

ABSTRACT
Mycobacterium tuberculosis is a critical threat to human health due to the increased prevalence of rifampin resistance (RMPr). Fitness defects have been observed in RMPr mutants with amino acid substitutions in the β subunit of RNA polymerase (RNAP). In clinical isolates, this fitness defect can be ameliorated by the presence of secondary mutations in the double-psi β-barrel (DPBB) domain of the β′ subunit of RNAP. To identify factors contributing to the fitness defects observed in vivo, several in vitro RNA transcription assays were utilized to probe initiation, elongation, termination, and 3′-RNA hydrolysis with the wild-type and RMPr M. tuberculosis RNAPs. We found that the less prevalent RMPr mutants exhibit significantly poorer termination efficiencies relative to the wild type, an important factor for proper gene expression. We also found that several mechanistic aspects of transcription of the RMPr mutant RNAPs are impacted relative to the wild type. For the clinically most prevalent mutant, the βS450L mutant, these defects are mitigated by the presence of secondary/compensatory mutations in the DPBB domain of the β′ subunit.
KEYWORDS: RNA polymerase kinetics, antibiotic resistance, tuberculosis
INTRODUCTION
Mycobacterium tuberculosis remains a substantial threat to human health. Currently rifampin (RMP), a semisynthetic rifamycin (Rif) derivative, is a key component of the primary treatment for tuberculosis (1). Rifampin acts by inhibiting the M. tuberculosis RNA polymerase (RNAP) (2, 3). Mutations in the rifamycin-resistance determining region (RRDR) of the β subunit of RNAP (from rpoB) comprise 95% of point mutations which confer resistance to RMP (4, 5). The most predominant mutations in the β subunit, βD435V, βH445Y, and βS450L (note we use M. tuberculosis numbering, these correspond to βD516V, βH526Y, and βS531L in the Escherichia coli RNAP), together account for 86% of RMP-resistant (RMPr) mutations found in clinical isolates (2, 5). The most prevalent mutation associated with rifamycin resistance is βS450L (6), which accounts for 40 to 75% of all rifamycin-resistant (Rifr) mutants in clinical reports (6–9).
The development of resistance to antibiotics usually comes at some cost to the organism's fitness (10, 11). A significant fitness defect, coupled with prolonged exposure to antibiotics, creates an environment where selective pressure to compensate for fitness defects can lead to the development of secondary mutations. There have been extensive studies correlating the prevalence of RMPr mutations in M. tuberculosis clinical isolates with the fitness costs of rifamycin resistance (12–15). Most of these studies employed either competition assays or growth rate measurements to determine whether a resistance mutation causes a fitness defect (16). Though many of the studies have tried to identify correlations between fitness and genotype, differences in genetic background and artificial growth conditions may convolute how fitness is presented. In addition, pleiotropic effects from drug resistance mutations can further complicate interpretation of direct effects from a particular mutation. In contrast, the intrinsic changes in enzyme function in vitro can be interpreted as a direct effect of a particular mutation.
Laboratory-evolved strains carrying the same RMPr point mutations have been observed to exhibit various fitness defects. Interestingly, strains carrying the βS450L mutation tend to exhibit the least fitness defect compared to other RMPr mutants in competition studies. The minimal observed fitness defect for this particular mutant was used as a rationale for explaining its prevalence in clinical isolates (12). However, a number of clinical isolates containing the βS450L mutation were observed to have various fitness defects despite sharing the same RMP resistance allele. This led to the discovery of secondary mutations, predominantly at the interface of the α and β′ subunits of RNAP, a majority of which are found in the double-psi beta-barrel (DPBB) domain of the β′ subunit (rpoC, Fig. 1), which houses the catalytic site of RNAP, including the magnesium-coordinating motif, DFDGD. We refer to these secondary mutations as “compensatory mutations” for their ability to ameliorate/compensate for the fitness defects of the primary RMPr mutants. The presence of these compensatory mutations is overwhelmingly, but not exclusively associated with the βS450L RMPr mutation (75%); approximately 9% of the compensatory mutations were observed in the α subunit (rpoA) (16, 17). The β′V483G and β′F452L mutations are the most prevalent mutations associated with the βS450L mutant and have been observed in all of the studies mentioned above, suggesting that they arose by convergent evolution and are physiologically relevant to the organism (12).
FIG 1.

Compensatory mutation sites. (A) Double-psi β-barrel (DPBB) of M. tuberculosis RNAP is highlighted with the DFDGD motif and catalytic Mg2+ (dark blue) and compensatory mutations (cyan). The compensatory mutations used in this study are denoted in magenta (PDB 5UHA). (B) Sequence alignment of M. tuberculosis, E. coli, and T. thermophilus DPBB. Stars above sequence alignment are compensatory mutations. The red stars indicate the compensatory mutations in this study.
There is an incomplete understanding regarding the disproportionate representation of particular RMPr mutations in clinical isolates despite the efficacy of these mutations to decrease the susceptibility of RNAP to RMP (18). Our study provides a molecular foundation for the enrichment of the βS450L mutant in clinical isolates and elucidates fitness defects associated with this mutant which likely drive the pressure to develop compensatory mutations. Three M. tuberculosis trans-acting transcription factors—CarD, NusA, and NusG—were also studied to determine the extent of alteration of the intrinsic properties of each RMPr mutant in physiologically relevant contexts. Several studies of RMPr E. coli have shown that mutations in the RRDR have differential effects on transcription initiation, elongation, and termination (19–22); however, these mutants have usually been selected from phenotypic screens of laboratory evolved RMPr mutants and differ from those observed in clinical settings. Until now, these effects have not been biochemically characterized in clinically relevant M. tuberculosis RMPr RNAPs. These studies provide insight into aspects of transcription which are critical to the M. tuberculosis viability.
RESULTS
RMPr mutant RNAPs form unstable open-promoter complexes (RPo) which are stabilized by compensatory mutations in the β′ subunit.
