

Abstract
The phylum Apicomplexa contains a group of protozoa causing diseases in humans and livestock. Plasmodium spp., the causative agent of malaria, contains a mitochondrion that is very divergent from that of their hosts. The malarial mitochondrion is a clinically validated target for the antimalarial drug atovaquone, which specifically blocks the electron transfer activity of the bc1 complex of the mitochondrial electron transport chain (mtETC). Most mtETC proteins are nuclear-encoded and imported from the cytosol, but three key protein subunits are encoded in the Plasmodium mitochondrial genome: cyt b, COXI, and COXIII. They are translated inside the mitochondrion by mitochondrial ribosomes (mitoribosomes). Here, we characterize the function of one large mitoribosomal protein in Plasmodium falciparum, PfmRPL13. We found that PfmRPL13 localizes to the parasite mitochondrion and is refractory to genetic knockout. Ablation of PfmRPL13 using a conditional knockdown system (TetR-DOZI-aptamer) caused a series of adverse events in the parasite, including mtETC deficiency, loss of mitochondrial membrane potential (Δψm), and death. The PfmRPL13 knockdown parasite also became hypersensitive to proguanil, a drug proposed to target an alternative process for maintaining Δψm. Surprisingly, transmission EM revealed that PfmRPL13 disruption also resulted in an unusually elongated and branched mitochondrion. The growth arrest of the knockdown parasite could be rescued with a second copy of PfmRPL13, but not by supplementation with decylubiquinone or addition of a yeast dihydroorotate dehydrogenase gene. In summary, we provide first and direct evidence that mitoribosomes are essential for malaria parasites to maintain the structural and functional integrity of the mitochondrion.
Keywords: malaria, mitochondria, ribosome, plasmodium, parasitology, mitochondrial DNA (mtDNA), apicomplexan, mitochondrial ribosome, mitoribosome, mRPL13, ribosomal protein, mitochondrion, mitochondrial genome
Introduction
Malaria remains a severe infectious disease in the tropical and subtropical regions of the world, causing millions of clinical cases and taking the lives of half a million people in 2016 (1). The causative agents, parasites of Plasmodium spp., have three genomes localized in three distinct cellular compartments, the nucleus, the apicoplast, and the mitochondrion. The nuclear genome is composed of 14 chromosomes (23 mega base pairs) and encodes ∼5500 proteins (2). The apicoplast is a relict plastid derived from primary and secondary endosymbiosis, which contains a circular bacterial like DNA (35 kb), encoding ∼30 proteins, 2 rRNA genes (16S and 23S), and a complete set of tRNAs required for protein translation within this organelle (3, 4).
The mitochondria of all eukaryotic organisms are believed to be derived from one single common ancestor, an α-protobacterium (5). In apicomplexan parasites, the mitochondrial genomes tend to be small, 6–11 kb in length (6, 7). In one genus, Cryptosporidium, the mitochondrial genome has been entirely lost (8). The mitochondrial DNA (mtDNA)2 of malaria parasites is only 6 kb, encoding three proteins of the mitochondrial electron transport chain (mtETC): cytochrome (cyt) b, cyt c oxidase subunits I and III (COX I and III) (6, 9, 10). These three proteins are translated by the mitochondrial ribosomes (mitoribosomes). The 6 kb mtDNA also encodes mitochondrial rRNA genes; however, these rRNAs are highly fragmented (∼30 small pieces of 20–200 bp) and scattered throughout the mitochondrial genome (11–13). It remains entirely unknown how these many small rRNA pieces come together to form a working mitoribosomal complex. Another striking feature of the malarial mtDNA is that it does not encode any tRNA genes, and mitochondrial tRNAs have to be imported from the cytosol (14).
To translate genes encoded on three distinctive genomes localized in three cellular compartments, Plasmodium parasites utilize three types of ribosomes. The cytosolic 80S ribosomes are numerous in the parasite cytoplasm and were easily detected by transmission EM studies carried out more than 50 years ago (15). Their structure has just been resolved by cryo–electron microscopy (cryo-EM) (16, 17). The ribosomes in the apicoplast and mitochondrion of malaria parasites are both prokaryotic type; yet, they possess unique features that are distinct from bacterial ribosomes or organellar ribosomes of other eukaryotes (18). It has been shown that active protein translation occurs in the apicoplast, which renders malaria parasites sensitive to several antibiotics (19–22). In contrast, the mitochondrial protein translation system in Plasmodium has remained an enigma for a long time. On one hand, direct evidence for mitochondrial protein translation in the parasites or in the entire Apicomplexa phylum is lacking, but on the other hand, the essentiality of the mtETC (23, 24) and the appearance of cyt b mutations conferring atovaquone resistance (25) strongly imply that the mitochondrial ribosomes are active.
In this study, we have characterized one conserved large subunit (LSU) protein of mitoribosomes in Plasmodium falciparum (PfmRPL13, PF3D7_0214200). It has been shown that L13 is a critical component involved in the first step of ribosome LSU assembly in bacteria (26) and is one of the eight LSU subunits required to form a minimal subribosomal particle maintaining peptidyltransferase activity in Thermus aquaticus (27). Here, for the first time, we provide direct evidence that PfmRPL13 is essential for malaria parasites, highlighting the significance of mitoribosomes for parasite physiology and survival.
Results
PfmRPL13 is essential in asexual blood stages of P. falciparum
PF3D7_0214200 (designated as PfmRPL13) is annotated as the putative mitochondrial ribosomal protein L13 in P. falciparum (www.plasmodb.org)3 (47). To compare the sequence similarity (or divergence) of mitochondrial ribosomal L13 proteins from various organisms, we performed a multiple sequence alignment and maximum likelihood phylogenetic analysis. As shown in Fig. S1, PfmRPL13 is closely related to its orthologues in other apicomplexan parasites, but shares much less sequence similarity to other mitoribosomal L13 proteins. PfmRPL13 and Toxoplasma mRPL13 share 55% identical residues, but the percent identity between the P. falciparum and human proteins is 23%, as calculated from the alignment (Fig. S1A). As might be expected then, in the phylogenetic results the apicomplexan mRPL13s form a separate, well-supported clade within the overall mitochondrial L13 grouping (Fig. S1B).
To verify the subcellular localization of PfmRPL13 in P. falciparum, we integrated a copy of PfmRPL13 tagged with triple HA (3HA) or GFP into the genome of Dd2attB parasites (28). Interestingly, Dd2attB transfected with the pLN-PfmRPL13-GFP construct grew very slowly and did not express GFP, as determined by fluorescence microscopy and Western blotting (data not shown), suggesting that expression of the PfmRPL13-GFP fusion protein was silenced by some unknown mechanism(s). However, Dd2attB transfected with the pLN-PfmRPL13-3HA construct expressed PfmRPL13-3HA. Immunofluorescence assay showed that PfmRPL13-3HA was localized in the parasite mitochondria with almost no signal in other cellular compartments (Fig. 1). Importantly, Dd2attB-PfmRPL13-3HA grew normally as compared with the parental line (data not shown). Based on its evolutionary sequence conservation (29) and mitochondrial localization, we conclude that PF3D7_0214200 is very likely to be the mitoribosomal large subunit protein L13 in P. falciparum.
Figure 1.
