discarded. A feature group is considered an in-source fragment if it's precursor m/z is found in the assigned MS/MS identification in-silico spectrum for any co-eluting feature group. Occasionally, LC-MS feature extraction tools will misidentify the M+1 isotope as the M0 isotope. LipiDex removes these errors by searching for co-eluting peaks whose m/z differs by the mass of a neutron within a user-defined mass tolerance.
Lipid grouping
Lipid grouping refers to the collapsing of redundant lipid identifications and modulation of the structural resolution embedded in the lipid identification (sum vs. molecular composition) based on the assigned MS/MS identifications. In this way, the minimum set of lipid identifications which supports the observed features and MS/MS are reported. Common lipid grouping scenarios are outlined in Figure S2. After lipid grouping, feature groups are identified with the sum composition (i.e. PC 34:1) when the MS/MS spectra support multiple isobaric identifications (Figure S2C) or contain significant co-fragmentation of isobaric lipids (Figure S2E). Lipids identifications are given the molecular composition (i.e. PC 16:0_18:1) if, after combining redundant adducts, the MS/MS spectra contain fatty-acid identifying fragments (Figure S2A, Figure S2D) with minimal co-fragmentation.
Retention time filtering
Lipid retention times (RT) for a specific lipid class roughly approximate a normal distribution in reversed-phase chromatographic separation. In LipiDex, lipid identifications are optionally filtered via retention time using an LC system-agnostic outlier detection. First, apex retention times for all identified feature groups are extracted and grouped together by lipid class. If there are at least four identifications for the specific lipid class, the median absolute RT deviation from the median (MAD) is calculated. MAD represents a more robust method for outlier detection than the standard deviation alone (Leys et al., 2013). Any identification which falls outside of a user-defined multiple of this value (3.5 by default) is considered to be a spurious identification and removed. In cases where there are very few identified lipids from a given class or non-endogenous lipid internal standards are used, true identifications may fall outside the class retention time window. In these or other instances of erroneous outlier detection, the user can either toggle the retention time filtering off or find the removed lipid in the Unfiltered Peak List CSV file.
Hap1 Collection
HAP1 cells were maintained in high glucose (25 mM) Iscove's Modified Dulbecco's Medium supplemented with 10% (v/v) fetal bovine serum and penicillin-streptomycin (IMDM, FBS, PS) in a humidified incubator (37 °C, 5% CO2). Cells were subcultured by trypsinization (0.05% Trypsin-EDTA). Two million HAP1 cells were plated into a 10 cm dish with IMDM, FBS, PS (37 °C, 10 mL) and incubated (37 °C, 5% CO2) for 46 h (until the cultures were ∼50– 60% confluent). The media was gently aspirated and replaced with glucose-free DMEM, FBS, PS with galactose (10 mM) and incubated (37 °C, 5% CO2, 24 h). Immediately prior to cell harvest, the media was aspirated and the cells were gently washed with PBS (∼2 mL, ∼20 °C). The PBS wash was aspirated, PBS (700 μL, 4 °C) was added, and the cells were harvested by scraping and transferring the resuspended cells into a 1.5 mL tube on ice. The cells were isolated by centrifugation (5000 g, 1 min, 4 °C). The supernatant was aspirated, and the cell pellets were snap frozen in N2(l) and stored at −80 °C.
Yeast Collection
An individual colony of yeast was used to inoculate a starter culture (3 mL pABA−,G, D) and incubated (30 °C, 230 rpm, 10–15 h). Media (100 mL media at ambient temperature in a sterile 250 mL Erlenmeyer flask) was inoculated with 2.5×106 yeast cells and incubated (30 °C, 230 rpm). Samples were harvested 25 h after inoculation. 1×108 yeast cells were harvested by centrifugation (3,000 g, 3 min, 4 °C), the supernatant was removed, and th e cell pellet was flash frozen in liquid nitrogen. Pellets were stored at −80 °C prior to extraction.