Single-round transcription assays performed on supercoiled plasmid DNA template containing the M. tuberculosis rRNA promoter (rrnA3) upstream of the synB terminator (23) were used to determine open-complex half-lives in the presence and absence of CarD (Fig. 2), a transcriptional regulator which is known to stabilize the rrnA3 open-promoter complex. The open-promoter complex half-life (t1/2 = 1.5 min) for the wild-type (WT) M. tuberculosis RNAP determined in the absence of CarD under these conditions is consistent with previous reports in the literature (t1/2 = 2 min) (24). Each of the three predominant RMPr-inducing mutations reduced the stability of the RNAP open-promoter complexes, having half-lives ∼50% of that for the WT. The compensatory mutations, β′F452L and β′V483G, provide stabilizing effects (20 to 30% longer half-life) on the WT open-promoter complex. These compensatory mutations also mitigated the defect in open-promoter complex stability for the βS450L mutant. Open-promoter complex half-lives were not evaluated for the βD435V and βH445Y mutants with compensatory mutations because these mutants are not observed in clinical isolates. Similar to in vivo observations, the β′F452L mutation partially recovered this defect (from 50 to 75% of WT) and the β′V483G mutation fully recovered the stability defect, bringing the open-complex half-life for the βS450Lβ′V483G double mutant essentially equal to that of the WT (25).
FIG 2.
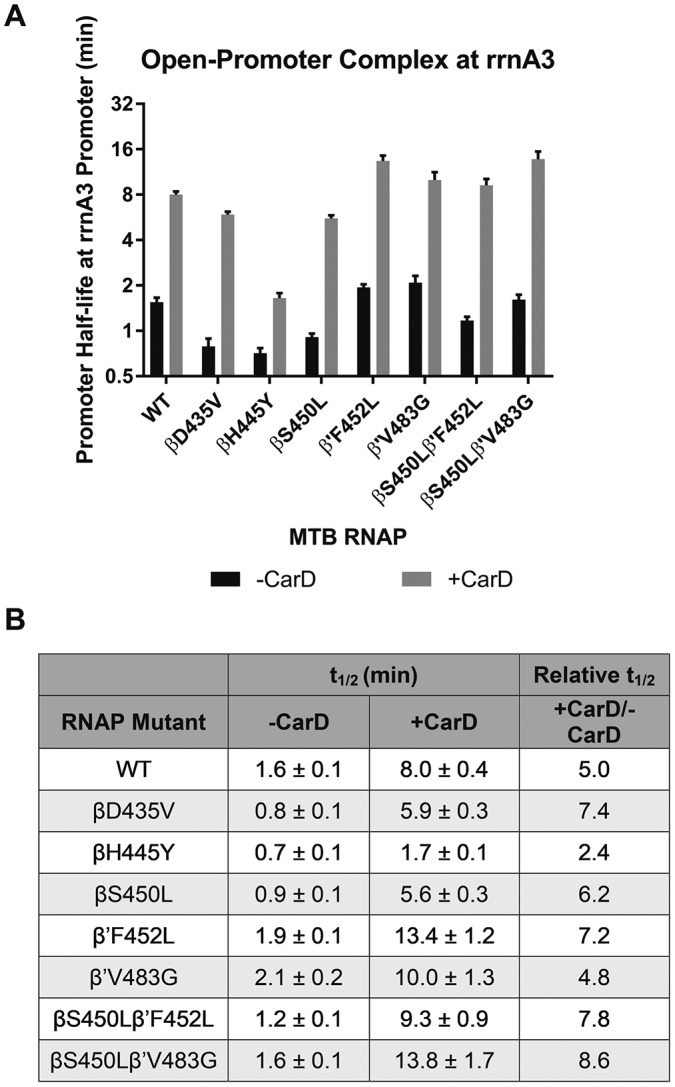
(A) Bar graph of open-promoter complex half-lives at the M. tuberculosis rrnA3 promoter in the presence and absence of transcription factor CarD. The promoter half-life times are in log2 scale. MTB, M. tuberculosis. (B) Table of open-promoter complex half-lives of M. tuberculosis RNAP variants in presence and absence of CarD.
In the presence of CarD the WT open-promoter complex was stabilized roughly 5-fold (Fig. 2). CarD also stabilized all RNAP mutant half-lives 5- to 8-fold, with the exception of the βH445Y mutant, which exhibited only a 2.4-fold increase in half-life. A similar trend in open-complex half-life across the RNAP variants is observed in the presence of CarD. Whether CarD is present or not, all RNAPs containing a compensatory mutation in the β′ subunit have a half-life greater than that observed for WT. (Data represented in Fig. 2B are derived from data in Fig. S3 in the supplemental material.)
Compensatory mutations in the β′ subunit mitigate the defect in elongation rate for the βS450L RMPr mutant.
Elongation rates for the WT and each of the M. tuberculosis RNAP mutants were determined using single-round transcription assays on a linear template containing the rrnA3 promoter (Fig. 3; see also Fig. S4 in the supplemental material). Runoff transcripts from this template produce a 398-nucleotide RNA. The template was modified by substitution of A10 and A11 each with T to allow for formation of a stalled elongation complex at the +19 position (EC19). βD435V and βH445Y RMPr RNAPs have greater elongation rates relative to the WT, whereas the βS450L mutant has a significantly slower elongation rate (Fig. 3; see also Fig. S4 in the supplemental material). Again, the presence of a compensatory mutation in the β′ subunit was able to substantially mitigate this defect (Fig. 3; see also Fig. S4 in the supplemental material). Each compensatory mutation on its own resulted in an increase in elongation rate relative to WT. It is noteworthy that there are no significant pause sites along the transcribed sequence downstream of the stalled +19 site. This allows the observed elongation rates to be attributed to processivity (phosphodiester bond formation) and not confounded by time spent at stalled positions along the template (see Fig. S4 in the supplemental material).
FIG 3.

(A) Plots used to determine the elongation rate for each of the RNAPs in this study. Density of runoff transcript was plotted against time. The time at which 50% of elongating RNAPs reach runoff length was divided by the length of the transcribed region to determine the average elongation rate. MTB, M. tuberculosis. (B) Summary of M. tuberculosis RNAP mutants' elongation rates determined from the curves in panels A and B.
The βS450L mutation improves termination efficiency, while the βD435V and βH445Y mutations impair termination efficiency at the tuf and metK terminators.