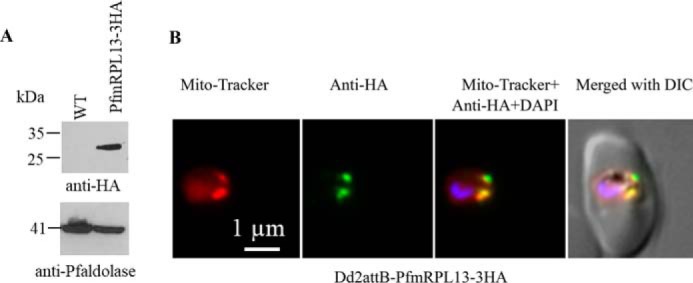
Expression and localization of PfmRPL13 in P. falciparum. A, expression of PfmRPL13–3HA in Dd2attB parasites was confirmed by Western blotting. P. falciparum aldolase (41 kDa) served as a loading control. B, PfmRPL13–3HA was localized in the parasite mitochondria as revealed by immunofluorescence assays.
To verify whether PfmRPL13 is essential for parasite growth in asexual blood stages, we attempted to knock it out in two WT lines, D10 and NF54, via a CRISPR/Cas9-mediated approach (see “Materials and methods” in the supporting information). Transfections in these lines were performed with circular or linearized template plasmids along with pUF1-Cas9 and selected with either a single drug for the template plasmid or two drugs for both the template and Cas9 plasmids (“Materials and methods”). In cultures transfected with a circular template plasmid, negative selection against maintenance of the plasmid was also performed (“Materials and methods”). However, despite multiple attempts, gene knockout was unsuccessful (data not shown). Because it has been shown that the only essential role of the mtETC in asexual blood stages is to provide an electron sink for the parasite dihydroorotate dehydrogenase (DHODH), which is essential for pyrimidine biosynthesis (23), we hypothesized that PfmRPL13 might be dispensable in the yeast DHODH (yDHODH) transgenic parasites, which do not require a functional mtETC. Knockout approaches as described above were carried out in the NF54attB-yDHODH-GFP line which carries a fusion protein of yDHODH and GFP. Gene knockout in this line was not successful after many attempts, however. In summary, PfmRPL13 is refractory to gene disruption, suggesting that PfmRPL13 is likely to be essential.
Because our knockout studies were unable to definitely determine the essentiality of PfmRPL13 in the parasites, we utilized a recently developed translational knockdown approach, the TetR-DOZI-aptamer system (30). In this system, protein translation is conditionally regulated by addition of anhydrotetracycline (aTc) (ON) or withdrawal of aTc (OFF). As shown in Fig. 2A, the TetR-DOZI-aptamer system was inserted into the genomic locus by double crossover recombination facilitated by CRISPR/Cas9 (“Materials and methods”). We transfected D10 WT parasites with a linearized template vector and two circular guider RNA (gRNA) constructs (“Materials and methods”), selected with blasticidin and aTc, and obtained transgenic parasites 3 weeks post electroporation. As shown in Fig. 2B, the genomic locus of PfmRPL13 was confirmed to be correctly modified in the transgenic parasites by diagnostic PCR analysis. We named this line D10-PfmRPL13-KD (knockdown). It has been shown that protein knockdown efficiency in the TetR-DOZI-aptamer system is determined by aptamer copy number, but aptamers are not very stable (31). To delineate aptamer copy numbers in D10-PfmRPL13-KD, the PCR product of 5′ integration (lane 1 in Fig. 2B) was sequenced and all 8 aptamer copies were intact (data not shown).
Figure 2.

Genetic ablation of PfmRPL13 through the TetR-DOZI-aptamer system facilitated with CRISPR/Cas9. A, the TetR-DOZI-aptamer system (30) was inserted into the PfmRPL13 genetic locus through a double crossover recombination strategy facilitated by a CRISPR/Cas9 system. TetR-DOZI-RLuc-Bsd is the combined gene for a fusion protein of tetracycline repressor with development of zygote inhibited (DOZI), followed by Renilla luciferase and blasticidin deaminase. 8xapt stands for 8 copies of TetR-binding aptamers. 5′HR and 3′HR are upstream and downstream homologous regions of the PfmRPL13 gene. The green box next to 5′HR indicates shield mutations that keep amino acid sequences intact but change nucleotide sequences to avoid repetitive cutting by Cas9. The dark blue box next to the green box is the 3HA tag. The template plasmid was fully digested with EcoRV before electroporation. Two gRNAs (**) adjacent to the stop codon of PfmRPL13 were cloned into the pAIO plasmid individually. The black bars underneath WT and integrated gene models indicate exons of the gene whereas dashed lines indicate the introns. B, the genotype of the PfmRPL13 knockdown line was verified by diagnostic PCR analysis. D10 genomic DNA served as a control. The sequences of primers P1–P4 are shown in Table S1 and their approximate positions are marked in (A). Lanes 1, 4, and 7 are products from PCR reactions with template DNA from the D10-PfmRPL13-KD line; lanes 2, 5, and 8 with DNA from D10 WT; lanes 3, 6, and 9 are no DNA controls. In lane 7, the fragment to be amplified was too big to work in PCR (>11 kb). C, a representative growth curve of the PfmRPL13 knockdown line grown in the presence or absence of aTc (250 nm). Cultures were split 1:5 on days 2, 4, 6, and 8. Parasitemia was determined under a light microscope by counting more than 1000 red blood cells in each Giemsa-stained thin blood smear. Growth index is the cumulative -fold expansion which is the multiplication of parasitemia and split factors over the time course. A split factor is 5 in a 1:5 split. Data shown are the mean ± S.D. of biological replicates. This experiment has been repeated more than 10 times. aTc, anhydrotetracycline. D, expression of PfmRPL13–3HA in the knockdown parasites maintained with and without aTc for 8 days was examined by Western blotting. The molecular mass of PfmRPL13–3HA is 28 kDa. P. falciparum aldolase (41 kDa) served as a loading control. E, MitoTracker staining patterns of healthy versus parasites with depolarized mitochondria. F, quantification of mitochondrial staining patterns in the PfmRPL13 knockdown line maintained with or without aTc, respectively. Green bars, tubular. Red bars, diffused. Data shown are averaged from n = 3 experiments and analyzed by a Student's t test. **, p < 0.01; *, p < 0.05.