Hap1 Cell, Yeast, and Mouse Liver Lipid Extraction
Yeast cell pellets, mouse liver homogenates (100 μg), and HAP1 cell pellets were thawed on ice and each sample was mixed with glass beads (0.5 mm diameter, 100 μL). CHCI3/MeOH (1:1, v/v, 4 °C) (900 μL) was added and vortexed (2 × 30 s). HCI (1 M, 200 μL, 4 °C) was added and vortexed (2 × 30 s). The samples were centrifuged (5,000 g, 2 min, 4 °C) to complete phase separation. 500 μL of the organic phase was transferred to a clean tube and dried under Ar(g). The remaining organic residue was reconstituted in ACN/IPA/H2O (65:30:5, v/v/v) (100 μL) for LC-MS analysis.
Human Plasma Lipid Extraction
Human plasma (Innovative Research, (20 μL) was thawed on ice and vortexed. MeOH (4 °C) (225 μL) was added and vortexed (30 s). Methyl tert-butyl ether (750 μL, 4 °C) was added and vortexed (30 s). The samples were mixed using an orbital shaker (6 mins) to extract lipids. H20 (225 μL, 4 °C) was added and vortexed (30 s) to induce ph ase separation. Samples were centrifuged (14,000 g, 2 min, 4 °C) to complete phase separation. 200 μL of the organic phase was transferred to a clean vial and dried in a vacuum centrifuge (45 min). The organic residue was reconstituted in MeOH/Toluene (9:1, v/v) (100 μL) for LC-MS/MS analysis.
LC-MS/MS Acquisition
Sample analysis was performed on an Acquity CSH C18 column held at 50 °C (2.1 × 100 mm × 1.7 μm particle size; Waters Corporation) using a Vanquish Binary Pump (400 μL/min; Thermo Scientific). Mobile phase A consisted of 10 mM ammonium acetate in ACN/H2O (70:30, v/v) containing 250 μL/L acetic acid. Mobile phase B consisted of 10 mM ammonium acetate in IPA/ACN (90:10, v/v) with the same additives. Initially, mobile phase B was held at 2% for 2 min and then increased to 30% over 3 min. Mobile phase B was then further increased to 85% over 14 min and then raised to 99% over 1 min and held for 7 min. The column was then re-equilibrated for 5 min before the next injection. Ten microliters of lipid extract were injected by a Vanquish autosampler (Thermo Scientific). The LC system was coupled to a Q Exactive HF mass spectrometer (Thermo Scientific) by a HESI II heated ESI source (Thermo Scientific). The MS was operated in positive and negative mode during sequential injections collecting both MS1 and MS2 spectra. For detailed MS acquisition parameters, reference Table S3.
Quantifications and Statistical Analysis
LC-MS/MS Data Analysis
The resulting LC-MS data were processed using Compound Discoverer 2.0 (Thermo Scientific). For a detailed description of all data processing parameters used, reference Table S4. Briefly, all MS1 scans were extracted (Select Spectra node) and aligned (Align Retention Times node) across all LC-MS/MS experiments. Chromatographic features were subsequently detected from the aligned scans (Detect Unknown Compounds node) and grouped according to common m/z and elution profile (Group Unknown Compounds node). Each LC-MS/MS experiment was then re-searched to minimize feature groups which contained missing values (Fill Gaps node). Finally, feature groups which were present in the blank injection were removed (Mark Background Compounds node). To generate an unaligned dataset, MS1 scans were extracted (Select Spectra node) and Chromatographic features were subsequently detected from the aligned scans (Detect Unknown Compounds node) using the same parameters as the aligned dataset. MS/MS spectra were converted to the MGF format using Proteowizard 3.0 (ProteoWizard Software Foundation v3.0) and searched against the LipiDex_HCD_Acetate, LipiDex_HCD_Hydroxy, and LipiDex_HCD_ULCFA libraries using Spectrum Searcher. In Peak Finder, lipid spectral matches were assigned to chromatographic features via precursor m/z and retention time. Lipid identifications were further filtered via MS/MS spectral purity, retention time, and chromatographic profile to remove artifacts and redundant identifications.
NIST MS/MS Dot Product Optimization Dataset
MS/MS lipid reference standard spectra which were collected under similar MS/MS conditions as those used in the LC-MS/MS dataset were extracted from the NIST Tandem Mass Spectral Library. For the HCD dataset, 82 phospho-, glycero-, and sphingo-lipid MS/MS spectra which were fragmented using higher-energy collisional dissociation (HCD) and a collision energy between 40-50 eV were extracted. For the CID dataset, 89 phosho-, glycero-, and sphingo-lipid MS/MS spectra which were fragmented using ion trap collision induced dissociation were extracted. In Spectrum Searcher, the spectra were analyzed using the dot-product equation described above with varying mass and intensity weights (0.1 to 3.0) and the search parameters listed in Table S3. Lipid identifications were deemed correct if they identified the correct fatty acid moieties or, if in the case of indistinguishable spectra (i.e. PC [M+H]+ spectra), the correct sum identification.