Termination efficiency was determined by single-round transcription on a linear template containing the tuf or the metK terminator downstream of a strong E. coli consensus promoter (23). For both the metK and the tuf terminators, there are two adjacent termination sites (consistent with the reported termination pattern for the tuf terminator [23]), the bands for both of which were included in the calculation of termination efficiency (Fig. 4). The βD425V and βH445Y RMPr were less able than the WT to terminate at either terminator. RNAPs containing the βS450L mutation overall were clearly the most efficient at intrinsic termination. The presence of either the β′F452L or β′V483G mutation decreased the termination efficiency of the WT RNAPs. However, both double mutants (βS450L/β′F452L and βS450L/β′V483G) retained higher termination efficiencies than the WT, albeit lower than the βS450L single mutant.
FIG 4.

Summary of termination studies at the tuf and metK terminators with all M. tuberculosis RNAPs included in this study in the presence or absence of NusA and NusG.
trans-Acting termination factors, NusA and NusG (Rv0639), were also included in termination assays. NusA stimulates termination (26). Rv0639 was annotated as the NusG homolog in M. tuberculosis; however, characterization of the protein indicates that it diverges from the canonical function observed for NusG in E. coli by stimulating termination in M. tuberculosis (23, 27). M. tuberculosis rv0639/NusG had various effects on termination depending on the terminator and RNAP mutant. At the tuf terminator, the RNAPs containing no RMPr mutations exhibited the strongest increases in termination efficiencies upon addition of NusG relative to other RNAPs. This was not the case at the metK terminator. At the metK terminator, RNAPs containing the βS450L mutation demonstrated the greatest, albeit modest relative to the tuf terminator, increases in termination efficiencies relative to no NusG, although this increase was less than that for the WT with the tuf terminator. NusA, which has been shown to increase termination efficiency in vitro for E. coli (28, 29), had essentially no effect on termination efficiencies in this study with the M. tuberculosis RNAP.
Compensatory mutations in the DPBB enhance RNA primer hydrolysis.
Single-nucleotide hydrolysis of and incorporation into an RNA8:DNA26 heteroduplex scaffold were monitored by denaturing polyacrylamide gel electrophoresis (PAGE; see Fig. S5 in the supplemental material) (30). We observed various degrees (all much smaller than incorporation) of hydrolysis of the 3′ UMP of the RNA8 oligonucleotide to a 7-mer product (Fig. 5). This likely happened during the 10-min preincubation prior to the addition of GTP. RNAP double mutants with the βS450L mutation and a compensatory mutation in the β′ subunit had elevated levels of primer hydrolysis to the 7-mer, with the β′V483G mutant (both with and without the βS450L mutation) having the greatest propensity for hydrolysis. The βD435V and βH445Y RMPr mutants displayed lower levels of hydrolysis, while the βS450L mutant displayed a similar proclivity for hydrolysis to that of the WT (Fig. 5; see also Fig. S5 in the supplemental material). Upon addition of GTP, the RNA8 is extended to the 9-mer. (Any hydrolysis of the 9-mer back to the 8-mer would be immediately corrected by addition of another G due to the excess of GTP [800-fold] present.) The percentage of active (e.g., catalytically competent) complex in a given preparation of RNAP was determined as the sum of the extension to the 9-mer and hydrolysis to the 7-mer RNA. M. tuberculosis RNAPs used in this study had activities ranging from 45 to 77% (see Fig. S5; see also Table S2 in the supplemental material).
FIG 5.
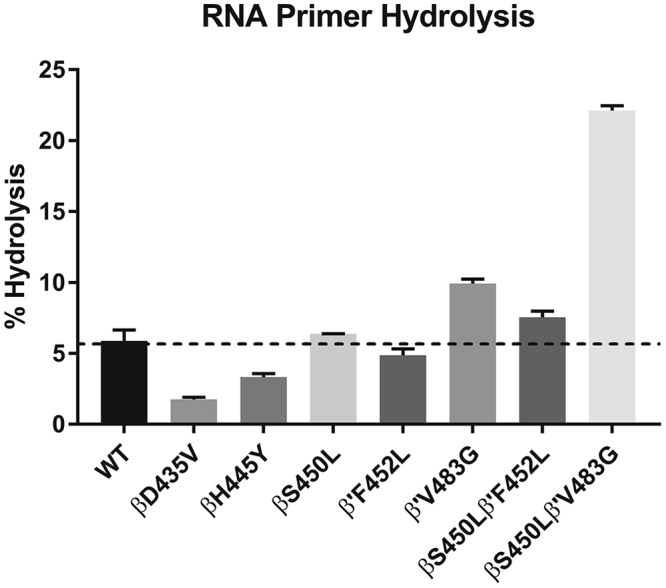
Percent hydrolysis of each RNAP in this study.
DISCUSSION
Rifampin-resistant (RMPr) mutations in the rifamycin-resistance determining region (RRDR) have been observed to inflict various fitness defects on M. tuberculosis, as evidenced by reductions in growth rates. Here, we attempted to find a molecular basis for these defects and identify why compensating mutations arise in the β′ subunit (rpoC). We found that RMPr mutations in the β subunit of M. tuberculosis RNAP (βD435V, βH445Y, and βS450L) all share similar reductions in open complex (RPo) stabilities relative to WT RNAP. The addition of CarD, a transcription factor, increased RPo stabilities 5- to 8-fold (2.4-fold for the βH445Y mutant) but did not significantly alter the trends across the mutants (Fig. 2B). The βH445Y M. tuberculosis RNAP, which exhibits the greatest reduction in RPo stability relative to the WT, has a significantly altered RMP binding pocket conformation (18). This may reduce the ability of CarD to fully stabilize the RPo complex for this mutant.
The RMPr mutations in the β subunit are adjacent to Fork Loop 2 (FL2), which has been observed to adopt multiple conformations, one where it interacts with the leading edge of the transcription bubble and others where it makes contacts with the bridge helix (31, 32). In the E. coli RNAP crystal structures with RMP, FL2 of the βS531L (βS450L in M. tuberculosis numbering) mutant was disordered, reducing contacts with RMP. A disordered FL2 was not observed in the WT E. coli RMP complex structure (18), suggesting that a defect in the conformational stability of FL2 is likely a contributor to the destabilization of the RPo observed in the RMPr mutants.