The expression of PfmRPL13 was then controlled by addition and removal of aTc in the culture. To rule out any possibility that aTc itself would interfere with parasite growth, the WT D10 line was exposed to media supplemented with aTc (250 nm) and no aTc for 2 weeks but no differences between the two were noticed (data not shown), suggesting that aTc did not alter parasite growth. To determine the effect of knocking down PfmRPL13 on parasite survival, D10-PfmRPL13-KD parasites grown under aTc were enriched by a Percoll gradient, washed thoroughly, and exposed to medium with or without aTc (250 nm) for several intraerythrocytic developmental cycles (IDCs) (“Materials and methods”). As shown in Fig. 2C, D10-PfmRPL13-KD exhibited a severe growth arrest when aTc was removed for three cycles or more. When aTc was removed for more than four cycles, the parasitemia dropped down to a negligible level. To assess parasite growth long term, the aTc-minus culture of D10-PfmRPL13-KD was maintained up to 1 month (split weekly), and growth arrest was maintained for the entire period (data not shown). In the first two cycles post aTc removal, however, the differences between aTc-plus and aTc-minus cultures were not significant under microscopic examination. We then compared their growth rates through [3H]hypoxanthine incorporation assays by titrating aTc in a serial dilution (“Materials and methods”). As shown in Fig. S2, in the first cycle, D10-PfmRPL13-KD incorporated similar levels of [3H]hypoxanthine in low and high concentrations of aTc; however, in the second cycle, the knockdown parasite grown at the lowest aTc concentration (0.24 nm) had a 40% reduction in [3H]hypoxanthine incorporation compared with that of a high aTc (250 nm) culture. In thin blood smears, morphologically deteriorating parasites were observed when aTc was removed for two cycles and the number of morphologically unhealthy parasites increased substantially after removal of aTc for three or more cycles (Fig. S3).
To determine PfmRPL13 expression levels in the knockdown parasites, a knockdown assay was set up using the same protocol as described above. Protein samples were collected every 2 days over four IDCs and examined by Western blotting (“Materials and methods”). As shown in Fig. 2D, PfmRPL13 expression was substantially diminished after aTc was removed for just one cycle. We then qualitatively monitored Δψm using MitoTracker in the knockdown parasites maintained with or without aTc (“Materials and methods”). As shown in Fig. 2E, a healthy mitochondrion in a late trophozoite has a tubular structure with MitoTracker staining largely confined to the organelle (Tubular, top panel), whereas a sick parasite was unable to constrain MitoTracker staining to the mitochondrion, and it diffused throughout the cytosol (Diffused, bottom panel). We quantified the percentages of Tubular (healthy) versus Diffused (sick) MitoTracker staining patterns in the PfmRPL13 knockdown parasites over four IDCs. As shown in Fig. 2F, D10-PfmRPL13-KD parasites grown with aTc continuously present maintained healthy mitochondria, with very few parasites showing a diffused pattern. However, when aTc was removed, the percentage of parasites with diffused staining in the PfmRPL13 knockdown parasites increased dramatically in the third and fourth cycles. These data suggest that PfmRPL13 and, hence, functional mitoribosomes were critical to maintain mitochondrial membrane potential and parasite health. Taken together, our data strongly imply that PfmRPL13 is essential for parasite growth and survival in the asexual blood stages of P. falciparum. In addition, our results agree with the recent genome-wide gene disruption study carried out in the rodent parasite, which revealed the essentiality of mitochondrial ribosomal protein L13 in P. berghei (32).
Genetic ablation of PfmRPL13 leads to mtETC deficiency and proguanil hypersensitivity
Because the mtETC is the recipient of the three proteins translated by mitoribosomes in malaria parasites, it is critical to evaluate the function of the mtETC in the PfmRPL13 knockdown parasites. To do that, we directly measured the bc1 complex enzymatic activity in vitro by an assay measuring its ability to reduce oxidized cyt c (a model of Q cycle (33) was depicted in Fig. 3A). We isolated mitochondria from D10-PfmRPL13-KD parasites maintained under aTc continuously and in the absence of aTc for two cycles and four cycles (“Materials and methods”). As shown in Fig. 3B, when aTc was removed for two cycles, there was a ∼20% reduction in the bc1 complex enzymatic activity (p < 0.05); however, after aTc was removed for four cycles, the bc1 complex enzymatic activity was diminished by 70% (p < 0.001). These data provide direct evidence that knocking down PfmRPL13 results in defects in the bc1 complex. It has been shown that whereas yDHODH transgenic parasites are resistant to atovaquone and other bc1 complex inhibitors, they become hypersensitive to proguanil when their mtETC is chemically inhibited (23). To explore whether mtETC malfunction in the PfmRPL13 knockdown parasites would also make these parasites hypersensitive to proguanil, we cultured the parasites in the absence of aTc for three IDCs and performed a [3H]hypoxanthine incorporation assay. As shown in Fig. 3C, in the WT D10 line, there was no change in proguanil EC50s in the presence or absence of aTc (250 nm), suggesting that aTc did not affect proguanil sensitivity. However, in the knockdown parasite, there was a 55-fold increase in proguanil sensitivity when PfmRPL13 was genetically abolished. The EC50 of proguanil in D10-PfmRPL13-KD parasites was 9.56 μm with aTc present, whereas it was reduced to 0.17 μm when aTc had been previously removed for three cycles. The yDHODH transgenic line was used as a positive control in the assay (23). As expected, the yDHODH line exhibited proguanil hypersensitivity when the parasite was added with 50 nm atovaquone (Fig. 3C). Remarkably, the level of proguanil hypersensitivity in our PfmRPL13 knockdown parasites (55-fold) was comparable with that of the yDHODH line under atovaquone treatment (62-fold). These data suggest that PfmRPL13 genetic ablation caused a severe mtETC deficiency in a degree that was similar to that triggered by atovaquone inhibition. To rule out the possibility that hypersensitivity to proguanil in the D10-PfmRPL13-KD line was merely a result of poor parasite growth in the absence of aTc, we tested its sensitivity to other antimalarial compounds including PA21A092 (34), a PfATP4 disruptor, and artemisinin. As shown in Fig. S4, in the presence or absence of aTc, D10-PfmRPL13-KD parasites exhibited the same levels of sensitivity to PA21A092 and artemisinin, respectively. Taken together, these data strongly suggest that proguanil hypersensitivity in the PfmRPL13 knockdown parasites was specifically induced by ablation of PfmRPL13 (see “Discussion”).
Figure 3.

Reduced cytochrome bc1 complex enzymatic activity and increased proguanil sensitivity in PfmRPL13 knockdown parasites. A, a schematic presentation of the Q cycle in the bc1 complex, modified from Ref. 33. In the assay, the rate of reduction of oxidized cyt c by reduced Qd catalyzed by the bc1 complex was measured in mitochondria isolated from P. falciparum cultures with a light-scatter rejecting UV/VIS spectrometer (OLIS, Bogart, GA). B, the cyt c reduction activity of the bc1 complex is dramatically reduced in the PfmRPL13 knockdown parasites. In each measurement, activity of the bc1 complex was determined by recording the absorbance change at 550 nm per microgram of protein in the mix. The bc1 enzymatic activity of the knockdown cultures was normalized to that of the culture maintained constantly under aTc. Statistical analysis is done by a Student's t test in n = 3 experiments. **, p < 0.001; *, p < 0.05. C, hypersensitivity to proguanil in the PfmRPL13 knockdown parasites. The knockdown parasites were grown for three cycles in the absence of aTc before exposure to proguanil. EC50 values of proguanil ([μm]) are D10 aTc ON (10.7), D10 aTc OFF (10.6), yDHODH line Atv OFF (6.99), yDHODH line Atv ON (0.11), PfmRPL13 aTc ON (9.56), PfmRPL13 aTc OFF (0.17). Atv, atovaquone. Data shown are the mean ± S.D. of three replicates and are representative of n = 5 independent experiments.