Lipid Data Analyzer 2 Comparison Dataset
To effectively compare the MS/MS identification performance of LipiDex against the recently published Lipid Data Analyzer 2 (Hartler et al., 2017), a dataset comprising 7 LC-MS/MS analyses of a lipid reference standard mix was downloaded from the LDA 2 software homepage (http://genome.tugraz.at/lda2/data/ControlExperiment1/Exp1_QExactive.zip) and analyzed using both LDA 2 and LipiDex. Lipids were identified in LDA 2 using precursor mass matching parameters of 2 matching isotopes, the Q Exactive positive and negative mass lists from the Biological Experiment dataset (http://genome.tugraz.at/lda2/data/Biological/Bio_QExactive.zip), and the OrbiTrap_exactive_neg and OrbiTrap_exactive_MS/MS rule sets. In LipiDex, MS/MS files were searched against the Lipidex_Formic spectral library using a 0.02 Th MS1 search tolerance, 0.02 MS2 Th search tolerance, 1 max search results returned, and a 61.0 Th MS2 low mass cutoff. To directly compare MS/MS identification performance, the spectral identifications from both programs were matched to the LDA 2-generated feature list and compared at the sum composition and molecular composition level for all lipid class excluding the deuterium-labelled TG standards which are not present included in both programs' libraries For reference standard feature identifications, identifications between LipiDex and LDA 2 were matched if both programs returned the same molecular composition identification. For identification of all lipid features, identifications were matched if both programs returned identical sum lipid identifications. MS/MS signal-to-noise was calculated for each spectrum using the ratio of the most intense spectral peak to the median spectral peak intensity (Yang et al., 2014).
In-silico Co-fragmentation Dataset
Lipid co-fragmentation was simulated by mixing experimental isobaric lipid spectra in-silico across varying rations and analyzing the resulting spectra using the spectral deconvolution algorithm. MS/MS identifications were generated from LC-MS/MS data as above and only high-scoring and high-purity MS/MS (dot-produce>890, purity>98%, maximum intensity>1.0E5) were retained for in-silico mixing. Isobaric spectra were then added together and their intensities modulated to mix spectra at various ratios (1:5, 1:4, 1:3, 1:2, 1:1, 2:1, 3:1, 4:1, and 5:1). The mixed spectra were then re-searched using Spectrum Searcher to generate spectral purity values.
Data and Software Availability
LipiDex source code, compiled program, manuscript datasets, and test dataset are available via GitHub: http://www.ncqbcs.com/resources/software/
Proteowizard for MS/MS file conversion is available here: http://proteowizard.sourceforge.net/downloads.shtml
Compound Discoverer 2.0 for feature finding is available here: https://www.thermofisher.com/order/catalog/product/OPTON-30783
MZmine 2 for feature finding is available via GitHub: http://mzmine.github.io/
Raw MS data files for multi-tissue dataset available via Chorus (ID: 1409): https://chorusproject.org
Supplementary Material
Figure S1: Omics Software Publications, Related to STAR Methods
Figure S2: Major Data Processing Features of Open Source Lipidomics Packages, Related to STAR Methods
Figure S3: Lipid Fragmentation Modelling using Fragmentation Templates, Related to STAR Methods
Figure S4: Dot-Product Weighting Factor Optimization for ion trap CID, Related to Figure 1
Figure S5: Identification Performance Comparison to Lipid Data Analyzer 2, Related to STAR METHODS
Figure S6: Spectral Deconvolution Logic Flow, Related to Figure 1
Figure S7: Common Lipid Grouping Scenarios and Final Identifications, Related to Figure 2
Figure S8: Lipid Retention Time Outlier Filtering, Related to Figure 2
Table S1. LipiDex Lipid Library Composition, Related to Figure 1
Table S2. LipidBlast Lipid Library Composition, Related to Figure 1
Table S3. LC-MS/MS Analysis Parameters, Related to Figure 2
Table S4. Data Processing Parameters, Related to Figure 2
Highlights.