Interestingly both compensatory mutations studied, β′F452L and β′V483G, had a stabilizing effect on RPo half-life for the βS450L mutant. The degree of stabilization by the β′F452 and β′V483G mutations partially and fully complemented the defect observed for the βS450L mutation, consistent with the growth rate measurements of M. smegmatis expressing recombinant M. tuberculosis rpoB-rpoC alleles with these mutations (see Fig. 7A) (25). The switch region of RNAP aids in the closure of the RNAP clamp around the DNA (Fig. 6) (33, 34). Mutations in the switch region have been shown to negatively affect open-promoter complex stability via weakening the strength of the RNAP clamp on the DNA (34–37). Compensatory mutations in the DPBB domain may induce conformational changes in the switch region to rectify defects in RPo stability caused by the RMPr mutations in the β subunit through interaction with the template strand (Fig. 6A and B, left panel).
FIG 7.

(A) Similarity between open-promoter complex and growth, as well as elongation rate and competitive fitness, observed for the clinically relevant genotypes *, competitive fitness and growth were determined by Song et al. (25). (B) Inverse correlation between elongation rate and termination efficiency observed for the M. tuberculosis RNAP variants in this study.
FIG 6.

Structure of Mycobacterium smegmatis RNAP with full transcription bubble (PDB 5VI5) (51). (A) Highlighted structural motifs that contribute to effects observed on transcription. (B) Perspective of RNA-DNA heteroduplex region from the perspective of the compensatory mutations in the DPBB (left panel) and the Rifr mutations (right panel).
Surprisingly, despite being the most prevalent of the RMPr mutants present in isolates the βS450L mutant showed the greatest defect in its elongation rate, whereas the βD435V and βH445Y RMPr mutants increased the rate of elongation relative to WT (Fig. 3). Mutations in this region of the β subunit have been shown to have various effects on the elongation rate (19). A laboratory-evolved RMPr mutation in the E. coli RNAP, βI572N, resulted in an attenuated elongation rate, while mutation of the same amino acid to a threonine had no effect on the elongation rate (19). There are other instances in which mutations in this region of the β subunit effect elongation rate (increases and decreases relative to the WT) (19, 38–42). The RMPr mutations in this study are clustered around the heteroduplex region on the RNA side of the RNA-DNA heteroduplex (Fig. 6). Mutations in this pocket affect the steric and surface (polarity/electrostatic) parameters of the RNA exit channel and have the potential for either weakening or strengthening interactions between the residues within the rifamycin-binding cleft and the extending nascent RNA (18). As one consequence, mutations in the RMP cleft could have signification effects on translocation of the RNAP and elongation rate.
The RMPr mutations in the fork domain have been shown to cause slippage along homopolymeric template DNA with E. coli and S. cerevisiae RNAP (19, 43, 44). It is thought that this occurs from weakening of the clamp, causing spontaneous melting of the heteroduplex region (19). The compensatory mutations, β′F452L and β′V483G, occur adjacent to the switch region which is part of that clamp responsible for closure around the RNA-DNA heteroduplex (Fig. 6). These mutations may stimulate elongation rate via maintenance of elongation stability. Interestingly, as noted above for RPo stability, the relative elongation rates for the mutants containing the βS450L in this study also mirror the relative competitive fitness observed in M. smegmatis (Fig. 7) (25). Production of rRNA in vivo is coupled to growth rate and organism fitness (45). It is possible that slow-growing bacteria such as M. tuberculosis, which has an elongation rate 15 times slower than that of E. coli, could present fitness defects that are reflective of reduced production of rRNA (23).
“Backtracking” is a process where the RNA-DNA hybrid region translocates in the reverse direction, most often in the process of “proofreading” (46, 47). Weakened RNA-DNA hybrid regions are associated with an increased ability for the heteroduplex to backtrack (48, 49), preventing incorporation of any new nucleoside triphosphate (NTP) and rendering the 3′ nucleotide susceptible to hydrolysis. We observed that the mutations studied have various effects on the hydrolysis of the 3′ nucleotide of the RNA8 oligonucleotide (Fig. 5). Consistent with observations of RNAPs that are deficient in backtracking (faster RNAPs are less prone to backtracking), diminished exonuclease activity was observed for the βD435V and βH445Y mutants (48). In contrast, the βS450L/β′V483G mutation dramatically increases hydrolysis of 3′-UMP from RNA8 while also decreasing the elongation rate. The RMPr mutations (on the RNA side of the RNA-DNA hybrid) may affect the RNA-DNA hybrid such that forward translocation is increased, resulting in increased elongation rates and reduced backtracking and hydrolysis. While the compensatory mutations (which occur on the DNA side of the heteroduplex) may have similar effects on the RNA-DNA hybrid.
RNAPs which do not efficiently recognize termination signals will continue transcribing neighboring genes. Our observations indicate that mutant RNAPs carrying the βS450L mutation are not just more efficient at intrinsic termination than the other RMPr mutants studied but also more efficient than the WT RNAP. Although the presence of a single compensatory mutation yields a slight decrease in termination efficiency, the double mutants maintain termination efficiencies which are still greater than the WT at both the tuf and the metK terminators (Fig. 4). As previously reported, M. tuberculosis NusA had no effect on the termination efficiencies, despite observed effects in an E. coli-based transcription system. However, M. tuberculosis NusG did increase the termination efficiencies for many of the mutant RNAPs tested, though not all. It had limited effects on termination when termination efficiencies were very low or very high. We also observed that the elongation rate and termination efficiency are inversely correlated (Fig. 7B), which is consistent with previous reports (23, 40). Our results suggest that this aspect of transcription may be the driving factor for initial RMPr mutant selection. The observed changes in termination efficiencies are likely driven primarily by changes in elongation rates; thus, the elongation rate may be the predominant factor in genotype selection. Although many factors contribute to the fitness of the organism, these intrinsic properties of the RMPr mutants will likely have a major impact on fitness and clinical prevalence.
Our study explored the molecular basis for the fitness defects of certain RMPr mutants observed in M. tuberculosis clinical isolates. We found that there are distinct mechanistic and kinetic differences for each RMPr mutant. These studies provide insight into the mechanistic framework for the evolution of secondary, compensatory mutations observed in the β′ subunit of RNA polymerase. We conclude that a critical factor is the balance between elongation rate and termination efficiency and that this likely explains the prevalence of the βS450L mutation and contributes to the presence of compensatory mutations. Another factor which seems to drive secondary mutations in the β′ subunit is their effect on RPo stability. Further studies which probe the structural consequences of the RMPr mutant fitness defect compensation by compensatory mutations in the β′ subunit are needed to confirm the exact mechanism of action of these mutations.