PfmRPL13 knockdown results in unusual morphological changes in the mitochondria
Next, we used transmission EM (TEM) to assess any morphological alterations in the knockdown parasites. To exclude the possibility that aTc causes changes in parasite morphology, D10 WT parasites were cultured with and without aTc for 2 weeks and their morphologies were examined by TEM (“Materials and methods”). As shown in Fig. S5, with or without aTc, D10 parasites displayed very similar mitochondrial morphologies. Mitochondria in both samples appeared to be circular structures in TEM 2D sections. Because each malaria parasite has just one single tubular mitochondrion (35), it is more likely to obtain cross-sections (circular) than longitudinal sections (tubular) during TEM 2D sample slicing. Mitochondrial morphologies similar to those of D10 were observed in the PfmRPL13 knockdown parasites maintained continuously in medium with aTc; in each TEM section, there were, again, a small number of circular structures present (Fig. 4). In contrast, when aTc was removed for three cycles, it seemed that more mitochondrial pieces were detected on each TEM section of the knockdown parasite (Fig. 4). In addition, many of the mitochondrial pieces appeared as long tubular structures, sometimes exhibiting branching (Fig. 4). To the best of our knowledge, such elongated and branched mitochondria in trophozoite stages of P. falciparum have not been reported previously in TEM studies in the literature. In all, these data revealed interesting morphological changes in parasite mitochondria when PfmRPL13 expression was genetically diminished.
Figure 4.
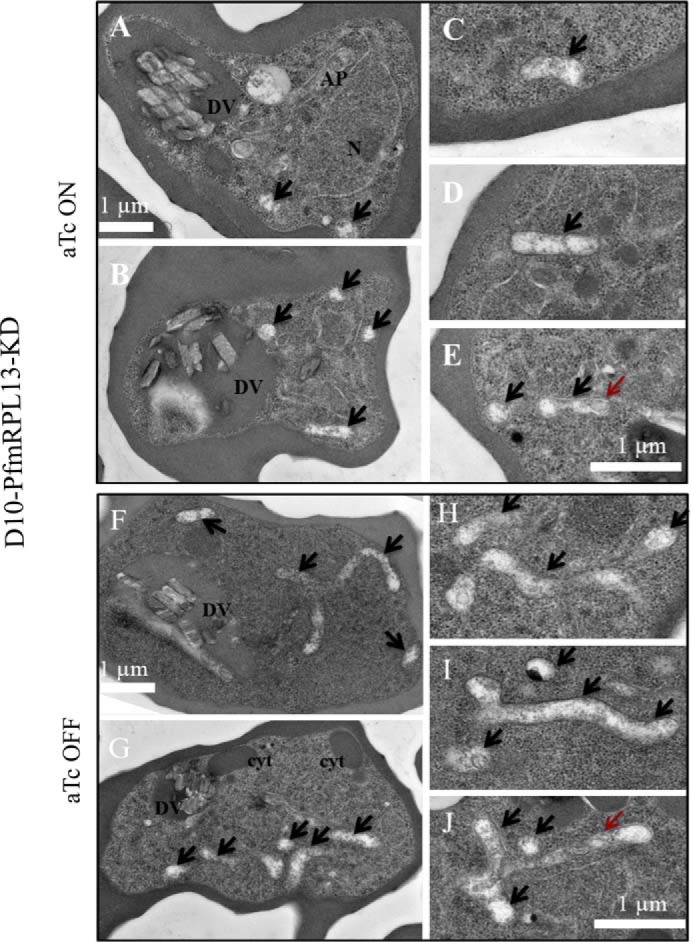
Morphological changes of mitochondria in the PfmRPL13 knockdown parasites maintained with or without aTc under transmission EM. A–E, TEM images of PfmRPL13 knockdown parasites maintained constantly under aTc. DV, digestive vacuole. AP, apicoplast. N, nucleus. F–J, TEM images of PfmRPL13 knockdown parasites grown without aTc for three cycles. DV, digestive vacuole. Cyt, cytostome. Black arrows indicate mitochondrial sections. Red arrows indicate internal membranes within the mitochondrial matrix. Each panel shows either an entire (A, B, F, G) or a partial (C–E, H–J) image of TEM.
Efforts to rescue PfmRPL13 knockdown parasites grown in the absence of aTc
To gain an understanding of the mechanisms that led to parasite demise when PfmRPL13 was knocked down, we attempted to rescue the knockdown parasites with three different approaches. Previous studies in P. falciparum have shown that mtETC inhibition by atovaquone can be partially rescued with addition of ubiquinone analogs such as decylubiquinone (Qd) (36). As shown in Fig. 5A, however, addition of Qd (50 μm) failed to restore growth of D10-PfmRPL13-KD parasites when aTc had been removed, suggesting that mtETC defects caused by PfmRPL13 knockdown could not be chemically rescued by Qd. Because inhibition of the mtETC by atovaquone can be fully suppressed by expression of a yeast DHODH gene (23), we hypothesized that provision of the yDHODH gene in the knockdown parasites might overcome mtETC defects caused by PfmRPL13 disruption. We then performed a second transfection of the knockdown parasites with an episomal plasmid carrying yDHODH-GFP, resulting in the double transgenic line D10-PfmRPL13-KD-yDHODH-GFP (“Materials and methods”). Expression of the fused yDHODH-GFP protein was confirmed with a fluorescence microscope and Western blotting (data not shown). As shown in Fig. 5B, in the absence of aTc, the double transgenic line displayed moderate growth in the first four cycles (short term) but still succumbed to death significantly after five cycles or longer without aTc (long term). These data suggested that, in the long term, yDHODH expression still failed to rescue mtETC defects resulting from PfmRPL13 ablation. We then complemented D10-PfmRPL13-KD with an episomal PfmRPL13 tagged with 3Myc, resulting in another double transgenic line, D10-PfmRPL13-KD-RL13Myc (“Materials and methods”). Expression of PfmRPL13Myc was confirmed by Western blotting in the double transgenic line maintained with or without aTc for four cycles (data not shown). As shown in Fig. 5C, D10-PfmRPL13-KD-RL13Myc grew equally well in the presence or absence of aTc for a long term. To verify aptamer copy number in the endogenous PfmRPL13 locus of the double transgenic line, D10-PfmRPL13-KD-RL13Myc, we checked its 5′ integration site by PCR and sequencing and eight aptamer copies were still intact (data not shown). Therefore, in the absence of aTc, the endogenous PfmRPL13 was not translated but the double transgenic line was sustained by the episomal expression of PfmRPL13 to maintain growth. Clearly, genetic ablation of PfmRPL13 in the parasite was only rescued with provision of complementing episomal PfmRPL13.
Figure 5.

Efforts to rescue growth of the PfmRPL13 knockdown line in the absence of aTc via three approaches. A, Qd was unable to rescue growth of the knockdown parasites in the absence of aTc. B, provision of episomal copies of yDHODH in the knockdown parasites failed to fully rescue growth without aTc over the long term. C, supplementing the knockdown parasites with episomal copies of PfmRPL13–3Myc successfully rescued growth in the absence of aTc. For all conditions, parasitemia and growth index were defined using the same method as descried in Fig. 2C. Data shown are the mean ± S.D. of n = 3 biological replicates.