A new open-source software suite for LC-MS/MS lipidomics data analysis
Flexible in-silico libraries accurately model fragmentation of diverse lipid types
Accurate quantitation of co-isolated and co-eluting isobaric lipids
Streamlined downstream data filtration for high-confidence lipid identifications
Acknowledgments
We gratefully acknowledge support from the National Institutes of Health Grant P41 GM108538 (awarded to J.J.C), the Department of Energy DE-FC02-07ER64494, and the Morgridge Institute for Research Metabolism Theme.
Footnotes
Author Contributions: P.D.H. developed the software package, curated spectral libraries, performed mass spectrometry analysis, and analyzed lipidomics data. P.D.H. and J.D.R. designed the study and interpreted the results. J.J.C. directed and supervised all aspects of the study. All authors contributed to writing and critical review of the manuscript.
Declaration of Interests: The authors declare no competing interests.
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aicheler F, Li J, Hoene M, Lehmann R, Xu G, Kohlbacher O. Retention Time Prediction Improves Identification in Nontargeted Lipidomics Approaches. Anal Chem. 2015;87:7698–7704. doi: 10.1021/acs.analchem.5b01139. [DOI] [PubMed] [Google Scholar]
- Cajka T, Fiehn O. Comprehensive analysis of lipids in biological systems by liquid chromatography-mass spectrometry. TrAC - Trends Anal Chem. 2014;61:192–206. doi: 10.1016/j.trac.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cajka T, Fiehn O. Toward Merging Untargeted and Targeted Methods in Mass Spectrometry-Based Metabolomics and Lipidomics. Anal Chem. 2016;88:524–545. doi: 10.1021/acs.analchem.5b04491. [DOI] [PubMed] [Google Scholar]
- Collins JR, Edwards BR, Fredricks HF, Van Mooy BAS. LOBSTAHS: An Adduct-Based Lipidomics Strategy for Discovery and Identification of Oxidative Stress Biomarkers. Anal Chem. 2016;88:7154–7162. doi: 10.1021/acs.analchem.6b01260. [DOI] [PubMed] [Google Scholar]
- Hartler J, Triebl A, Ziegl A, Trötzmüller M, Rechberger GN, Zeleznik OA, Zierler KA, Torta F, Cazenave-Gassiot A, Wenk MR, et al. Deciphering lipid structures based on platform-independent decision rules. Nat Methods. 2017;14:1171–1174. doi: 10.1038/nmeth.4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert AS, Richards AL, Bailey DJ, Ulbrich A, Coughlin EE, Westphall MS, Coon JJ. The One Hour Yeast Proteome. Mol Cell Proteomics. 2014;13:339–347. doi: 10.1074/mcp.M113.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Koo I, Wei X, Zhang X. A method of finding optimal weight factors for compound identification in gas chromatography-mass spectrometry. Bioinformatics. 2012;28:1158–1163. doi: 10.1093/bioinformatics/bts083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind T, Liu KH, Lee DY, DeFelice B, Meissen JK, Fiehn O. LipidBlast in silico tandem mass spectrometry database for lipid identification. Nat Methods. 2013;10:755–758. doi: 10.1038/nmeth.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind T, Okazaki Y, Saito K, Fiehn O. LipidBlast templates as flexible tools for creating new in-silico tandem mass spectral libraries. Anal Chem. 2014;86:11024–11027. doi: 10.1021/ac502511a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koelmel JP, Kroeger NM, Ulmer CZ, Bowden JA, Patterson RE, Cochran JA, Beecher CWW, Garrett TJ, Yost RA. LipidMatch: an automated workflow for rule-based lipid identification using untargeted high-resolution tandem mass spectrometry data. BMC Bioinformatics. 