MATERIALS AND METHODS
Plasmid and DNA manipulation.
Plasmid pMt-rrnA3 contains the M. tuberculosis rrnA3 promoter region −66 to 351. DNA was amplified from H37Rv genomic DNA (+1468189 to +1468605) and subcloned into pTZ18U using PciI and EcoRI restriction sites using primers Mt rrnA3 −66 AMP FOR and Mt rrnA3 +351 AMP REV (see Table S1 in the supplemental material). pMt-rrnA3-synB was produced by insertion of the synB terminator derived from synthetic oligonucleotides (IDT Technologies) with flanking XbaI restriction sites (23). The pMt-rrnA3 derivative, pEC19, was produced by mutagenesis of both A10 and A11 (relative to the rrnA3 TSS) to thymine using rrnA3 EC19 FOR and rrnA3 EC19 REV. pEC26-Tuf was generated by digestion of pMt-rrnA3 with PciI and MscI to remove the M. tuberculosis rrnA3 promoter. The E. coli consensus promoter which lacks cytosine until the +26 position, relative to the transcription start site, was subcloned using synthetic oligonucleotides. The M. tuberculosis tuf and metK terminators were subcloned into pEC26 using synthetic oligonucleotides containing the terminator sequences with flanking XhoI restriction sites. Plasmid used for promoter studies were purified using a Qiagen Miniprep kit. PCR amplified DNA fragments were generated from pEC19 (elongation studies) and pEC26-Tuf or pEC26-metK (termination studies) using primers EC19 AMP FOR and EC AMP REV for the elongation studies template and EC26 AMP FOR and EC AMP REV for the termination studies. PCR products were purified on a 2% agarose gel and extracted with a Qiagen gel extraction kit. Plasmid DNA and PCR fragments were then further purified by phenol-chloroform extraction, followed by ethanol precipitation. DNA was stored in water at −20°C.
Expression constructs for M. tuberculosis CarD (Rv3583c), NusA (Rv2841c), and NusG (Rv0639) were produced similarly. Each gene was amplified from H37Rv genomic DNA using primers in Table S1 in the supplemental material. All genes were subcloned into a modified pET19b construct containing a N-terminal decahistidine tag, followed by the Precission protease recognition cleavage site LEVLFQ/GP (pET19bpps). CarD and NusA were subcloned into the pET19bpps vector using NdeI and BamHI restriction sites. NusG was subcloned into pET19bpps using the NdeI and XhoI restriction sites.
Protein purification.
M. tuberculosis RNAP was prepared as previously reported (18). CarD was prepared as follows. BL21(DE3) E. coli containing pET19bpps-CarD were grown to an optical density at 600 nm (OD600) of 0.8. Protein expression was induced by addition of 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside). Cells were incubated at 28°C for 4 h prior to harvesting via centrifugation. Cells were lysed via sonication in 20 mM Tris-HCl (pH 8.0), 300 mM NaCl, 5% glycerol, 5 mM β-mercaptoethanol (β-ME), 1 mM phenylmethylsulfonyl fluoride (PMSF), 1× Roche cOmplete ULTRA protease cocktail, 200 U of DNase I, and 1 mg/ml lysozyme. Clarified lysate was passed through a 0.22-μm-pore size filter and applied to a HisTrap HP column (GE Healthcare). CarD was eluted over linear gradient to 500 mM imidazole. Fractions containing decahistadine-tagged CarD were collected and Precission protease (GE Healthcare) was added. CarD was dialyzed overnight at 4°C in 10 mM Tris-HCl (pH 8.0), 500 mM NaCl, 0.1 mM EDTA, 5% glycerol, and 5 mM β-ME. CarD was then passed through 1 ml of glutathione S-transferase (GST)–agarose and concentrated prior to application to a HiPrep Sephacryl S-200 HR column (GE Healthcare) run in 20 mM Tris-HCl (pH 8.0), 200 mM NaCl, 0.1 mM EDTA, 5% glycerol, and 1 mM dithiothreitol (DTT). Purified CarD was stored at −80°C.
NusA and NusG were prepared identically. E. coli BL21(DE3) cells containing either pET19bpps-NusA or pET19bpps-NusG were grown to an OD600 of 0.4 and induced by the addition of 0.5 mM IPTG. Cells were incubated overnight at 16°C. Cells were lysed via sonication in 20 mM Tris-HCl (pH 8.0), 300 mM NaCl, 5 mM imidazole, 5% glycerol, 5 mM β-ME, 0.1% Triton X-100, 1 mM PMSF, 1× Roche cOmplete ULTRA protease cocktail, 200 U of DNase I, and 1 mg/ml lysozyme buffer. Clarified lysate was passed through a 0.22-μm-pore size filter and then applied to a HisTrap HP column, and proteins were eluted by a linear gradient to 500 mM imidazole. Fractions containing either Nus protein were collected and Precission protease was added. Proteins were dialyzed into 10 mM Tris-HCl (pH 8.0), 50 mM NaCl, 5 mM β-ME, 5% glycerol, and 0.1 mM EDTA and passed through 1 ml of GST-agarose before application to a 6-ml Source 15Q column. Fractions containing Nus proteins were collected and dialyzed into 20 mM Tris-HCl (pH 8.0), 100 mM NaCl, 0.1 mM EDTA, 1 mM DTT, and 50% glycerol. Proteins were stored at −80°C.
Determination open-promoter complex half-life.
The open-promoter complex half-life was determined by single-round transcription. Supercoiled plasmid template containing −66 to +350 of the transcription start site of the M. tuberculosis rrnA3 promoter, followed by the synB terminator, was used for this assay. M. tuberculosis RNAP (30 nM) was preincubated with pMt-rrnA3-SynB (10 ng/μl) for 30 min at 37°C to form the open complex in 40 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 30 mM KCl, 1 mM DTT, 0.1 mM EDTA, and 25 μg/ml bovine serum albumin (BSA). When present, CarD was added to a final concentration of 2 μM and incubated for an additional 5 min. Heparin was then added to 50 μg/ml, and aliquots taken at various time points were added to the NTP mixture (1 mM GTP, 200 μM ATP, 200 μM UTP, 10 μM CTP, 0.15 μCi/μl [α32P]CTP [final concentration]). Reactions were allowed to continue for 15 min before the addition of 2× RNA loading buffer (95% formamide, 0.02% sodium dodecyl sulfate, 0.01% bromophenol blue, 0.005% xylene cyanol, 0.5 mM EDTA). Reactions were resolved using 6% acrylamide–8 M urea PAGE. Gels were imaged, and bands were quantified by phosphorimaging (Molecular Dynamics; ImageQuant software). The fraction of complex at each time point was normalized to the zero time point and fitted to a linear plot. The half-life was determined by calculating the time at which half of the remaining complex was present. All reactions were conducted in triplicate with the errors representing the standard deviation of the slope of the line.