Discussion
With the recent decline in the efficacy of artemisinin-based combination therapies (37), there is an urgent need to discover new antimalarial drugs. The mtETC of the parasite is absolutely essential throughout its complex life cycle. The mtETC is critical both to sustain the essential pyrimidine biosynthesis pathway (23) and to maintain the mitochondrial membrane potential (38). The malarial mtETC has been the focus of many endeavors seeking novel antimalarial drugs (39), yet the structure and function of mitochondrial ribosomes (mitoribosomes), which translate critical protein subunits of the mtETC, remain entirely uncharacterized. In this study, we found that one mitochondrial ribosomal large subunit protein L13 (PfmRPL13) was localized to the mitochondrion (Fig. 1) and was essential for growth of asexual blood stage parasites (Fig. 2). Knockdown of PfmRPL13 resulted in a series of adverse events in the parasite, including loss of Δψm (Fig. 2F), mtETC deficiency (Fig. 3B), and hypersensitivity to proguanil (Fig. 3C). Remarkably, PfmRPL13 knockdown also led to unusual mitochondrial morphologies (Fig. 4). For the first time, we provided direct evidence that mitoribosomes are essential in asexual blood stages of malaria parasites. Our genetically tagged parasite line will also be a good tool to study structural and functional aspects of mitoribosomes in the future.
The origin of all eukaryotic mitochondria can be traced back to the integration of an α-protobacterium into the proto-eukaryote ancestor (5, 40). From this single symbiotic event, the common ancestor of all eukaryotic organisms has evolved into the myriad extant cells and organisms with many distinct morphologies and functions that allow survival in drastically different environmental milieus. Needless to say, mitochondria are key players in this ongoing evolutionary adaptation, because they are critical for maintaining cellular bioenergetics, redox balance, signaling, and even life and death decisions (41). However, in apicomplexan parasites, which cause numerous diseases in human and livestock, the mitochondrion has been streamlined by loss of many functions that are important in mammalian mitochondria, for example, fatty acid catabolism, amino acid degradation, steroid biosynthesis, and initiation of apoptosis (35). Nevertheless, the mitochondrion is still absolutely essential in every stage of the parasite's life cycle (39). Even Cryptosporidium, which has lost the mitochondrial genome, has a mitosome (mitochondrion derivative organelle) that harbors at least one essential biochemical pathway—iron-sulfur cluster biogenesis (8, 42). To maintain protein import and critical biochemical pathways, mitochondria and mitochondrion-related organelles require an electrochemical proton gradient (composed of ΔpH + Δψm) across the mitochondrial inner membrane. In malaria parasites, the primary mechanism to sustain Δψm, the principle component of the proton gradient, is fulfilled by the mtETC (24, 38). Whereas most mtETC proteins are nuclear-encoded and imported into the mitochondrion, three core components are encoded on the 6 kb mtDNA: cyt b, COXI, and COXIII. The mitochondrial ribosomes and protein translation system are therefore critical to produce these three mtETC components. In this study, we have shown that PfmRPL13 ablation in P. falciparum results in mtETC deficiency (Fig. 3B), which leads to the loss of Δψm (Fig. 2F) and parasite death (Fig. 2C). For the first time, we have provided direct evidence that mitochondrial ribosomes are essential in malaria parasites. It is likely that a domino effect follows PfmRPL13 ablation: 1) reduced translation efficiency in mitochondrial ribosomes; 2) shortage of cyt b, COXI, and COXIII proteins; 3) failure to assemble functional mtETC complexes; 4) significant reduction of Δψm; 5) inability to complete pyrimidine biosynthesis, and 6) ultimately parasite death. In the future, we plan to further characterize PfmRPL13 knockdown parasites to better understand the detailed mechanisms leading to parasite collapse.
One proposed alternative pathway to maintain Δψm in malaria parasites is rotation of the F0F1 ATP synthase complex (Complex V) in the reverse direction, hydrolyzing ATP (−4 charge) to ADP (−3 charge) (23). One ADP (in the matrix) is then exchanged with one cytosolic ATP, moving one net negative charge across the mitochondrial inner membrane. In addition, ATP hydrolysis by intact coupled Complex V results in the extrusion of protons across the membrane in the opposite direction, providing a further contribution to Δψm. In the yDHODH transgenic parasites, Δψm remained substantially intact even after a complete inhibition of the mtETC by atovaquone, but was rapidly disrupted when proguanil was added subsequently (23). The synergistic effect of atovaquone and proguanil in collapsing Δψm suggests that proguanil disrupts the alternative Δψm pathway that is likely maintained by Complex V. Remarkably, PfmRPL13 knockdown parasites exhibited proguanil hypersensitivity to a similar degree as yDHODH transgenic parasites under atovaquone inhibition (Fig. 3C). These results suggest that PfmRPL13 knockdown caused a severe defect in the mtETC, which rendered parasites reliant on the alternative pathway (proguanil-sensitive) to maintain Δψm.
Ablation of PfmRPL13 resulted in a significant morphological change in the parasite mitochondria (Fig. 4). It has been well known that the mitochondrion of P. falciparum in the asexual blood stages is a tubular structure with very few or no cristae on the inner membrane (35). However, in our PfmRPL13 knockdown parasites, many long tubular and even branched mitochondria were observed in the TEM 2D sections, although they still remained cristae-deficient (Fig. 4). The mechanisms that led to these unusual morphological changes in the mitochondria of knockdown parasites remain unknown at present. Because there is only one mitochondrion per parasite, it appears that the mitochondrion of the knockdown parasite has undergone a tremendous “growth” in mass and volume. We speculate that mtETC deficiency and/or other mitochondrial defects that result from PfmRPL13 ablation may have triggered a mitochondrial biogenesis pathway(s) to compensate for the loss of mitochondrial components or functions. On the other hand, because these are TEM 2D images, the likelihood that the unusual structures were caused by mitochondrial fragmentation cannot be ruled out. Clearly, further investigation is needed to address these hypotheses.
The results of rescue experiments indicate that the mtETC defects caused by PfmRPL13 knockdown cannot be fully released by addition of the yeast DHODH gene (Fig. 5B), even though yeast DHODH completely restored parasite growth under atovaquone inhibition (23). It has been shown that the bc1 complex of the yDHODH transgenic line remained fully functional (23). It is likely that atovaquone inhibits the electron transfer activity of the bc1 complex by binding to the Qo site of cyt b, but leaving the complex physically intact. In our PfmRPL13 knockdown parasites, however, the mtETC defects seem to be more severe. Mitoribosome deficiency caused by PfmRPL13 knockdown would be expected to compromise both the electron transfer activity and the physical integrity of the mtETC. Although the mtETC can be metabolically bypassed by expression of the yDHODH gene (23), the physical components of the mtETC cannot be supplemented by this exogenous gene. In the PfmRPL13 knockdown parasites, it is likely that loss of integrity of mtETC and Δψm causes a wide range of detrimental defects in the mitochondrion. For example, the mitochondrial iron–sulfur cluster biogenesis pathway, dependent on a functional Δψm, will be deprived, which in turn is detrimental to the mitochondrion and the rest of the parasite. Indeed, our knockdown line was only able to be rescued by complementation with episomal expression of PfmRPL13 (Fig. 5C). These data further support that in addition to its enzymatic activities, physical integrity of the mtETC is essential for mitochondrial physiology.