2017;18:331. doi: 10.1186/s12859-017-1744-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhl C, Tautenhahn R, Böttcher C, Larson TR, Neumann S. CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal Chem. 2012;84:283–289. doi: 10.1021/ac202450g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiecien NW, Bailey DJ, Rush MJP, Cole JS, Ulbrich A, Hebert AS, Westphall MS, Coon JJ. High-Resolution Filtering for Improved Small Molecule Identification via GC/MS. Anal Chem. 2015;87:8328–8335. doi: 10.1021/acs.analchem.5b01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe CP, Stefely JA, Jochem A, Hutchins PD, Wilson GM, Kwiecien NW, Coon JJ, Wickens M, Pagliarini DJ. Multi-omics Reveal Specific Targets of the RNA-Binding Protein Puf3p and Its Orchestration of Mitochondrial Biogenesis. Cell Syst. 2017;6:125–135. doi: 10.1016/j.cels.2017.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leys C, Ley C, Klein O, Bernard P, Licata L. Detecting outliers: Do not use standard deviation around the mean, use absolute deviation around the median. J Exp Soc Psychol. 2013;49:764–766. [Google Scholar]
- Liebisch G, Vizcaíno JA, Köfeler H, Trötzmüller M, Griffiths WJ, Schmitz G, Spener F, Wakelam MJO. Shorthand notation for lipid structures derived from mass spectrometry. J Lipid Res. 2013;54:1523–1530. doi: 10.1194/jlr.M033506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahieu NG, Patti GJ. Systems-Level Annotation of a Metabolomics Data Set Reduces 25 000 Features to Fewer than 1000 Unique Metabolites. Anal Chem. 2017;89:10397–10406. doi: 10.1021/acs.analchem.7b02380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahieu NG, Spalding JL, Patti GJ. Warpgroup: Increased precision of metabolomic data processing by consensus integration bound analysis. Bioinformatics. 2015;32:268–275. doi: 10.1093/bioinformatics/btv564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Institute of Standards and Technology. NIST Tandem Mass Spectral Library. 2012. [Google Scholar]
- Nesvizhskii AI, Aebersold R. Interpretation of Shotgun Proteomic Data. Mol Cell Proteomics. 2005;4:1419–1440. doi: 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- Orešič M, Simell S, Sysi-Aho M, Näntö-Salonen K, Seppänen-Laakso T, Parikka V, Katajamaa M, Hekkala A, Mattila I, Keskinen P, et al. Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in children who later progress to type 1 diabetes. J Exp Med. 2008;205:2975–2984. doi: 10.1084/jem.20081800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer A, Phapale P, Chernyavsky I, Lavigne R, Fay D, Tarasov A, Kovalev V, Fuchser J, Nikolenko S, Pineau C, et al. FDR-controlled metabolite annotation for high-resolution imaging mass spectrometry. Nat Methods. 2016;14:57–60. doi: 10.1038/nmeth.4072. [DOI] [PubMed] [Google Scholar]
- Patti GJ, Yanes O, Siuzdak G, Kind T, Niehaus TD, Broadbelt LJ, Hanson AD, Fiehn O, Tyo KEJ, Henry CS, et al. Innovation: Metabolomics: the apogee of the omics trilogy. Nat Rev Mol Cell Biol. 2012;13:263–269. doi: 10.1038/nrm3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluskal T, Castillo S, Villar-Briones A, Orešič M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics. 2010;11:395. doi: 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reidenbach AG, Kemmerer ZA, Aydin D, Jochem A, Mcdevitt MT, Hutchins PD, Wilkerson EM, Stark JL, Stefely JA, Johnson IE, et al. Conserved lipid and small molecule modulation of COQ8 reveals regulation of the ancient UbiB family. 2017 doi: 10.1016/j.chembiol.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads TW, Burhans MS, Chen VB, Hutchins PD, Rush MJP, Clark JP, Stark JL, McIlwain SJ, Eghbalnia HR, Pavelec DM, et al. Caloric Restriction Engages Hepatic RNA Processing Mechanisms in Rhesus Monkeys Resource Caloric Restriction Engages Hepatic RNA Processing Mechanisms in Rhesus Monkeys. Cell Metab. 2018;27:1–12. doi: 10.1016/j.cmet.2018.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheubert K, Hufsky F, Petras D, Wang M, Nothias LF, Dührkop K, Bandeira N, Dorrestein PC, Böcker S. Significance estimation for large scale metabolomics annotations by spectral matching. Nat Commun. 2017;8 doi: 10.1038/s41467-017-01318-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatter DA, Aldrovandi M, O'Connor A, Allen SM, Brasher CJ, Murphy RC, Mecklemann S, Ravi S, Darley-Usmar V, O'Donnell VB. Mapping the Human Platelet Lipidome Reveals Cytosolic Phospholipase A2 as a Regulator of Mitochondrial Bioenergetics during Activation. Cell Metab. 