Determination of elongation rate.
Elongation rate was determined by a single-round transcription assay on a linear template. M. tuberculosis RNAP (120 nM) was preincubated with linear DNA (30 nM), EC19, in 40 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 10 mM KCl, 1 mM DTT, 0.1 mM EDTA, and 25 μg/ml BSA for 15 min at 37°C. Stalled-elongation complex was formed by incubating 10 μM GTP, 10 μM UTP, 2 μM CTP, and 0.3 μCi/μl [α32P]CTP for 10 min. Transcription was resumed by adjusting the all NTPs to 100 μM and the addition of 50 μg/ml heparin. Aliquots were removed at the time points denoted in the figures and stopped by the addition of 2× RNA loading buffer. Reactions were resolved using 8% acrylamide–8 M urea PAGE and processed as described above. Elongation rates were determined by quantifying the density of runoff product over time, which was fit to a standard Boltzmann sigmoidal curve. Average elongation rates were determined as the point at which 50% of runoff product is formed and are presented in nucleotides per second. All experiments were conducted in triplicate.
Determination of termination efficiency.
Termination efficiencies were determined by single-round transcription assays on linear templates. M. tuberculosis terminators tuf and metK were identified by genomic profiling of M. tuberculosis intrinsic terminators (50). M. tuberculosis RNAP (50 nM) was preincubated with linear DNA (25 nM), EC26-metK, or EC26-Tuf in 40 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 10 mM KCl, 1 mM DTT, 0.1 mM EDTA, and 25 μg/ml BSA for 15 min at 37°C. Stalled elongation complexes were formed by incubating 10 μM GTP, 10 μM ATP, 2 μM UTP, and 0.1 μCi/μl [α-32P]UTP for 10 min. When used, NusA and NusG were added to 1 μM and allowed to incubate for 10 min. Transcription was restarted by addition of 50 μg/ml heparin and adjusting each NTP concentration to 200 μM. Reactions were allowed to continue for 30 min before 2× RNA loading buffer was added. Reactions were resolved using 6% acrylamide–8 M urea PAGE and processed as described above. Termination efficiencies were calculated by dividing the density of terminated products by the sum of the terminated and runoff products in each lane.
Primer extension assays.
Determination of the concentration of active RNAP and percent of RNA hydrolysis was conducted using a DNA-RNA scaffold that mimics the transcription elongation complex (30). First, an 8-nucleotide RNA (IDT Technologies) was labeled at the 5′ end with [γ-32P]GTP (Perkin-Elmer) using T4 polynucleotide kinase (New England BioLabs). The T4 polynucleotide kinase was then heat inactivated. The DNA-RNA heteroduplex scaffolds were prepared by mixing equimolar amounts of template DNA26 (5′-CTGAAATCGACATCGCCGCTCAACAT-3′) and 5′-labeled RNA8 (5′-GCGGCGAU-3′), and heating to 85°C for 10 min, followed by slow cooling to room temperature. The elongation complex was formed by mixing equimolar RNAP with the DNA-RNA heteroduplex scaffold (500 nM) in reaction buffer at 28°C for 10 min. The reaction mixture consisted of 40 mM Tris-HCl (pH 7.9), 40 mM potassium glutamate, 0.1 mM EDTA, 10 mM MgCl2, 25 μg/ml BSA, and 1 mM DTT; both reaction mixtures contained 15 mU of inorganic pyrophosphatase (New England BioLabs). The RNA extension reaction was conducted for 10 min after adding the next incoming GTP (400 μM final concentration) to the elongation complex. RNA extension was stopped by adding an equal volume of 2× RNA loading buffer. RNAs were resolved by gel-electrophoresis in a 20% acrylamide–8 M urea PAGE gel and processed as described above.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by NIH grant R01-AI110780 (G.A.G.) and the University of Michigan, College of Pharmacy.
We thank Katsu Murakami and Catherine Sutherland (Pennsylvania State University) and members of the Garcia lab for critical reading and helpful suggestions about the manuscript.
M.A.S. and G.A.G. conceived the project and wrote the manuscript. M.A.S. and F.S.U. performed all experiments. G.A.G. supervised the project and secured funding. All authors analyzed the results and approved the final version of the manuscript.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00164-18.