In summary, we provide direct evidence that the mitochondrial ribosomes in the human malaria parasites are essential. Disruption of mitoribosomes by ablation of a critical protein subunit causes a chain of adverse events and eventual parasite demise. Our data highlight the significance of the mitochondrial protein translation machinery, which is evidently critical to sustain mitochondrial function, structure, and physiology.
Experimental procedures
Plasmid construction
For localization studies, the coding region of P. falciparum mitochondrial ribosomal subunit L13 (PfmRPL13, PF3D7_0214200) was cloned into the pLNmRL2 vectors, bearing either a 3HA or GFP tag (43). For CRISPR/Cas9-mediated knockout studies, two homologous regions and one gRNA of PfmRPL13 were cloned into the pL6 vector, which was kindly provided by Dr. Lopez-Rubio (44). The type II Streptococcus pyogenes Cas9 was encoded in the pUF1-Cas9 vector (44). For CRISPR/Cas9-mediated knockdown studies, the TetR-DOZI-aptamer system was utilized, which was kindly provided by Dr. Jacquin Niles (30). The original pMG75-ATP4-aptamer vector (30) was reconstructed to remove the attP sites and to replace the single HA tag with a triple HA tag. The pAll-In-One (pAIO) pre-gRNA vector was kindly provided by Dr. Spillman and Dr. Beck (31), which contained Cas9 fused with yDHODH (23) and elements for expressing a gRNA. For complementation studies, a second transfection was performed in the PfmRPL13 knockdown parasites, introducing episomal plasmids of yDHODH-GFP (23) or PfmRPL13–3Myc encoded on a vector that had a human dihydrofolate reductase selectable marker. Details for other cloning procedures are available in the supporting information.
Parasite lines, parasite culture, and transfections
See supporting information for details.
Immunofluorescence assay
Parasite samples (50 μl) were prelabeled with 60 nm MitoTracker Red CMXRos (M7512, Life Technologies by Thermo Fisher Scientific), fixed with 4% formaldehyde/0.0075% glutaraldehyde 1 h at 37 °C, permeabilized with 0.1% Triton X-100/PBS for 10 min, treated with 0.1 mg/ml NaBH4 for 5 min and blocked with 5% BSA/PBS before primary and secondary antibody incubations. The HA probe (sc-7392, Santa Cruz Biotechnology) and an Alexa Fluor 488 conjugated anti-mouse secondary antibody (A-21141, Life Technologies by Thermo Fisher Scientific) were used at 1:350 dilution for overnight at 4°. All other steps followed the published protocol (43). The parasites were visualized with an Olympus epifluorescence microscope and images were processed using SlideBook software.
Western blotting
See supporting information for details.
Parasite growth curves and knockdown experiment
Parasite cultures were tightly synchronized with several rounds of alanine treatment (0.5 m alanine/10 mm HEPES, pH 7.6) and trophozoite/schizont stages were isolated using a Percoll gradient (89428–524, GE Healthcare Life Sciences). The highly enriched parasites were thoroughly washed with medium and inoculated into new cultures with each 10 μl parasitized pellet diluted in 10 ml medium containing 1 ml blood. Cultures were maintained in the presence and absence of 250 nm anhydrotetracycline (Millipore Sigma) and were split 1:5 every 2 days. At each split, samples were collected for thin blood smears and protein analysis by Western blotting. Parasitemia was determined by counting at least 1000 RBCs under a Leica light microscope.
MitoTracker staining and quantification
The knockdown experiment was set up as described above. In every 2 days, aliquots of parasitized RBCs (∼10 μl pellet) were taken from cultures maintained with or without aTc, re-suspended in 200 μl medium containing 10 nm MitoTracker Red CMXRos and incubated for 30 min. The parasites were washed three times with 1× PBS, re-suspended in a small volume of medium (∼15 μl) and visualized under an Olympus epifluorescence microscope. For each condition, a total of 200 or more parasites were examined within 15 min.
Mitochondrion preparation (Mito Prep)
Mitochondria of P. falciparum cultures were isolated according to a published protocol (45). Briefly, parasites were tightly synchronized and expanded to a large volume (∼2 liters). Cultures were harvested at late trophozoite stage and lysed with 0.05% saponin. The pellet was washed, re-suspended in a MESH buffer and disrupted using a N2 cavitation bomb (45). The cell debris was removed by a low speed centrifugation and the supernatant was passed through a MACS Cell Separation Column (Miltenyi Biotec) at 4 °C. The eluted material was pelleted by centrifugation, aliquoted, and stored at −80 °C. Details are listed in the supporting information.
bc1 complex enzymatic activity measurement
Cytochrome c reductase activity was assayed by a minor modification of published methods (45, 46). The assay was performed at 35 °C in a stirred cuvette with a total volume of 300 μl, which contained 5 μl Mito Prep sample, 100 μm decylubiquinol, 100 μm horse heart cytochrome c (Millipore Sigma), 0.1 mg/ml n-Docecyl β-d-Maltoside, 60 mm HEPES, pH 7.4, 10 mm sodium malonate, 1 mm EDTA, and 2 mm KCN. The reduction of cyt c was recorded at 550 nm with a CLARiTY VF integrating spectrophotometer (OLIS, Bogart, GA). The short chain ubiquinol analog decylubiquinol was prepared by reducing decylubiquinone in DMSO with sodium borohydride and acidifying the mixture with concentrated HCl. Aliquots were stored under argon at −80 °C. Protein concentration of Mito Prep samples was determined by a Bio-Rad colorimetric assay using a spectrometer (Spectronic Genesys 5) at the wavelength of 595 nm.
[3H]Hypoxanthine growth inhibition assay
Growth inhibition assays using [3H]hypoxanthine incorporation were performed in 96-well plates as described previously (36), and data were analyzed by GraphPad Prism 6. See the supporting information for details.
Magnetic enrichment of parasites and transmission EM
aTc was removed from PfmRPL13 knockdown parasites for three intraerythrocytic cycles. Parasites were enriched by a MACS Cell Separation Column in a magnetic apparatus. See the supporting information for other details.
Author contributions
H. K. and A. B. V. conceptualization; H. K. resources; H. K. and J. M. M. data curation; H. K. software; H. K. and M. W. M. formal analysis; H. K. supervision; H. K. and A. B. V. funding acquisition; H. K. validation; H. K., S. D., J. M. M., and M. W. M. investigation; H. K. visualization; H. K., S. D., J. M. M., and M. W. M. methodology; H. K. writing-original draft; H. K. project administration; H. K., S. D., J. M. M., M. W. M., and A. B. V. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Dr. Jose Juan Lopez-Rubio (Institut Pasteur, France), Dr. Jacquin Niles (MIT, United States), Dr. Natalie Spillman (The University of Melbourne, Australia), and Dr. Joshua Beck (Iowa State University, United States) for sharing plasmid vectors. We are grateful to Dr. Suresh Ganesan, a former member of Dr. Niles' group, for his assistance in constructing pMG75 for double crossover recombination. We thank Dr. Wandy Beatty at the Molecular Microbiology Imaging Facility at Washington University in St. Louis for conducting TEM studies. Dr. Lawrence Bergman (Drexel University College of Medicine) replaced the single HA tag of pMG75-ATP4 with a triple HA tag.