2016;23:930–944. doi: 10.1016/j.cmet.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefely JAJA, Licitra F, Laredj L, Reidenbach AGAG, Kemmerer ZAZA, Grangeray A, Jaeg-Ehret T, Minogue CECECE, Ulbrich A, Hutchins PDPD, et al. Cerebellar Ataxia and Coenzyme Q Deficiency through Loss of Unorthodox Kinase Activity. Mol Cell. 2016;63:608–620. doi: 10.1016/j.molcel.2016.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein SE, Scott DR. Optimization and Testing of Mass-Spectral Library Search Algorithms for Compound Identification. J Am Soc Mass Spectrom. 1994;5:859–866. doi: 10.1016/1044-0305(94)87009-8. [DOI] [PubMed] [Google Scholar]
- Taguchi R, Ishikawa M. Precise and global identification of phospholipid molecular species by an Orbitrap mass spectrometer and automated search engine Lipid Search. J Chromatogr A. 2010;1217:4229–4239. doi: 10.1016/j.chroma.2010.04.034. [DOI] [PubMed] [Google Scholar]
- Tsugawa H, Cajka T, Kind T, Ma Y, Higgins B, Ikeda K, Kanazawa M, VanderGheynst J, Fiehn O, Arita M. MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat Methods. 2015;12:523–526. doi: 10.1038/nmeth.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsugawa H, Ikeda K, Arita M. The importance of bioinformatics for connecting data-driven lipidomics and biological insights. Biochim Biophys Acta - Mol Cell Biol Lipids. 2017a;1862:762–765. doi: 10.1016/j.bbalip.2017.05.006. [DOI] [PubMed] [Google Scholar]
- Tsugawa H, Ikeda K, Tanaka W, Senoo Y, Arita M, Arita M. Comprehensive identification of sphingolipid species by in silico retention time and tandem mass spectral library. J Cheminform. 2017b;9:1–12. doi: 10.1186/s13321-017-0205-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veling MT, Reidenbach AG, Freiberger EC, Kwiecien NW, Hutchins PD, Drahnak MJ, Jochem A, Ulbrich A, Rush MJP, Russell JD, et al. Multi-omic Mitoprotease Profiling Defines a Role for Oct1p in Coenzyme Q Production. Mol Cell. 2017;68:970–977e11. doi: 10.1016/j.molcel.2017.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velsko IM, Overmyer KA, Speller C, Klaus L, Collins MJ, Loe L, Laurent, Frantz AF, Sankaranarayanan K, Lewis CM, et al. The dental calculus metabolome in modern and historic samples. Metabolomics. 2017;1 doi: 10.1007/s11306-017-1270-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Neta P, Stein SE. Quality control for building libraries from electrospray ionization tandem mass spectra. Anal Chem. 2014;86:6393–6400. doi: 10.1021/ac500711m. [DOI] [PubMed] [Google Scholar]
- Yore MM, Syed I, Moraes-Vieira PM, Zhang T, Herman MA, Homan EA, Patel RT, Lee J, Chen S, Peroni OD, et al. Discovery of a class of endogenous mammalian lipids with anti-diabetic and anti-inflammatory effects. Cell. 2014;159:318–332. doi: 10.1016/j.cell.2014.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Omics Software Publications, Related to STAR Methods
Figure S2: Major Data Processing Features of Open Source Lipidomics Packages, Related to STAR Methods
Figure S3: Lipid Fragmentation Modelling using Fragmentation Templates, Related to STAR Methods
Figure S4: Dot-Product Weighting Factor Optimization for ion trap CID, Related to Figure 1
Figure S5: Identification Performance Comparison to Lipid Data Analyzer 2, Related to STAR METHODS
Figure S6: Spectral Deconvolution Logic Flow, Related to Figure 1
Figure S7: Common Lipid Grouping Scenarios and Final Identifications, Related to Figure 2
Figure S8: Lipid Retention Time Outlier Filtering, Related to Figure 2
Table S1. LipiDex Lipid Library Composition, Related to Figure 1
Table S2. LipidBlast Lipid Library Composition, Related to Figure 1
Table S3. LC-MS/MS Analysis Parameters, Related to Figure 2
Table S4. Data Processing Parameters, Related to Figure 2