REFERENCES
- 1.World Health Organization. 2016. Global tuberculosis report 2016. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst SA. 2001. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 104:901–912. doi: 10.1016/S0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- 3.Gill SK, Garcia GA. 2011. Rifamycin inhibition of WT and Rif-resistant Mycobacterium tuberculosis and Escherichia coli RNA polymerases in vitro. Tuberculosis 91:361–369. doi: 10.1016/j.tube.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Andre E, Goeminne L, Cabibbe A, Beckert P, Kabamba Mukadi B, Mathys V, Gagneux S, Niemann S, Van Ingen J, Cambau E. 2017. Consensus numbering system for the rifampicin resistance-associated rpoB gene mutations in pathogenic mycobacteria. Clin Microbiol Infect 23:167–172. doi: 10.1016/j.cmi.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Ramaswamy S, Musser JM. 1998. Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuberc Lung Dis 79:3–29. doi: 10.1054/tuld.1998.0002. [DOI] [PubMed] [Google Scholar]
- 6.Poudel A, Nakajima C, Fukushima Y, Suzuki H, Pandey BD, Maharjan B, Suzuki Y. 2012. Molecular characterization of multidrug-resistant Mycobacterium tuberculosis isolated in Nepal. Antimicrob Agents Chemother 56:2831–2836. doi: 10.1128/AAC.06418-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imperiale BR, Zumarraga MJ, Weltman G, Zudiker R, Cataldi AA, Morcillo NS. 2012. First evaluation in Argentina of the GenoType® MTBDRplus assay for multidrug-resistant Mycobacterium tuberculosis detection from clinical isolates and specimens. Rev Argent Microbiol 44:283–289. [PubMed] [Google Scholar]
- 8.Rahmo A, Hamdar Z, Kasaa I, Dabboussi F, Hamze M. 2012. Genotypic detection of rifampicin-resistant M. tuberculosis strains in Syrian and Lebanese patients. J Infect Public Health 5:381–387. doi: 10.1016/j.jiph.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 9.Tang K, Sun H, Zhao Y, Guo J, Zhang C, Feng Q, He Y, Luo M, Li Y, Sun Q. 2013. Characterization of rifampin-resistant isolates of Mycobacterium tuberculosis from Sichuan in China. Tuberculosis 93:89–95. doi: 10.1016/j.tube.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 10.Andersson DI, Levin BR. 1999. The biological cost of antibiotic resistance. Curr Opin Microbiol 2:489–493. doi: 10.1016/S1369-5274(99)00005-3. [DOI] [PubMed] [Google Scholar]
- 11.Sundqvist M. 2014. Reversibility of antibiotic resistance. Ups J Med Sci 119:142–148. doi: 10.3109/03009734.2014.903323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gagneux S, Long CD, Small PM, Van T, Schoolnik GK, Bohannan BJ. 2006. The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science 312:1944–1946. doi: 10.1126/science.1124410. [DOI] [PubMed] [Google Scholar]
- 13.Billington OJ, McHugh TD, Gillespie SH. 1999. Physiological cost of rifampin resistance induced in vitro in Mycobacterium tuberculosis. Antimicrob Agents Chemother 43:1866–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mariam DH, Mengistu Y, Hoffner SE, Andersson DI. 2004. Effect of rpoB mutations conferring rifampin resistance on fitness of Mycobacterium tuberculosis. Antimicrob Agents Chemother 48:1289–1294. doi: 10.1128/AAC.48.4.1289-1294.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gagneux S. 2009. Fitness cost of drug resistance in Mycobacterium tuberculosis. Clin Microbiol Infect 15(Suppl 1):S66–S68. doi: 10.1111/j.1469-0691.2008.02685.x. [DOI] [PubMed] [Google Scholar]
- 16.Gygli SM, Borrell S, Trauner A, Gagneux S. 2017. Antimicrobial resistance in Mycobacterium tuberculosis: mechanistic and evolutionary perspectives. FEMS Microbiol Rev 41:354–373. doi: 10.1093/femsre/fux011. [DOI] [PubMed] [Google Scholar]
- 17.Comas I, Borrell S, Roetzer A, Rose G, Malla B, Kato-Maeda M, Galagan J, Niemann S, Gagneux S. 2012. Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat Genet 44:106–110. doi: 10.1038/ng.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Molodtsov V, Scharf NT, Stefan MA, Garcia GA, Murakami KS. 2017. Structural basis for rifamycin resistance of bacterial RNA polymerase by the three most clinically important RpoB mutations found in Mycobacterium tuberculosis. Mol Microbiol 103:1034–1045. doi: 10.1111/mmi.13606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou YN, Lubkowska L, Hui M, Court C, Chen S, Court DL, Strathern J, Jin DJ, Kashlev M. 2013. Isolation and characterization of RNA polymerase rpoB mutations that alter transcription slippage during elongation in Escherichia coli. J Biol Chem 288:2700–2710. doi: 10.1074/jbc.M112.429464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou YN, Jin DJ. 1998. The rpoB mutants destabilizing initiation complexes at stringently controlled promoters behave like “stringent” RNA polymerases in Escherichia coli. Proc Natl Acad Sci U S A 95:2908–2913. doi: 10.1073/pnas.95.6.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin DJ, Walter WA, Gross CA. 1988. Characterization of the termination phenotypes of rifampicin-resistant mutants. J Mol Biol 202:245–253. doi: 10.1016/0022-2836(88)90455-X. [DOI] [PubMed] [Google Scholar]
- 22.McDowell JC, Roberts JW, Jin DJ, Gross C. 1994. Determination of intrinsic transcription termination efficiency by RNA polymerase elongation rate. Science 266:822–825. doi: 10.1126/science.7526463. [DOI] [PubMed] [Google Scholar]
- 23.Czyz A, Mooney RA, Iaconi A, Landick R. 2014. Mycobacterial RNA polymerase requires a U-tract at intrinsic terminators and is aided by NusG at suboptimal terminators. mBio 5:e00931. doi: 10.1128/mBio.00931-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hubin EA, Fay A, Xu C, Bean JM, Saecker RM, Glickman MS, Darst SA, Campbell EA. 2017. Structure and function of the mycobacterial transcription initiation complex with the essential regulator RbpA. Elife 6:e22520. doi: 10.7554/eLife.22520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song T, Park Y, Shamputa IC, Seo S, Lee SY, Jeon HS, Choi H, Lee M, Glynne RJ, Barnes SW, Walker JR, Batalov S, Yusim K, Feng S, Tung CS, Theiler J, Via LE, Boshoff HI, Murakami KS, Korber B, Barry CE III, Cho SN. 2014. Fitness costs of rifampicin resistance in Mycobacterium tuberculosis are amplified under conditions of nutrient starvation and compensated by mutation in the beta′ subunit of RNA polymerase. Mol Microbiol 91:1106–1119. doi: 10.1111/mmi.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mondal S, Yakhnin AV, Sebastian A, Albert I, Babitzke P. 2016. NusA-dependent transcription termination prevents misregulation of global gene expression. Nat Microbiol 1:1–8. doi: 10.1038/nmicrobiol.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kalyani BS, Kunamneni R, Wal M, Ranjan A, Sen R. 2015. A NusG paralogue from Mycobacterium tuberculosis, Rv0639, has evolved to interact with ribosomal protein S10 (Rv0700) but not to function as a transcription elongation-termination factor. Microbiology 161:67–83. doi: 10.1099/mic.0.083709-0. [DOI] [PubMed] [Google Scholar]