This work was supported by National Institutes of Health Career Transition Award K22, K22AI127702 (to H. K.) and National Institutes of Health Award R01 AI028398 (to A. B. V.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S5 and Table S1 and supporting information.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- mtDNA
- mitochondrial DNA
- mtETC
- mitochondrial electron transport chain
- cyt
- cytochrome
- LSU
- large subunit
- cryo-EM
- cryo–electron microscopy
- DHODH
- dihydroorotate dehydrogenase
- aTc
- anhydrotetracycline
- gRNA
- guider RNA
- IDC
- intraerythrocytic developmental cycle
- TEM
- transmission EM
- Qd
- decylubiquinone
- TetR
- tetracycline repressor
- 3HA
- triple HA.
References
- 1. World Health Organization (2017) World malaria report 2017. WHO Publications, Geneva, Switzerland [Google Scholar]
- 2. Gardner M. J., Hall N., Fung E., White O., Berriman M., Hyman R. W., Carlton J. M., Pain A., Nelson K. E., Bowman S., Paulsen I. T., James K., Eisen J. A., Rutherford K., Salzberg S. L., et al. (2002) Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419, 498–511 10.1038/nature01097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wilson R. J., Denny P. W., Preiser P. R., Rangachari K., Roberts K., Roy A., Whyte A., Strath M., Moore D. J., Moore P. W., and Williamson D. H. (1996) Complete gene map of the plastid-like DNA of the malaria parasite Plasmodium falciparum. J. Mol. Biol. 261, 155–172 10.1006/jmbi.1996.0449 [DOI] [PubMed] [Google Scholar]
- 4. Goodman C. D., and McFadden G. I. (2014) Ycf93 (Orf105), a small apicoplast-encoded membrane protein in the relict plastid of the malaria parasite Plasmodium falciparum that is conserved in Apicomplexa. PLoS One 9, e91178 10.1371/journal.pone.0091178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gray M. W. (2014) The pre-endosymbiont hypothesis: A new perspective on the origin and evolution of mitochondria. Cold Spring Harb. Perspect. Biol. 6, a016097 10.1101/cshperspect.a016097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vaidya A. B., Akella R., and Suplick K. (1989) Sequences similar to genes for two mitochondrial proteins and portions of ribosomal RNA in tandemly arrayed 6-kilobase-pair DNA of a malarial parasite. Mol. Biochem. Parasitol. 35, 97–107 10.1016/0166-6851(89)90112-6 [DOI] [PubMed] [Google Scholar]
- 7. Hikosaka K., Kita K., and Tanabe K. (2013) Diversity of mitochondrial genome structure in the phylum Apicomplexa. Mol. Biochem. Parasitol. 188, 26–33 10.1016/j.molbiopara.2013.02.006 [DOI] [PubMed] [Google Scholar]
- 8. Puiu D., Enomoto S., Buck G. A., Abrahamsen M. S., and Kissinger J. C. (2004) CryptoDB: the Cryptosporidium genome resource. Nucleic Acids Res. 32, D329–D331 10.1093/nar/gkh050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suplick K., Akella R., Saul A., and Vaidya A. B. (1988) Molecular cloning and partial sequence of a 5.8 kilobase pair repetitive DNA from Plasmodium falciparum. Mol. Biochem. Parasitol. 30, 289–290 10.1016/0166-6851(88)90098-9 [DOI] [PubMed] [Google Scholar]
- 10. Feagin J. E. (1992) The 6-kb element of Plasmodium falciparum encodes mitochondrial cytochrome genes. Mol. Biochem. Parasitol. 52, 145–148 10.1016/0166-6851(92)90046-M [DOI] [PubMed] [Google Scholar]
- 11. Feagin J. E., Werner E., Gardner M. J., Williamson D. H., and Wilson R. J. (1992) Homologies between the contiguous and fragmented rRNAs of the two Plasmodium falciparum extrachromosomal DNAs are limited to core sequences. Nucleic Acids Res. 20, 879–887 10.1093/nar/20.4.879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Suplick K., Morrisey J., and Vaidya A. B. (1990) Complex transcription from the extrachromosomal DNA encoding mitochondrial functions of Plasmodium yoelii. Mol. Cell. Biol. 10, 6381–6388 10.1128/MCB.10.12.6381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Feagin J. E., Harrell M. I., Lee J. C., Coe K. J., Sands B. H., Cannone J. J., Tami G., Schnare M. N., and Gutell R. R. (2012) The fragmented mitochondrial ribosomal RNAs of Plasmodium falciparum. PLoS One 7, e38320 10.1371/journal.pone.0038320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sharma A., and Sharma A. (2015) Plasmodium falciparum mitochondria import tRNAs along with an active phenylalanyl-tRNA synthetase. Biochem. J. 465, 459–469 10.1042/BJ20140998 [DOI] [PubMed] [Google Scholar]
- 15. Aikawa M. (1966) The fine structure of the erythrocytic stages of three avian malarial parasites, Plasmodium fallax, P. lophurae, and P. cathemerium. Am. J. Trop. Med. Hyg. 15, 449–471 10.4269/ajtmh.1966.15.449 [DOI] [PubMed] [Google Scholar]
- 16. Wong W., Bai X. C., Brown A., Fernandez I. S., Hanssen E., Condron M., Tan Y. H., Baum J., and Scheres S. H. (2014) Cryo-EM structure of the Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetine. Elife 3, 03080 10.7554/eLife.03080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sun M., Li W., Blomqvist K., Das S., Hashem Y., Dvorin J. D., and Frank J. (2015) Dynamical features of the Plasmodium falciparum ribosome during translation. Nucleic Acids Res. 43, 10515–10524 10.1093/nar/gkv991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gupta A., Shah P., Haider A., Gupta K., Siddiqi M. I., Ralph S. A., and Habib S. (2014) Reduced ribosomes of the apicoplast and mitochondrion of Plasmodium spp., and predicted interactions with antibiotics. Open Biol. 4, 140045 10.1098/rsob.140045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chaubey S., Kumar A., Singh D., and Habib S. (2005) The apicoplast of Plasmodium falciparum is translationally active. Mol. Microbiol. 56, 81–89 10.1111/j.1365-2958.2005.04538.x [DOI] [PubMed] [Google Scholar]
- 20. Goodman C. D., Pasaje C. F. A., Kennedy K., McFadden G. I., and Ralph S. A. (2016) Targeting protein translation in organelles of the Apicomplexa. Trends Parasitol. 32, 953–965 10.1016/j.pt.2016.09.011 [DOI] [PubMed] [Google Scholar]
- 21. Dahl E. L., and Rosenthal P. J. (2007) Multiple antibiotics exert delayed effects against the Plasmodium falciparum apicoplast. Antimicrob. Agents Chemother. 51, 3485–3490 10.1128/AAC.00527-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Camps M., Arrizabalaga G., and Boothroyd J. (2002) An rRNA mutation identifies the apicoplast as the target for clindamycin in Toxoplasma gondii. Mol. Microbiol. 43, 1309–1318 10.1046/j.1365-2958.2002.02825.x [DOI] [PubMed] [Google Scholar]
- 23. Painter H. J., Morrisey J. M., Mather M. W., and Vaidya A. B. (2007) Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature 446, 88–91 10.1038/nature05572 [DOI] [PubMed] [Google Scholar]