- 28.Schmidt MC, Chamberlin MJ. 1987. NusA protein of Escherichia coli is an efficient transcription termination factor for certain terminator sites. J Mol Biol 195:809–818. doi: 10.1016/0022-2836(87)90486-4. [DOI] [PubMed] [Google Scholar]
- 29.Yakhnin AV, Babitzke P. 2002. NusA-stimulated RNA polymerase pausing and termination participates in the Bacillus subtilis trp operon attenuation mechanism in vitro. Proc Natl Acad Sci U S A 99:11067–11072. doi: 10.1073/pnas.162373299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Temiakov D, Anikin M, McAllister WT. 2002. Characterization of T7 RNA polymerase transcription complexes assembled on nucleic acid scaffolds. J Biol Chem 277:47035–47043. doi: 10.1074/jbc.M208923200. [DOI] [PubMed] [Google Scholar]
- 31.Kettenberger H, Armache KJ, Cramer P. 2004. Complete RNA polymerase II elongation complex structure and its interactions with NTP and TFIIS. Mol Cell 16:955–965. doi: 10.1016/j.molcel.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 32.Meyer PA, Ye P, Suh MH, Zhang M, Fu J. 2009. Structure of the 12-subunit RNA polymerase II refined with the aid of anomalous diffraction data. J Biol Chem 284:12933–12939. doi: 10.1074/jbc.M809199200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gnatt AL, Cramer P, Fu J, Bushnell DA, Kornberg RD. 2001. Structural basis of transcription: an RNA polymerase II elongation complex at 3.3 A resolution. Science 292:1876–1882. [DOI] [PubMed] [Google Scholar]
- 34.Pupov D, Miropolskaya N, Sevostyanova A, Bass I, Artsimovitch I, Kulbachinskiy A. 2010. Multiple roles of the RNA polymerase β′ SW2 region in transcription initiation, promoter escape, and RNA elongation. Nucleic Acids Res 38:5784–5796. doi: 10.1093/nar/gkq355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rutherford ST, Villers CL, Lee JH, Ross W, Gourse RL. 2009. Allosteric control of Escherichia coli rRNA promoter complexes by DksA. Genes Dev 23:236–248. doi: 10.1101/gad.1745409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wiesler SC, Burrows PC, Buck M. 2012. A dual switch controls bacterial enhancer-dependent transcription. Nucleic Acids Res 40:10878–10892. doi: 10.1093/nar/gks844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang N, Schafer J, Sharma A, Rayner L, Zhang X, Tuma R, Stockley P, Buck M. 2015. Mutations in RNA polymerase bridge helix and switch regions affect active-site networks and transcript-assisted hydrolysis. J Mol Biol 427:3516–3526. doi: 10.1016/j.jmb.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petushkov I, Pupov D, Bass I, Kulbachinskiy A. 2015. Mutations in the CRE pocket of bacterial RNA polymerase affect multiple steps of transcription. Nucleic Acids Res 43:5798–5809. doi: 10.1093/nar/gkv504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tavormina PL, Reznikoff WS, Gross CA. 1996. Identifying interacting regions in the beta subunit of Escherichia coli RNA polymerase. J Mol Biol 258:213–223. doi: 10.1006/jmbi.1996.0244. [DOI] [PubMed] [Google Scholar]
- 40.Tavormina PL, Landick R, Gross CA. 1996. Isolation, purification, and in vitro characterization of recessive-lethal-mutant RNA polymerases from Escherichia coli. J Bacteriol 178:5263–5271. doi: 10.1128/jb.178.17.5263-5271.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jin DJ, Gross CA. 1991. RpoB8, a rifampicin-resistant termination-proficient RNA polymerase, has an increased Km for purine nucleotides during transcription elongation. J Biol Chem 266:14478–14485. [PubMed] [Google Scholar]
- 42.Heisler LM, Suzuki H, Landick R, Gross CA. 1993. Four contiguous amino acids define the target for streptolydigin resistance in the beta subunit of Escherichia coli RNA polymerase. J Biol Chem 268:25369–25375. [PubMed] [Google Scholar]
- 43.Strathern J, Malagon F, Irvin J, Gotte D, Shafer B, Kireeva M, Lubkowska L, Jin DJ, Kashlev M. 2013. The fidelity of transcription: RPB1 (RPO21) mutations that increase transcriptional slippage in S. cerevisiae. J Biol Chem 288:2689–2699. doi: 10.1074/jbc.M112.429506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu X, Martin CT. 2009. Transcription elongation complex stability: the topological lock. J Biol Chem 284:36262–36270. doi: 10.1074/jbc.M109.056820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu M, Dai X. 2018. On the intrinsic constraint of bacterial growth rate: M. tuberculosis's view of the protein translation capacity. Crit Rev Microbiol 2018:1–10. doi: 10.1080/1040841X.2018.1425672. [DOI] [PubMed] [Google Scholar]
- 46.Bar-Nahum G, Epshtein V, Ruckenstein AE, Rafikov R, Mustaev A, Nudler E. 2005. A ratchet mechanism of transcription elongation and its control. Cell 120:183–193. doi: 10.1016/j.cell.2004.11.045. [DOI] [PubMed] [Google Scholar]
- 47.Kireeva ML, Nedialkov YA, Cremona GH, Purtov YA, Lubkowska L, Malagon F, Burton ZF, Strathern JN, Kashlev M. 2008. Transient reversal of RNA polymerase II active site closing controls fidelity of transcription elongation. Mol Cell 30:557–566. doi: 10.1016/j.molcel.2008.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nudler E, Mustaev A, Lukhtanov E, Goldfarb A. 1997. The RNA-DNA hybrid maintains the register of transcription by preventing backtracking of RNA polymerase. Cell 89:33–41. doi: 10.1016/S0092-8674(00)80180-4. [DOI] [PubMed] [Google Scholar]
- 49.Sosunov V, Sosunova E, Mustaev A, Bass I, Nikiforov V, Goldfarb A. 2003. Unified two-metal mechanism of RNA synthesis and degradation by RNA polymerase. EMBO J 22:2234–2244. doi: 10.1093/emboj/cdg193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mitra A, Angamuthu K, Nagaraja V. 2008. Genome-wide analysis of the intrinsic terminators of transcription across the genus Mycobacterium. Tuberculosis 88:566–575. doi: 10.1016/j.tube.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 51.Hubin EA, Lilic M, Darst SA, Campbell EA. 2017. Structural insights into the mycobacteria transcription initiation complex from analysis of X-ray crystal structures. Nat Commun 8:16072. doi: 10.1038/ncomms16072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.