- 24. Fry M., and Pudney M. (1992) Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4′-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80). Biochem. Pharmacol. 43, 1545–1553 10.1016/0006-2952(92)90213-3 [DOI] [PubMed] [Google Scholar]
- 25. Vaidya A. B., and Mather M. W. (2000) Atovaquone resistance in malaria parasites. Drug Resist. Updates 3, 283–287 10.1054/drup.2000.0157 [DOI] [PubMed] [Google Scholar]
- 26. Timsit Y., Acosta Z., Allemand F., Chiaruttini C., and Springer M. (2009) The role of disordered ribosomal protein extensions in the early steps of eubacterial 50S ribosomal subunit assembly. Int. J. Mol. Sci. 10, 817–834 10.3390/ijms10030817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Khaitovich P., Mankin A. S., Green R., Lancaster L., and Noller H. F. (1999) Characterization of functionally active subribosomal particles from Thermus aquaticus. Proc. Natl. Acad. Sci. U.S.A. 96, 85–90 10.1073/pnas.96.1.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nkrumah L. J., Muhle R. A., Moura P. A., Ghosh P., Hatfull G. F., Jacobs W. R. Jr., and Fidock D. A. (2006) Efficient site-specific integration in Plasmodium falciparum chromosomes mediated by mycobacteriophage Bxb1 integrase. Nat. Methods 3, 615–621 10.1038/nmeth904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smits P., Smeitink J. A., van den Heuvel L. P., Huynen M. A., and Ettema T. J. (2007) Reconstructing the evolution of the mitochondrial ribosomal proteome. Nucleic Acids Res. 35, 4686–4703 10.1093/nar/gkm441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ganesan S. M., Falla A., Goldfless S. J., Nasamu A. S., and Niles J. C. (2016) Synthetic RNA-protein modules integrated with native translation mechanisms to control gene expression in malaria parasites. Nat. Commun. 7, 10727 10.1038/ncomms10727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spillman N. J., Beck J. R., Ganesan S. M., Niles J. C., and Goldberg D. E. (2017) The chaperonin TRiC forms an oligomeric complex in the malaria parasite cytosol. Cell Microbiol. 19, 12719 10.1111/cmi.12719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bushell E., Gomes A. R., Sanderson T., Anar B., Girling G., Herd C., Metcalf T., Modrzynska K., Schwach F., Martin R. E., Mather M. W., McFadden G. I., Parts L., Rutledge G. G., Vaidya A. B., Wengelnik K., Rayner J. C., and Billker O. (2017) Functional profiling of a Plasmodium genome reveals an abundance of essential genes. Cell 170, 260–272.e8 10.1016/j.cell.2017.06.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hunte C., Palsdottir H., and Trumpower B. L. (2003) Proton motive pathways and mechanisms in the cytochrome bc1 complex. FEBS Lett. 545, 39–46 10.1016/S0014-5793(03)00391-0 [DOI] [PubMed] [Google Scholar]
- 34. Das S., Bhatanagar S., Morrisey J. M., Daly T. M., Burns J. M. Jr., Coppens I., and Vaidya A. B. (2016) Na+ influx induced by new antimalarials causes rapid alterations in the cholesterol content and morphology of Plasmodium falciparum. PLoS Pathog. 12, e1005647 10.1371/journal.ppat.1005647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vaidya A. B., and Mather M. W. (2009) Mitochondrial evolution and functions in malaria parasites. Annu Rev. Microbiol. 63, 249–267 10.1146/annurev.micro.091208.073424 [DOI] [PubMed] [Google Scholar]
- 36. Ke H., Morrisey J. M., Ganesan S. M., Painter H. J., Mather M. W., and Vaidya A. B. (2011) Variation among Plasmodium falciparum strains in their reliance on mitochondrial electron transport chain function. Eukaryot. Cell 10, 1053–1061 10.1128/EC.05049-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ashley E. A., Dhorda M., Fairhurst R. M., Amaratunga C., Lim P., Suon S., Sreng S., Anderson J. M., Mao S., Sam B., Sopha C., Chuor C. M., Nguon C., Sovannaroth S., Pukrittayakamee S., et al. (2014) Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 371, 411–423 10.1056/NEJMoa1314981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Srivastava I. K., Rottenberg H., and Vaidya A. B. (1997) Atovaquone, a broad spectrum antiparasitic drug, collapses mitochondrial membrane potential in a malarial parasite. J. Biol. Chem. 272, 3961–3966 10.1074/jbc.272.7.3961 [DOI] [PubMed] [Google Scholar]
- 39. Ke H., and Mather M. (2017) +Targeting mitochondrial functions as antimalarial regime, what is next? Curr. Clin. Micro. Rpt. 4, 175–191 10.1007/s40588-017-0075-5 [DOI] [Google Scholar]
- 40. Burger G., Gray M. W., Forget L., and Lang B. F. (2013) Strikingly bacteria-like and gene-rich mitochondrial genomes throughout jakobid protists. Genome Biol. Evol. 5, 418–438 10.1093/gbe/evt008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vafai S. B., and Mootha V. K. (2012) Mitochondrial disorders as windows into an ancient organelle. Nature 491, 374–383 10.1038/nature11707 [DOI] [PubMed] [Google Scholar]
- 42. Lill R., Srinivasan V., and Mühlenhoff U. (2014) The role of mitochondria in cytosolic-nuclear iron-sulfur protein biogenesis and in cellular iron regulation. Curr. Opin. Microbiol. 22, 111–119 10.1016/j.mib.2014.09.015 [DOI] [PubMed] [Google Scholar]
- 43. Balabaskaran Nina P., Morrisey J. M., Ganesan S. M., Ke H., Pershing A. M., Mather M. W., and Vaidya A. B. (2011) ATP synthase complex of Plasmodium falciparum: Dimeric assembly in mitochondrial membranes and resistance to genetic disruption. J. Biol. Chem. 286, 41312–41322 10.1074/jbc.M111.290973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ghorbal M., Gorman M., Macpherson C. R., Martins R. M., Scherf A., and Lopez-Rubio J. J. (2014) Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat. Biotechnol. 32, 819–821 10.1038/nbt.2925 [DOI] [PubMed] [Google Scholar]
- 45. Mather M. W., Morrisey J. M., and Vaidya A. B. (2010) Hemozoin-free Plasmodium falciparum mitochondria for physiological and drug susceptibility studies. Mol. Biochem. Parasitol. 174, 150–153 10.1016/j.molbiopara.2010.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Trumpower B. L., and Edwards C. A. (1979) Purification of a reconstitutively active iron-sulfur protein (oxidation factor) from succinate. Cytochrome c reductase complex of bovine heart mitochondria. J. Biol. Chem. 254, 8697–8706 [PubMed] [Google Scholar]
- 47. Aurrecoechea C., Brestelli J., Brunk B. P., Dommer J., Fischer S., Gajria B., Gao X., Gingle A., Grant G., Harb O. S., Heiges M., Innamorato F., Iodice J., Kissinger J. C., Kraemer E., et al. (2009) PlasmoDB: a functional genomic database for malaria parasites. Nucleic Acids Res. 37, D539–D543 10.1093/nar/gkn814 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.